
Abstract
Cats are unable to synthesise vitamin D3 (cholecalciferol) from sunlight and are, therefore, totally dependent on obtaining it from their diet. Under normal conditions, vitamin D3 is absorbed intact from the intestine in association with dietary fat. It is transported via chylomicrons to the liver, in association with vitamin D binding protein (VDBP), where it is hydroxylated to produce 25-hydroxycholecalciferol (25[HO]D3). This molecule is transported to the kidney where it undergoes a second hydroxylation in the mitochondria of the cells of the proximal convoluted tubules. This enzymatic step is tightly controlled (see below) and results in the formation of the active metabolite, 1,25(HO)D3 (calcitriol), which acts to increase levels of calcium and phosphate in the serum via its actions on the intestines, kidneys and bone. 1,2
In the intestine, calcitriol binds to vitamin D receptors in the cytoplasm of enterocytes, increasing calcium and phosphate transport from gut to serum. In the kidneys, calcitriol increases tubular resorption of calcium and phosphorus from the glomerular filtrate, while in bone it maintains levels of calcium and phosphorus sufficient to mineralise newly formed osteoid. 1–3 Calcitriol also regulates the production of some bone proteins produced by osteoblasts. 4
The synthesis of 1,25(HO)D3 is tightly controlled by the hormone itself, parathyroid hormone (PTH) and via regulation of the 1-α-hydroxylase and 24-hydroxylase enzymes. This maintains the 1,25(HO)D3 concentration in serum within a very tight range. Physiological levels of 25(HO)D3 are 500–1000 times those of 1,25(HO)D3 on a molar basis — and, as a consequence, the former acts as the storage form of vitamin D within the body. 1
Vitamin D deficiency results in the clinical syndrome of rickets in animals with an actively growing skeleton (eg, kittens and pups). The decreased concentrations of calcium and phosphate in extracellular fluid are so marked that new osteoid laid down by osteoblasts is not mineralised, resulting in characteristic clinical and radiographic rachitic features. Secondary hyperparathyroidism inevitably accompanies rickets, as a consequence of the reduced ionised calcium concentration in the serum.
Rickets can be produced by feeding a diet deficient in both vitamin D3 and calcium under extreme clinical or experimental conditions. 5–7 However, nutritional secondary hyperparathyroidism is by far the most common nutritional bone disease in small animals (including kittens), because even ‘all-meat’ diets contain sufficient vitamin D3 to prevent rickets. 3,5
Rickets unrelated to dietary deficiency has been recorded in cats, due to inborn errors in vitamin D3 metabolism. 8–11 Historically, cases have divided into (i) vitamin D-dependent type 1, caused by a defect in the gene encoding the enzyme 25-hydroxy-vitamin D3-1-α-hydroxylase (CYP27B1), 11 and (ii) vitamin D-dependent type 2, caused by a defect in the receptor gene (VDDR11) for the active 1,25(HO)D3 metabolite. 8–10 In reported type 1 cases, levels of 25(HO)D3 are in the reference interval (RI), but failure of the hydroxylation step in the renal tubules leads to low levels of 1,25(HO)D3 in serum. In type 2 cases, levels of 25(HO)D3 are also normal, but levels of 1,25(HO)D3 are increased due to ‘receptor resistance’.
We report for the first time a case of rickets in one of a litter of four Cornish Rex kittens associated with very low levels of both 25(HO)D3 and 1,25(HO)D3. The kitten's three littermates were clinically unaffected, and the whole litter had been fed a nutritionally complete commercial diet replete in vitamin D, calcium and phosphate. The kitten with rickets responded promptly to replacement calcitriol therapy.
Clinical report
History and physical findings
A 3-month-old female Cornish Rex kitten was presented for routine neutering. On questioning of the owner, it was reported to have a bunny-hopping gait, was showing reluctance to climb stairs and it strained excessively during defecation. Despite eating well and gaining weight, the kitten was smaller in stature and less active than its three siblings, which ambulated and interacted normally.
The litter of kittens had been bred and reared by the owner. Since weaning, they had been fed a nutritionally complete commercial dry diet (Hill's Feline Growth) and a commercial ‘wet’ food (Whiskas Kitten; Uncle Ben's).
Physical examination revealed bilaterally symmetrical bony swellings of the carpi (Fig 1), stifles and tarsi. Deep palpation of these regions was resented. Flexion and extension of the coxofemoral joints was likewise resented. The kitten was small (1.3 kg), but in good condition (body condition score 5/9). Although it was strongly ambulatory, it had a hunched, stilted gait. The clinical examination was otherwise unremarkable.

(a) Antebrachia of the affected kitten. (b) Close-up image showing prominent swelling in the carpal region (arrow)
Laboratory investigations
Blood was taken for routine biochemical analysis, with samples submitted from one of the unaffected siblings for the purposes of comparison (Table 1).
Results of routine serum biochemical analysis

Intervals marked with an asterisk are exceptions, and derived from Tomsa et al (1999) data from seven healthy control kittens 4–6 months old 3
RI = reference interval. Substantially abnormal values given in bold
Alkaline phosphatase activity was increased compared with the laboratory RI and the value for the unaffected sibling (522 U/l, RI <192 U/l); this was considered more likely to be of bone than hepatic origin. The affected kitten had hypophosphataemia, while its ‘normal’ littermate was slightly hyperphosphataemic. While the total serum calcium concentration of the affected kitten was within the veterinary laboratory's RI for adult cats, it was reduced compared with both the ‘control’ kitten and values determined for kittens of a similar age (Table 1). 3 Taking into account published values for total calcium and inorganic phosphate in growing kittens, the affected kitten was thus hypocalcaemic and hypophosphataemic.
Radiology
The kitten was premedicated with acepromazine (0.1 mg) and buprenorphine (0.015 mg) injected subcutaneously, and anaesthesia was induced using isoflurane (3%) in 100% oxygen via a close-fitting face mask. After intubation, the patient was maintained using isoflurane (1.5–2%) while radiographs were taken of the axial skeleton, skull and limbs.
Radiographs demonstrated marked overall osteopenia and reduced thickness of long bone cortices (Fig 2a and b). All long bone physes were markedly widened and flared, yielding a distinctive cup shape with mushroomed metaphyses. Spinal and skull radiographs were unremarkable. Radiographs of the unaffected kitten were taken for comparison (Fig 2c). None of the abnormalities noted in the affected kitten were evident in its sibling.

(a,b) Lateral and craniocaudal radiographs of the affected kitten's distal limb. Note the widened growth plates (white arrowheads) and mushroomed metaphyses (black arrowheads) of the distal radius and ulna. (c) A craniocaudal radiograph of a normal littermate is presented for comparison; note the greater overall bone density and the narrower physes
Presumptive diagnosis and confirmation
The physical, laboratory and radiographic findings were strongly suggestive of vitamin D deficiency (ie, rickets). To confirm this presumptive diagnosis and to try to determine its underlying biochemical basis, blood specimens from the affected kitten and a normal littermate were collected anaerobically, kept on ice and rapidly transferred to a human endocrinology reference laboratory for evaluation of ionised calcium, PTH and 25(HO)D3 and 1,25(HO)D3 concentrations. PTH concentrations were determined using an in-house, two-site immunoradiometric assay, while vitamin D analogues were analysed using standard radioimmunoassays.
Meloxicam (0.05 mg/kg PO daily) was administered to provide analgesia pending laboratory results. In addition, the client was advised to severely restrict the activity of the kitten to minimise the risk of pathological fractures developing.
The reference laboratory demonstrated reduced concentrations of ionised calcium, 25(HO)D3 and 1,25(HO)D3 in the affected kitten, in concert with an increased PTH concentration, consistent with a diagnosis of rickets and secondary hyperparathyroidism (Table 2). While both kittens had reduced 25(HO)D3 concentrations compared with the RI used for human patients, the affected kitten's levels were markedly lower than those of its sibling.
Endocrine data prior to, during and after therapy

Tomsa et al (1999) data from seven healthy control kittens 4–6 months old 3
Endocrine Laboratory, Royal North Shore Hospital
Determined on two separate occasions
RI = reference interval. Substantially abnormal values given in bold. NA = not assessed
Therapy
On the basis of the physical, radiological, biochemical and endocrinological findings, a confident diagnosis of rickets was made, and treatment instituted with 1,25(HO)D3 (calcitriol; 30 ng PO q24h with food).
There was a positive and unequivocal response to therapy. The owner reported rapid improvement in the kitten's activity level and mobility. It was soon able to move freely up and down stairs and no longer experienced problems defecating. Examination 4 weeks after commencing treatment revealed substantial growth of the kitten (from 1.3 to 2.3 kg). Swollen joint regions were less prominent, and flexion and extension did not elicit any pain. The remainder of the clinical examination was unremarkable.
Repeat radiographs revealed an overall increase in bone density, thickening of long bone cortices, progressive mineralisation of the radiolucent portions of the widened growth plates and a reduction in the metaphyseal cupping (Fig 3).

Craniocaudal radiographs of the affected kitten's distal limb before and during calcitriol therapy. Note how the diffuse osteopenia observed prior to therapy is not evident in the images taken during therapy; the bone cortices have also become much denser. The irregular widened growth plates, so prominent prior to therapy, have become progressively ‘filled in’ during therapy, while the mushroomed metaphyseal regions are gradually remodelled into a more normal appearance
The response was further monitored by serial determinations of ionised calcium concentration, and levels of 25(HO)D3 and PTH during therapy (Table 2). All analytes measured had improved, with the ionised calcium concentration at 4 weeks (and 8 weeks) being near-normal (and normal). Interestingly, the 25(HO)D3 at 4 weeks had increased substantially, and by 8 weeks was within the RI. The increase in 25(HO)D3 (from <10 to 40.7 nmol/l at 4 weeks and 132 nmol/l at 8 weeks) as a result of treatment was unexpected, as replacement therapy consisted of a 30 ng daily oral dose of its metabolite 1,25(HO)D3. The PTH concentration had normalised at 4 weeks, and was subnormal at 8 weeks.
The owner reported continued improvement, with the kitten behaving and ambulating normally, comparable to its littermates. At 8 weeks there was minimal bony swelling associated with joints, with no pain during manipulation through the full range of movement in any of the joints examined. Repeat radiographs (Fig 3c) revealed normal bone density, resolution of metaphyseal cupping and virtually normal physes. The ionised calcium concentration in serum remained comparable with that of the control littermate and within the RI for kittens, 3 25(HO)D3 concentrations had normalised, while the PTH level was low, suggesting perhaps supraphysiological concentrations of calcitriol due to therapy. Calcitriol supplementation was continued at the same absolute dose (in other words, at a reduced dose on a mg/kg basis as the kitten subsequently grew).
Follow-up
The kitten was rehomed interstate. Follow-up phone calls were made with the new owner and the attending veterinarian. When the kitten was 9 months old, no abnormalities were detected during physical examination and it appeared to have developed into a normal adult cat. The vet reported a single episode of forelimb lameness, possibly related to jumping from a height, but this was mild and did not require treatment; radiographs were not taken at that time.
Calcitriol supplementation was continued at 30 ng PO q24h until the cat was 12 months of age (in April 2010), when the drug was discontinued. Follow-up ionised calcium and 25(HO)D3 concentrations were determined a month later, at which point the ionised calcium was within the normal range and, interestingly, the 25(OH)D3 was high! It was suspected that this was a consequence of the kitten being fed a diet known to be high in vitamin D3 (Hill's Feline Growth). The kitten was temporarily changed onto an all-meat diet to ensure no exogenous vitamin D had been added to the ration, as is the case with commercially formulated rations. The ionised calcium and 25(OH)D3 levels were reassessed 1 month later, when the 25(OH)D3 levels were within the RI (Table 2).
It is anticipated that the cat will require no further therapy in relation to vitamin D and calcium homeostasis during its life, although we plan to measure appropriate analytes on a regular basis (Fig 4).

The cat at 22 months old (February 2011). Note the normally proportioned limbs, with no evidence of residual deformity
Discussion
There is no doubt that the affected kitten had rickets, as the pathognomonic clinical, radiological, biochemical and hormonal changes were all present, and there was a prompt and complete response to replacement therapy using calcitriol. It was fortunate that the diagnosis was made early, before irreparable damage (pathological fractures, angular limb deformity, pelvic collapse, spinal curvature) had been done to the rapidly growing skeleton of the kitten.
Calcitriol was selected over cholecalciferol for replacement therapy because dose rates for this vitamin D analogue were more freely available, and in case there was a defect in formation of this active hormone (as empiric therapy was started before the full results of the vitamin D assays were available). The dose of calcitriol was determined after referring to doses used previously in kittens and cats. A total oral daily dose of 30 ng proved completely effective in reversing all detectable biochemical derangements, without any undesirable sequelae such as hypercalcaemia, hyperphosphataemia and metastatic calcification of tissues.
A nutritional cause for rickets can be excluded on the basis of the patient's diet, and the absence of clinical features in its three littermates fed the same diet. Growing kittens fed a diet with adequate cholecalciferol (3.1 μg/kg) require less than 6 g calcium per kg diet on a dry matter basis. 7 The affected kitten and its littermates were fed Hill's Feline Growth (a kibble type ration designed for kittens) and a commercial ‘wet’ kitten food. This dry food contains 1.22% calcium (12.2 g Ca/kg) and 1246 IU/kg cholecalciferol (31 μg/kg; ie, 10 times the minimum requirement), while the calcium to phosphate ratio of both diets was in the vicinity of 1.2:1. The fact that this diet provides more than adequate calcium and cholecalciferol levels, and that the littermates were clinically unaffected (with normal or near-normal concentrations of vitamin D analogues, calcium, phosphate and PTH), taken together, demonstrates diet was not a factor in this case of rickets.
Interestingly, 25(HO)D3, the storage form of vitamin D3, was decreased as well as the active metabolite 1,25(HO)D3. Considering the diet had adequate vitamin D3, the most plausible explanations for this observation would be:
Inadequate intestinal absorption of vitamin D3;
A defect in transfer of vitamin D3 to the liver (perhaps involving defective or reduced concentrations of VDBP); or
Insufficient hepatic hydroxylation of vitamin D, resulting in decreased formation of 25(HO)D3 and, as a consequence, 1,25(HO)D3.
As the kitten produced normal stools (without steatorrhoea) and had no other evidence of intestinal dysfunction, vitamin D3 malabsorption would seem unlikely. Traditional thinking would suggest that defective hydroxylation would also be unlikely, as there are at least six cytochrome P450 enzymes in the liver possessing 25-hydroxylase activity in humans, 12 of which two (CYP27A1 and CYP2R1) are conserved among species known to have an active vitamin D signalling pathway. 13 Thus, a genetic defect affecting the activity of only a single enzyme would theoretically not cause the observed reduction in 25(HO)D3 concentrations, given adequate dietary intake of vitamin D3. That said, a mutation in the CYP2R1 gene has been reported in a Nigerian child with rickets, 13 with reduced concentrations of 25(HO)D3 but normal concentrations of 1,25(HO)D3.
Although the findings in the kitten of the present report are somewhat consistent with a CYP2R1 defect, it is unclear why the 25(HO)D3 concentration would normalise with calcitriol therapy if this was the underlying genetic defect. Perhaps the problem in the kitten was actually a delayed maturation of enzymes in the liver possessing 25-hydroxylase activity, or temporarily low levels of VDBP, with calcitriol therapy providing immediate relief of the clinical signs while the postulated delay in these biochemical pathways corrected itself spontaneously. Alternatively, it is possible that the low levels of 25(HO)D3 may have resulted from overactive hepatic inactivation of absorbed dietary vitamin D (D Fraser, personal observation).
The increasing concentrations of 25(HO)D3 observed over the first 8 weeks after starting treatment suggest, with the benefit of hindsight, that a transient defect had self-corrected. Interestingly, 1 month after stopping calcitriol therapy, the patient had elevated 25(HO)D3 levels. At this time the kitten was being fed a diet containing 10 times the minimum requirement of vitamin D. Morris et al (1999) 7 noted a linear relationship between dietary cholecalciferol and plasma 25(HO)D3. When the kitten was temporarily transferred onto an all-meat diet (without exogenous vitamin D supplementation) and retested 1 month later, the 25(OH)D3 levels had normalised. Indeed, it appeared all abnormalities had self-corrected, suggesting the kitten would not require ongoing therapy.
Our kitten responded promptly to treatment with calcitriol and, at the time of writing, continues to progress well. While the exact mechanism of the vitamin D3 deficiency was not completely characterised, this case represents a novel form of rickets without precedence in the literature (Table 3). Presumably this kitten suffered from some transient inborn error of vitamin D metabolism, which may have arisen de novo during gestation, or been the result of a genetic defect inherited from its dam and/or sire. The 25(HO)D3 concentrations in the one littermate tested were somewhat low (in association with an increased serum PTH concentration and a marginal ionised calcium concentration), raising the possibility that this kitten may have been affected subclinically by the same problem as its rachitic littermate.
Laboratory findings in cats with rickets unrelated to nutritional deficiency as kittens
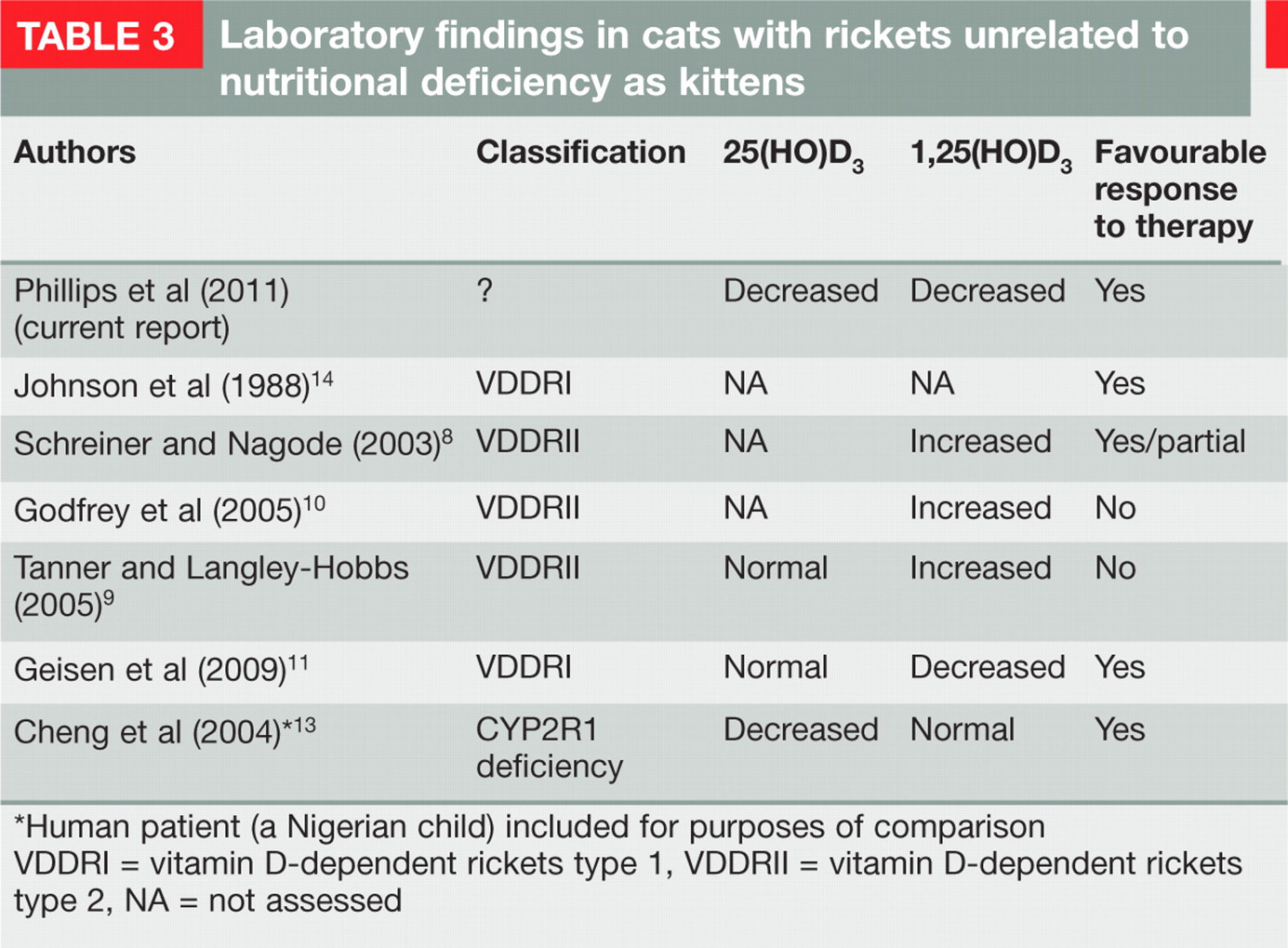
Human patient (a Nigerian child) included for purposes of comparison
VDDRI = vitamin D-dependent rickets type 1, VDDRII = vitamin D-dependent rickets type 2, NA = not assessed
The value of examining a full ‘calcium profile’ (ie, total and ionised calcium, phosphate, PTH, 25[HO]D3 and 1,25[HO]D3) via a human or veterinary reference laboratory is emphasised by this case, and a favourable prognosis in some kittens with rickets makes such investigations worthwhile. Even when financial limitations make detailed investigations impossible, trial therapy using a nutritionally complete diet and physiological doses of calcitriol or cholecalciferol is inexpensive and can result in a very satisfactory clinical response.
Footnotes
Acknowledgements
Richard Malik is supported by the Valentine Charlton Bequest of the Centre for Veterinary Education of the University of Sydney. Sharon Barton, the breeder of the affected cat, is thanked for her enthusiasm and cooperation. The subsequent owner of the cat and the attending veterinarian were generous with their time, allowing important follow-up blood tests and radiological studies.