
Abstract
CX3CR1 (fractalkine receptor) is important for sustaining normal microglial activity in the brain. Lack of CX3CR1 reportedly results in neurotoxic microglial phenotype in disease models. The objective of this study was to test the hypothesis that the absence of CX3CR1 worsens the outcome in cerebral ischemia. We observed significantly smaller (56%) infarcts and blood—brain barrier damage in CX3CR1-deficient (CX3CR1−/−) animals compared with CX3CR1 +/− and wild-type mice after transient occlusion of the middle cerebral artery (MCAo). Functional recovery of CX3CR1−/−animals was enhanced, while less number of apoptotic cells and infiltrating leukocytes were found in the ipsilateral hemisphere. Expression of IL-1β mRNA, protein, and interleukin (IL)-1Ra and tumor necrosis factor (TNF)-α mRNAs was lower in CX3CR1−/− mice, whereas no difference was observed in the number of IL-1β-expressing microglia or plasma IL-1β concentration. We observed early IL-1β expression in astrocytes in vivo after MCAo and after oxygen—glucose deprivation in vitro, which might contribute to the ischemic damage. Our findings indicate that lack of CX3CR1 does not result in microglial neurotoxicity after MCAo, but rather significantly reduces ischemic damage and inflammation. Reduced IL-1β and TNFα expression as well as decreased leukocyte infiltration might be involved in the development of smaller infarcts in CX3CR1−/− animals.
Introduction
The pathophysiology of ischemic stroke includes a sequence of inflammatory events that involves several central and peripheral cell types as well as large number of inflammatory molecules (McColl et al, 2007; Muir et al, 2007). Microglia are among the first candidates to contribute to ischemic damage, as these cells display very early activation in stroke and are capable of expressing potentially harmful molecules, such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and reactive oxygen species (Chong et al, 2005; Zheng and Yenari, 2004). Although the processes of microglial activation, proliferation, cytokine production, and phagocytosis have been studied extensively in animal models of cerebral ischemia, the exact function of these cells is not fully understood and several controversial findings have been reported. For example, inhibition of microglial activation by minocycline was shown to result in better outcome (Yenari et al, 2006; Yrjanheikki et al, 1998), whereas selective ablation of proliferating microglia worsened the outcome (Lalancette-Hebert et al, 2007) after experimental stroke. This might indicate that the large complexity of central ischemic processes as well as involvement of peripheral inflammatory events in stroke makes it difficult to study the exact microglial function in vivo.
CX3CR1 is the only known receptor of fractalkine (CX3CL1), and has been implicated in several inflammatory processes in the periphery, such as recruitment, adhesion, and vascular transmigration of leukocytes (Imai et al, 1997). In the brain, neurons constitutively express high levels of the chemokine CX3CL1, which has been suggested to be responsible for sustaining normal microglial activity through interaction with microglial CX3CR1 (Biber et al, 2007; Hanisch and Kettenmann, 2007). It was recently shown that CX3CR1 deficiency dysregulates microglial responses, resulting in microglial neurotoxicity in vivo in three different disease models using transgenic mice (Cardona et al, 2006), in which the cx3cr1 gene was replaced by a green fluorescent protein (GFP) reporter gene (Jung et al, 2000). Unlike peritoneal macrophages, microglia isolated from homozygote (CX3CR1−/−) mice expressed significantly more IL-1β in response to activation by lipopolysaccharides (LPS) than microglia derived from heterozygote (CX3CR1 +/−) mice (Cardona et al, 2006). Interleukin-1β was shown to contribute to the formation of ischemic damage through various mechanisms (Touzani et al, 1999, 2002) and microglia are considered to be its primary source in the brain (Gosselin and Rivest, 2007). Therefore, we hypothesized that lack of CX3CR1 might result in increased brain damage and cell death in cerebral ischemia. To this end, we subjected CX3CR1−/−, CX3CR1 +/− mice and their wild-type (WT) C57B16 littermates to transient middle cerebral artery occlusion (MCAo), to reveal the role of CX3CR1 deficiency in ischemic damage, neuronal apoptosis, and inflammation.
Materials and methods
Animals
Experiments were performed in adult male CX3CR1+/GFP, CX3CR1GFP/GFP, and C57B1/6H mice aged 12 to 16 weeks, weighing 26 to 28 g (n = 58). CX3CR1/GFP mice were obtained from the European Mouse Mutant Archive (EMMA), backcrossed for more than 10 generations to C57B1/6 (Jung et al, 2000). In these mice, the cx3cr1 gene was replaced by a GFP reporter gene such that heterozygote CX3CR1+/GFP (CX3CR1+/−) mice express GFP in cells that retain receptor function, whereas cells in homozygote CX3CR1GFP/GFP (CX3CR1−/−, knockout (KO)) mice are labeled with GFP and also lack CX3CR1. Animals had free access to food and water and were maintained under temperature, humidity, and light-controlled conditions (21°C ± 1°C, 65% humidity, 12-h light/12-h dark cycle, with lights on at 0700 hours). All procedures were conducted in accordance with the guidelines set by the European Communities Council Directive (86/609 EEC) and approved by the Institutional Animal Care and Use Committee of the Institute of Experimental Medicine.
Transient Middle Cerebral Artery Occlusion
Anesthesia was induced with 2% halothane and maintained in 1% halothane-air mixture. During surgery, the core temperature was monitored with a rectal probe and maintained at 37 ± 0.5°C, using a homeothermic blanket. Animals were exposed to transient MCAo for 60 mins, as described previously (Wheeler et al, 2003). Briefly, a nylon filament (tip diameter 180 μm) was introduced into the origin of the external carotid artery and advanced through the internal carotid artery to occlude the MCA. A total of 60 mins later, reperfusion was induced and animals were allowed to recover. Mice were subcutaneously injected with 1 mL of sterile saline daily and continuously monitored for neurologic symptoms. To investigate that heterozygote mice maintain CX3CR1 receptor function completely, they were compared with C57B1/6 mice (having two copies of the CX3CR1 gene) after MCAo. The volume of ischemic damage and blood—brain barrier (BBB) breakdown was not different at any reperfusion intervals.
After 4, 24, or 72 h of reperfusion, mice were anaesthetized and perfused transcardially with 10 mL saline followed by 40 mL 4% paraformaldehyde (pH = 7.4). After cryoprotection of brains in 20% sucrose-KPBS for 24 h, five alternate sets of 20 μm coronal brain sections were cut on a sliding microtome. All sections were collected in an antifreeze solution (30% ethylene glycol and 20% glycerol in phosphate-buffered saline) and stored at −20°C until processing.
Measurement of Infarct Volume
The volume of ischemic damage was measured using a modification of a method described previously (McColl et al, 2004). Briefly, areas of ischemic damage were identified on cresyl-violet-stained sections at eight neuro-anatomically defined coronal levels. Digitized images were created and the areas of damage measured using ImageJ software (NIH, Bethesda, MD, USA). The volume of damage was calculated by integration of areas of damage with the distance between coronal levels. The end points for integration were 2.9 mm (rostral limit) and −4.9 mm (caudal limit) regarding bregma. Volumes are expressed as a percentage of the total hemispheric volume.
Evaluation of Blood—Brain Barrier Permeability
The BBB damage was determined using a modified protocol of immunostaining for IgG (Richmon et al, 1998). Free floating sections were rinsed in 1% H2O2 for 10 mins to quench endogenous peroxidase activity, washed 3 × in KPBS, and blocked for 1 h in KPBS containing 0.3% Triton-X-100, 2% bovine serum albumin, and 5% normal horse serum (Vector Laboratories, Burlingame, CA, USA). After overnight incubation in biotinylated horse anti-mouse IgG (1:500; Vector Laboratories) at 4°C, the sections were rinsed in KPBS and incubated in avidin—biotin horseradish peroxidase complex (ABC; Vector Laboratories) for 1 h. Staining was visualized with 3,3-diaminobenzidine tetrachloride. The volume of BBB damage was calculated as described above.
Immunofluorescence and Immunoperoxidase Labeling
Double- or triple-labeling immunofluorescence was performed on free-floating brain sections or on astroglial cell cultures. After blocking in 2% normal donkey or goat sera (Vector Laboratories), the sections were incubated overnight at 4°C in various mixtures of the following primary antibodies: monoclonal mouse anti-GFP (1:1,000; Molecular Probes, Eugene, OR, USA); polyclonal rabbit anti-Ibal (1:1,000; Wako Chemicals, Neuss, Germany); monoclonal rat anti-mouse CD45 (1:250; Serotec, Oxford, UK); monoclonal rat anti-mouse NIMP-R14 (1:100; Hycult Biotechnology, Uden, The Netherlands); monoclonal mouse anti-GFAP (1:1,000; Sigma, Oxford, UK); polyclonal rabbit anti-glutamine synthetase (1:1,000; Sigma); polyclonal sheep anti-IL-1β (1:500; affinity purified; NIBSC); polyclonal goat anti-IL-1β (1:500; R&D Systems, Minneapolis, MN, USA); and polyclonal rabbit-anti Annexin V (1:500; Novus Biologicals, Littleton, CO, USA). The antigens were visualized with appropriate fluorochrome-conjugated secondary antisera (donkey anti-mouse Alexa 488, donkey anti-rabbit Alexa 488, donkey anti-mouse Alexa 594, donkey anti-rabbit Alexa 594, donkey anti-sheep Alexa 594, or donkey anti-goat Alexa 594; Molecular Probes) used in 1:500 dilutions for 2 h at room temperature. In some cases, immunostaining was visualized with an adequate biotinylated secondary antibody, followed by streptavidine-Alexa 350 or 594 conjugates. Microglial cells were also labeled with biotinylated tomato lectin (10 μg/mL) and streptavidine-Alexa 594. Some sections were counterstained with 2 μg/mL of diamidinophenylindole (Molecular Probes) for 1 min. Apoptotic cells were identified by using an affinity-purified antibody to detect endogenous Annexin V, which binds to phosphatidyl serine residues on the cell surface. The ratio of apoptotic neurons was evaluated by NeuN/Annexin V immunofluorescence as described by Cardona et al (2006). Sections were mounted onto gelatin-coated slides and coverslipped with Vectashield mounting medium (Vector Laboratories). Goat anti-IL-1β (gIL-1β) staining was also visualized by biotinylated rabbit anti-goat antisera and developed with 3,3-diaminobenzidine tetrachloride-nickel, with or without preincubation of gIL-1β with 180 ng/mL recombinant mouse IL-1β (R&D Systems).
Images were viewed using a Nikon Eclipse 6000 microscope equipped with Spot RTcolor digital camera (Diagnostic Instruments Inc., IL, USA). Confocal images were captured using Olympus BX-61 microscope. Double or triple-fluorescent images were generated using SPOT Advanced software and Adobe Photoshop. Quantification of fluorescent images and 3D reconstruction of confocal images were performed using the ImageJ software (NIH).
In Situ Hybridization Histochemistry
To monitor IL-1β mRNA, a riboprobe complementary to 373 to 940 nucleotides of the mouse IL-1β gene was transcribed from plasmid in the presence of 35S-UTP. Hybridization and autoradiographic techniques were modified following the method described by Simmons et al (1989). Tissue sections were mounted onto SuperFrost Ultra Plus (Menzer-Glazer) slides post-fixed with 4% paraformaldehyde, digested with Proteinase K (10 in 50 mmol/L Tris, pH = 8 and 5 mmol/L EDTA at 37°C, 5 mins), acetylated (0.25% acetic anhydride in 0.1 mol/L triethanolamine, pH = 8), and dehydrated. Hybridization mixture (50% formamide, 0.3 mol/L NaCl, 10 mmol/L Tris (pH = 8), 2 mmol/L EDTA, 1 × Denhardt's, 10% dextran sulfate, 0.5 mg/mL yeast tRNA) was pipetted onto the slides (100 μL, containing probe at 107d.p.m./mL) and hybridized overnight at 56°C. Sections were then rinsed in 4x SSC (1 × SSC: 0.15 mol/L NaCl and 15 mmol/L trisodiumcitrate buffer, pH = 7), digested with ribonuclease A (20 mg/mL in Tris-EDTA buffer with 0.5 mol/L NaCl at 37°C for 30 mins), gradually desalted, and washed in 0.1 × SSC at 65 to 75°C for 30 mins. Slides were dipped in NTB nuclear emulsion (Kodak) and exposed for 2 weeks, developed in D-19 developer, and lightly counterstained with cresyl violet.
Adhesive Removal Test
The motor response to sensory stimuli was measured with a stimulation test developed for rats (Marshall and Gotthelf, 1979; Schallert et al, 1983) and adopted for mice (Fleming et al, 2004). Small adhesive stimuli (50 mm2) were placed on the snout of the mouse in their home cages. The behavior was video-recorded and later analyzed by an observer blind to the treatment schedules. The following behavioral variables were assessed: the time to remove the stimulus and the number of attempts for tape removal. To remove the stimulus, animals would raise both forelimbs toward their face and swipe off the stimulus with both forepaws. Typically, both intact WT, CX3CR1+/−, and CX3CR1−/− mice make contact and remove the stimulus within 15 secs, without differences according to genotype. Each animal (n = 4/group) was tested 24, 48, and 72 h after MCAo. If the animal did not remove the stimulus within 60 secs, the experimenter removed it and the trial for the next mouse was initiated. On each experimental day, the average performance of three trials was calculated and used for later statistical analysis. The intertrial interval differed between 12 and 15 mins for the experimental subjects.
Sample Preparation and IL-1β ELISA
Blood samples were routinely collected from the right heart ventricle before transcardial perfusion (4, 24, and 72 h after MCAo) in the presence of K-EDTA (10 μL of 20% K-EDTA/1 mL blood). Samples were centrifuged at 8,000 r.p.m., aliquoted, and stored at −20°C.
Brain homogenates were prepared from CX3CR1−/−and +/− mice (n = 5/group) 24 h after 60 mins MCAo. One mouse among the CX3CR1+/− mice was excluded from further analysis because of surgical artifact. Animals were anaesthetized and the chest was opened. After blood sampling from the right ventricle, the brain was removed and immediately frozen in dry ice. The brainstem and cerebellum were removed, and the forebrain was dissected into hemispheres. The cortex was separated around the corpus callosum. To preserve the whole striatum, majority of the hypothalamus and thalamus was removed from the rest of the hemisphere. Brain samples of 50 mg were homogenized in 300 μL of sterile phosphate-buffered saline (pH = 7.4) containing protease inhibitors (Complete Mini; Roche, Mannheim, Germany), sonicated for 30 secs on ice, and centrifuged at 13,000 r.p.m. Supernatants were stored at −70°C until ELISA or quantitative real-time PCR measurement.
The concentration of IL-1β peptide in plasma samples and brain homogenates was measured using mouse IL-1β/IL-1F2 DuoSet kit (DY401; R&D Systems) according to the manufacturer's recommendations. Samples were diluted 1:1 in Tris-buffered saline (20 mmol/L Trizma base, 150 mmol/L NaCl, pH = 7.4) containing 0.1% bovine serum albumin and 0.05% Tween 20.
Quantitative Real-Time PCR
Total RNA was isolated from homogenates (same samples as used for IL-1β ELISA) of the ipsilateral-contralateral striatum and cortex, using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and then converted to cDNA by high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) on ABI StepOne instrument according to the manufacturer's instructions. Primers used for the comparative CT experiments were designed by the Primer Express 3.0 program. Gene expression was analyzed by ABI StepOne program. Primer sequences for the following genes are
GAPDH:
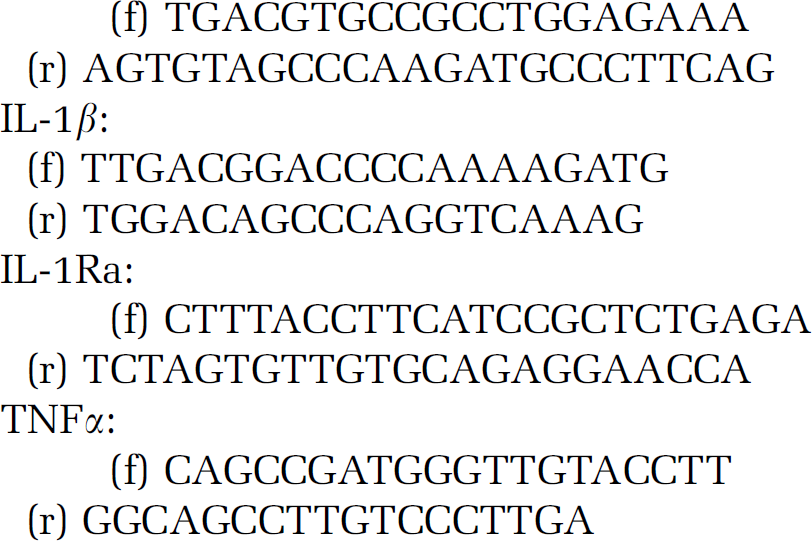
GAPDH message was used as internal control. Relative quantity of mRNAs was referred to corresponding brain sites of an intact CX3CR1−/− mouse. Melting curve analysis to confirm the identity of PCR products had been performed by using StepOne instrument's Software v.2.0 according to the instructions of the manufacturer.
Astroglial and Microglia-Enriched Cultures
Astrocytes were isolated from the forebrains of neonatal (P2) CD1 mice and transgenic mice expressing eGFP under the control of human GFAP promoter (Nolte et al, 2001) or in knockin constructs within the CX3CR1 gene (Jung et al, 2000). Meninges were removed and the tissue was minced with razor blades. The tissue pieces were subjected to enzymatic dissociation, using 0.05% w/v trypsin and 0.05% w/v DNase I (Sigma) for 10 mins at room temperature. The cells were plated onto poly-
Fluorescence-Activated Cell Sorting
Flow cytometry was undertaken using FACS Vantage flow cytometry analysis system (BD Biosciences). Single-cell suspensions were first gated on forward scatter, and, subsequently, within this population based on GFP expression. Non-GFP-expressing cells were used as negative control for background fluorescence. To obtain microglia-enriched cultures, GFP-positive microglia from CX3CR1−/− and +/− glial cultures were sorted by FACS 3 days before oxygen-glucose deprivation (OGD). To optimize survival of microglial cells after separation by FACS, cultures contained 7% to 10% of astrocytes.
To obtain pure astroglial cultures, we used two different approaches. In the first series of experiments, we used astrocytic cultures prepared from CX3CR1 +/− mice (Jung et al, 2000). The contaminating microglial (GFP-positive) cells were removed from the cultures by fluorescent cell sorting. In the second set of experiments, cultures were prepared from the forebrains of hGFAP-GFP mice (Nolte et al, 2001). Astrocytes expressing GFP were subjected to FACS before the experiments. In this way, we eliminated microglial cells in two different ways from the astocytic cultures. The sorted hGFAP-GFP-positive or CX3CR1-negative cells were replated onto poly-
Oxygen—Glucose Deprivation on Microglia-Enriched Cultures and on Cultured Astrocytes
Culture medium was replaced with sterile, deoxygenated HEPES (145 mmol/L NaCl, 3 mmol/L KCl, 1 mmol/L MgCl2, 2 mmol/L CaCl2, 10 mmol/L HEPES; 288 to 300 mOsm, pH = 7.2) without glucose, or with HEPES supplemented with 10 mmol/L glucose (control cultures). Astrocyte cultures were then placed into an airtight incubating chamber, flushed with a gas mixture of 94% N2, 5% CO2, and 1% O2 for 15 mins, and the chamber was closed and the cultures incubated for 3 h at 37°C. At the end of OGD, incubation medium (HEPES) was collected and the cultures were incubated in MEM. Supernatants from astrocyte cultures were collected 4 and 24 h after OGD and stored at −70°C. Astrocytes were harvested 4 and 24 h after hypoxia, centrifuged at 2,000 rpm, and placed in sterile phosphate-buffered saline (pH = 7.4) containing protease inhibitors (Complete Mini; Roche). Samples were then sonicated for 30 secs and stored at −70°C before IL-1β ELISA. Microglia-enriched cultures were harvested 4 h after 3 h of OGD and samples were treated as described above.
Statistical Analysis
Statistical analysis was performed using Student's t-test or one-way ANOVA (analysis of variance) followed by Bonferroni's post hoc test. Behavioral data were analyzed by repeated-measure ANOVA (the two factors were genotype and time). Where interaction between the two factors was observed, Fischer LSD post hoc comparisons were also run.
Results
Absence of CX3CR1 Results in Significantly Smaller Infarct Size, which is Associated with Smaller BBB Damage
Four hours after MCAo, mild pathologic changes were observed in the ipsilateral striatum of cresyl-violet-stained brain sections. Loss of staining intensity was obvious in the lateral globus pallidus in CX3CR1−/−, CX3CR1 +/−, and C57B16 (WT) animals. Blood—brain barrier damage identified by using immunostaining to mouse IgG was seen in the striatum as early as 4 h after MCAo, whereas one-third of CX3CR1 +/− and C57B16 mice displayed IgG labeling in some part of the ipsilateral cerebral cortex.
After 24 h of reperfusion, 23.1% ± 3.3% of the ipsilateral hemisphere displayed ischemic damage in CX3CR1−/− mice, compared with 36.0% ± 4.7% and 33.8% ± 5.9% in CX3CR1 +/− and WT animals, respectively (Figure 1A). The BBB breakdown at 24 h of reperfusion was smaller in CX3CR1−/− mice than that seen in CX3CR1 +/− mice (Figure 1B). Data were not significantly different at the 24-h time point using one-way ANOVA followed by Bonferroni's post hoc test.

CX3CR1−/− mice have smaller infarct size compared with mice expressing CX3CR1, which is associated with reduced BBB damage. (
A total of 72 h after MCAo, the volume of ischemic damage assessed by cresyl violet staining was significantly smaller in CX3CR1−/− mice than in mice expressing the fractalkine receptor (P < 0.05; 25.8% ± 4.0% in CX3CR1−/− mice versus 45.6% ± 2.2% in CX3CR1 +/− and 47.4% ± 2.9% in WT mice). In heterozygous and WT mice, 60% to 90% of the ipsilateral cerebral cortex in the territory of the MCA, as well as 50% to 70% of the ipsilateral hippocampus and thalamus, became ischemic by 72 h after MCAo, and sometimes the damage was also extended to the lateral hypothalamus (Figure 1C). In contrast, only two out of six CX3CR1−/− animals displayed ischemic damage in the hippocampus and thalamus, and the extent of cortical infarct was also significantly smaller. Corresponding to the ischemic damage, integrity of BBB was seriously affected in 50% to 90% of the ipsilateral cortex, thalamus, and hippocampus in all mice having functional CX3CR1, whereas in three out of six CX3CR1−/− animals, BBB damage was confined to the striatum and the hippocampus, the thalamus being only partially affected in the rest of the CX3CR1−/− animals (Figure 1D). A total of 72 h after MCAo, the volume of BBB damage in CX3CR1−/− mice was significantly smaller (28.0% ± 6.61%) compared with CX3CR1 +/−(54.5% ± 4.1%) and WT (45.7 ± 1.8) animals. To investigate if CX3CR1 deficiency had any direct effect on BBB disruption after MCAo, values of BBB damage were normalized to the infarct size. Values of BBB damage/ischemic damage ratio were between 0.88 and 1.19 in all groups, and no significant differences were observed among CX3CR1−/−, CX3CR1 +/−, and WT animals (1.00 ± 0.12, 1.07 ± 0.11, and 0.88 ± 0.06 after 24 h and 1.09 ± 0.1, 1.19 ± 0.21, and 0.96 ± 0.19 after 72 h of MCAo). These results indicate that fractalkine receptor deficiency had no direct effect on BBB disruption after MCAo.
CX3CR1−/− Mice Exhibit Lower Number of Apoptotic Cells in the Brain After MCAo, Compared with Animals Expressing CX3CR1
Annexin V immunohistochemistry was used to investigate whether marked CX3CR1-dependent differences seen in the size of ischemic damage and BBB breakdown are associated with different numbers of apoptotic cells in the brains of CX3CR1−/− and CX3CR1 +/− animals.
A total of 4 h after MCAo, only occasional Annexin V labeling was observed in the ipsilateral cerebral cortex in both genotypes and rarely in the striatum. Ependymal cells displayed Annexin V staining in the lateral and sometimes in the third ventricle.
A total of 24 h after MCAo, more Annexin V-positive cells were found in heterozygote mice than in CX3CR1−/− mice, except for the ischemic striatum, where the number of apoptotic cells was relatively low. Annexin V-positive cells appeared in the ischemic cortex (207 ± 134 cells/mm2 in CX3CR1−/−; 624 ± 443 cells/mm2 in CX3CR1 +/−mice) and several cells were observed in the nonischemic penumbral cortex as well as in the piriform area (Figures 2A and 2B). Marked differences were observed in the left hippocampus and the thalamus between the two genotypes (20 ± 33 versus 199 ± 127 cells/mm2 in the hippocampus; 57 ± 66 versus 564 ± 168 cells/mm2 in the thalamus of CX3CR1−/− and CX3CR1 +/− mice, respectively (P < 0.05)).

CX3CR1 +/− mice have more Annexin V-positive cells in the ipsilateral hemisphere after MCAo. (
At 72 h of reperfusion, the number of Annexin V-positive cells was much lower than that seen at 24 h (not shown). Late cellular death was observed mainly in the ipsilateral cerebral cortex and thalamus in heterozygote mice, whereas only two out of six CX3CR1−/− mice displayed Annexin V labeling in these areas.
To reveal the phenotype of apoptotic cells, Annexin V immunostaining was colocalized with tomato lectin, CD45, GFAP, GFP, and NeuN markers. Annexin V labeling was colocalized with NeuN in 40% to 70% of apoptotic cells. (Figure 2C). However, more intensive Annexin V-immunostaining was often accompanied with the reduction or complete loss of NeuN immunopositivity. Annexin V labeling was also seen in some microglial cells, astrocytes, and CD45-positive leukocytes and was often associated with tomato lectin-positive vascular elements (not shown).
In both CX3CR1−/− and heterozygote mice, microglia were consequently found attached to Annexin V-positive cells. High-resolution confocal microscopy revealed that microglia extended its processes toward apoptotic cells sometimes from a relatively large distance (Figure 2D; see Supplementary Video).
Adhesive Removal Test Indicates Better Recovery in KO Mice After MCAo
The number of attempts to remove the stimulus showed genotype-specific differences (Figure 3). CX3CR1−/− animals made a faster and more pronounced recovery after MCAo compared with heterozygote control ones. The overall difference between mice with different genetic background reached significance (Fgenotype(1,6) = 4.326, P < 0.083), the repeated-measure factor (Ftime(2,12) = 5.148, P < 0.03), and the interaction between the factors were significant (Finteraction(2,12) = 4.469, P < 0.04). With post hoc test, compared with the first postoperative day, when the number of attempts for tape removal was very low, CX3CR1−/− but not heterozygote animals showed a pronounced increase in the number of attempts, indicating a less-severe impairment and faster recovery after MCA manipulation in CX3CR1−/− animals.

The sensorimotor performance of mice with different CX3CR1 background after MCAo. The adhesive removal test was performed on postoperative days 1, 2, and 3. Both the adhesive removal latency (
The adhesive removal latencies tended to be lower in the CX3CR1−/− group compared with the heterozygote animals, but were not significant (Fgenotype(1,6) = 2.271, P < 0.18; Ftime(2,12) = 1.337, P < 0.30; Finteraction(2,12) = 1.522, P < 0.26). By the third postoperative day, half of the CX3CR1−/− animals were able to remove the adhesive tape compared with none in the heterozygote group.
GFP/CD45-Staining Reveals Less-Infiltrating Leukocytes in the Ipsilateral Hemisphere after MCAo in CX3CR1−/− Mice
The GFP-positive microglia in CX3CR1−/− and +/− mice were colocalized with Iba1 and tomato lectin in all animals examined, and the microglial expression of CD45 and CD34 markers increased in the ipsilateral hemisphere after MCAo. Astrocytes expressing GFAP or glutamine synthetase as well as neurons identified by NeuN expression were not positive to GFP in intact mice and in mice subjected to MCAo (not shown), confirming previous observations by others (Cardona et al, 2006).
To reveal infiltrating leukocytes in the brains of CX3CR1−/− and CX3CR1 +/− mice, CD45 immunostaining was used, which labels all leukocyte populations, including resident microglia of hematopoietic origin. CD45/GFP-positive microglia and macrophages are thereby discriminated from potentially infiltrating other leukocytes negative to CX3CR1 (GFP), which include mature neutrophils, eosinophils, T and B lymphocytes as well as most of the natural killer cells (Jung et al, 2000).
Microglia become widely activated in both genotypes after MCAo in the ipsilateral hemisphere. There was no significant difference in the phenotype and the number of GFP + /CD45 + microglia and macrophage cells between CX3CR1−/− and +/−animals in the ipsilateral hemisphere 24 h after MCAo. In contrast, the microglial cell number was significantly higher in the ipsilateral cerebral cortex of CX3CR1−/− animals (Figure 4A) than in that of heterozygote mice 72 h after MCAo (346 ± 58 versus 221 ± 57 cells/mm2, respectively; P < 0.05).
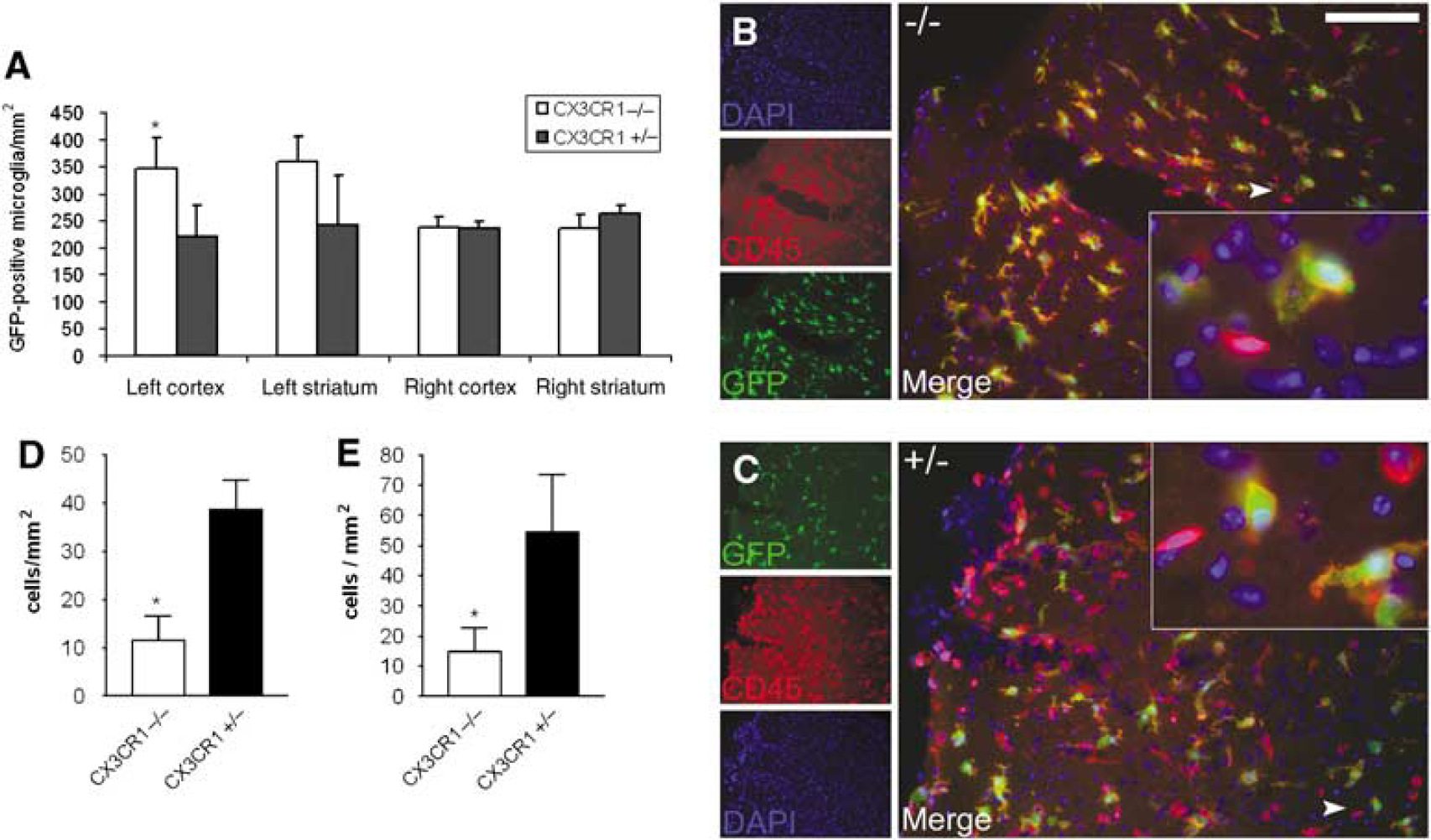
CX3CR1−/− mice have more microglia and less infiltrating CD45-positive leukocytes in the brain after MCAo. The figure shows the amount of GFP-positive microglia and CD45-positive/GFP-negative (possibly infiltrating) leukocytes in the ischemic hemisphere 72 h after MCAo. (
A total of 72 h after MCAo, both in ipsilateral cerebral cortex and striatum, CX3CR1−/− mice had significantly less GFP-/CD45 + leukocytes (Figures 4B–4E) than CX3CR1 +/− animals (11.6 ± 7.7 versus 38.6 ± 9.2 cells/mm2 in the cortex and 14.8 ± 12.3 versus 54.5 ± 25.6 cells/mm2 in the striatum, respectively; P < 0.05). About 50% to 70% of CD45 +/GFP leukocytes were neutrophils, expressing the NIMP marker (not shown).
IL-1β Production is Blunted in the Brain of CX3CR1−/−Mice after MCAo: a Possible Role for Astrocytes
IL-1β in plasma samples: Plasma IL-1β levels did not show any difference among CX3CR1−/−, CX3CR1 +/−, and C57B16 mice in either the 4, 24, or 72 h reperfusion intervals after MCAo. Only half of the animals (irrespective of genotype) had a detectable amount of IL-1β in the plasma samples after 4 and 24 h of reperfusion between 11.4 and 18.5 pg/mL and between 9.8 and 29.5 pg/mL, respectively. After 72 h of reperfusion, IL-1β was detectable only in 20% to 40% of the animals.
Microglial IL-1β production after MCAo: Using two different affinity-purified antibodies against IL-1β, we could not detect microglial cells expressing IL-1β in the brain 4 h after MCAo. A total of 24 h after ischemia, a relatively low number of IL-1β-positive microglia (not exceeding 15% of the total in either brain section) was detected in the ipsilateral cerebral cortex in both genotypes (Figures 5A, 5B, and 6A), and some of these cells were also seen in the ipsilateral thalamus and hippocampus in both genotypes. The average cell number was not significantly different between CX3CR1−/− and +/−mice (14.4 ± 14.2 versus 7.7 ± 10.1 cells/mm2 with sheep anti-IL-1β; 10.8 ± 8.1 versus 5 ± 7.3 cells/mm2 with goat anti-IL-1β antibody, respectively) in the cortex, and IL-1β-positive microglia were just occasionally observed in the ischemic striatum. In situ hybridization revealed IL-1β-expressing cells in the ipsilateral hemisphere at 24 h of reperfusion. In the ipsilateral cerebral cortex, the location and average number of cells containing elevated IL-1β mRNA (Figure 6B) corresponded to microglia showing IL-1β immunofluorescence (Figure 6A).
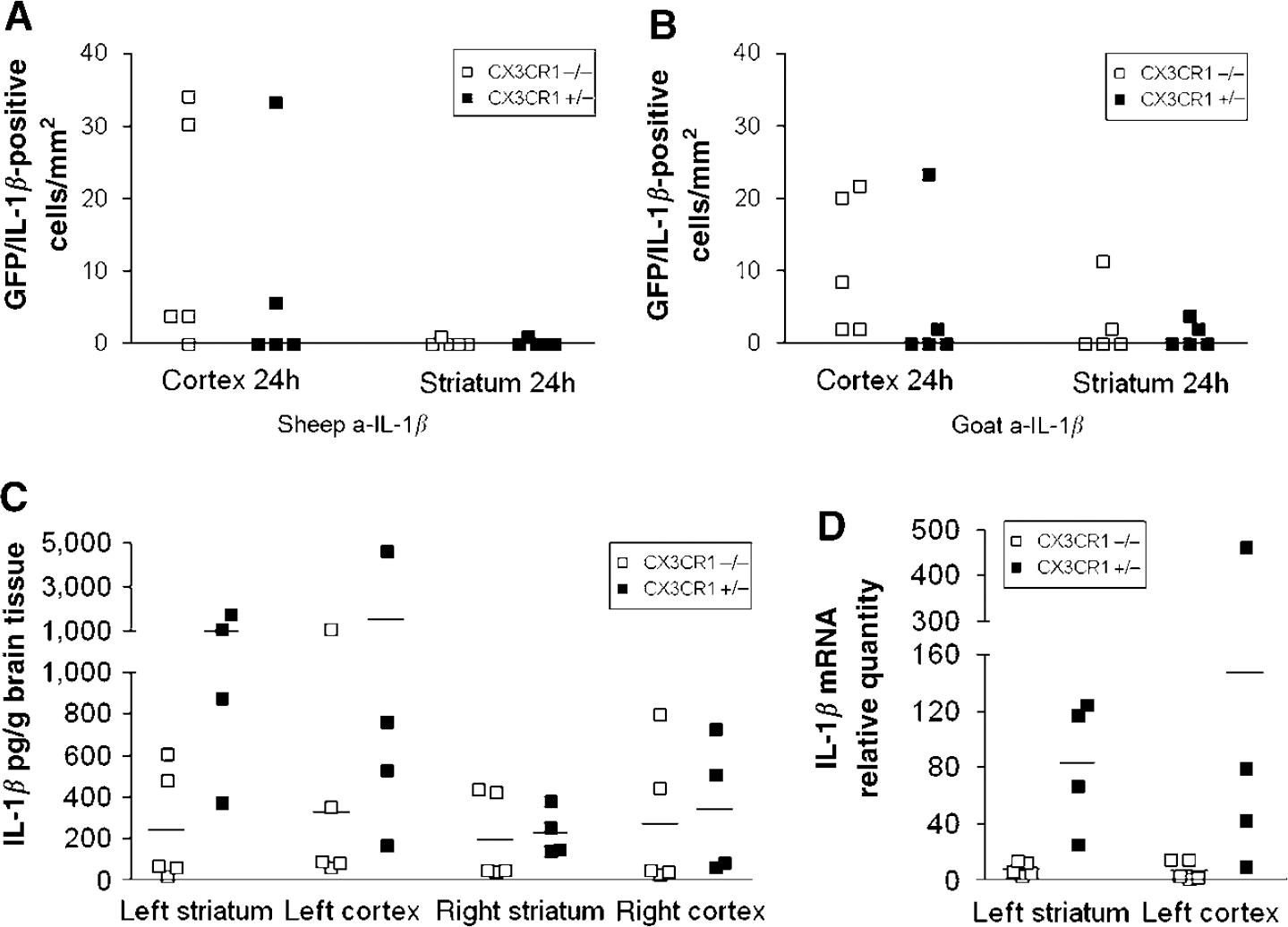
Interleukin-1β in microglia and in brain homogenates after MCAo. The figure shows the expression of IL-1β in microglia and the amount of IL-1β peptide and mRNA in brain homogenates of CX3CR1−/− and CX3CR1 +/− mice after MCAo. (

Distribution of IL-1β-positive microglia and astrocytes in the brain after MCAo. The IL-1β-positive cells in the brain after MCAo were revealed by immunofluorescence and in situ hybridization. (
By 72 h after MCAo, the number of IL-1β-positive microglia did not exceed 2 to 5 cells/mm2. No IL-1β-positive microglia were observed in the contralateral hemisphere.
IL-1β expression in brain homogenates: ELISA measurement revealed high IL-1β peptide levels 24 h after MCAo in the ipsilateral (left) hemisphere (Figure 5C). CX3CR1−/− mice exhibited significantly lower amount of IL-1β compared with CX3CR1 +/− animals in the left striatum (245 ± 125 versus 1025 ± 289 pg/mL, respectively; P < 0.05). This difference was also seen in the left cortex (330 ± 192 versus 1526 ± 1048 pg/mL, respectively). Elevated IL-1β peptide levels were also observed in the contralateral hemisphere in both genotypes after MCAo. Real-time PCR measurement revealed increased IL-1β mRNA levels in the ipsilateral hemisphere 24 h after MCAo. CX3CR1−/− mice showed a blunted elevation of IL-1β mRNA after MCAo, compared with CX3CR1 +/− mice (Figure 5D). The difference was significant in the left striatum (7.6 ± 2-fold increase in CX3CR1−/− versus 83.1 ± 23-fold increase in heterozygote mice; P < 0.05). Elevation of IL-1β mRNA after MCAo was also observed in the contralateral striatum and cortex in both genotypes compared with control (2.7 ± 0.7 in CX3CR1−/− versus 9.6 ± 10.4-fold increase in heterozygote in the right striatum, and 1.3 ± 0.3 in CX3CR1−/− versus 2.7 ± 0.6-fold increase in heterozygote in the right cortex).
Because the very low number of IL-1β-positive microglia found in the ipsilateral striatum, as well as the complete absence of these cells in the contralateral hemisphere, did not explain the results of IL-1β ELISA and real-time PCR measurements of brain homogenates, we investigated the possibility of IL-1β production by astrocytes.
IL-1β expression in astrocytes: As early as 4 h after MCAo, IL-1β-expressing GFP-negative cells were observed in the ipsilateral hemisphere in both CX3CR1 genotypes as well as in WT mice. A population of these cells located in the caudal striatum (sometimes near the ependyma of the lateral ventricle; Figure 6C) and the corpus callosum appeared to be GFAP- or glutamine synthetase-positive. Elongated IL-1β-positive cells were seen in the capsula interna, thalamus, and in the ventricular part of the dorsal ipsilateral thalamus and hippocampus expressing GFAP or glutamine synthetase (not shown). Some cells expressing IL-1β mRNA were also observed in these brain sites (Figure 6D).
A total of 24 h after MCAo, gIL-1β-positive astrocytes were seen in the ipsilateral hippocampus (Figure 6E) and thalamus in three out of five CX3CR1 +/− mice, which were only seen in one CX3CR1−/− animal. To confirm the specificity of labeling, gIL-1β antibody was preabsorbed with 180 ng/mL recombinant mouse IL-1β peptide before overnight incubation. Preabsorption prevented both astrocyte- and microglia-like IL-1β immunostaining in the brain parenchyma (Figure 6F) at 24 h, but was able to prevent cellular staining in 50% astrocytelike cells at 4 h. Neuronal-like gIL-1β staining was also observed 24 h after MCAo, even on the contralateral hemisphere, which was absent if preabsorption with recombinant mouse IL-1β was performed.
IL-1β expression in vitro after OGD in microglia-enriched cultures and in astrocytes: To investigate microglial IL-1β expression in vitro, microglia-enriched cultures were prepared (Figure 7A). A total of 4 h after OGD, elevated IL-1β expression was observed in cell lysates of microglia-enriched cultures derived from CX3CR1−/− and CX3CR1 +/−mice compared with control cultures. Culture supernatants (MEM) were devoid of IL-1β peptide 4 h after OGD, and only slight increase was observed in the incubation media (HEPES) compared with control cultures. No significant difference in IL-1β production was observed in cell lysates between CX3CR1−/− and CX3CR1 +/− microglia-enriched cultures (Figure 7B). To confirm early astrocytic IL-1β expression after OGD, astrocyte cultures were prepared from GFP/GFAP, CX3CR1 +/−, and CD1 mice. The IL-1β peptide was not detected in culture supernatants (MEM) 4 h after OGD, but incubation media (HEPES) taken minutes after the end of 3 h of hypoxia contained 15 to 30 pg/mL IL-1β in different cultures. Cell lysates of astrocytes contained elevated IL-1β levels compared with controls (Figure 7C) 4 h after OGD. Sorting GFP-positive astrocytes (from cultures derived from GFP/GFAP mice) or removing GFP-positive microglia (from cultures derived from CX3CR1 +/− mice) by FACS to get very pure astrocyte cultures did not have any affect on IL-1β production at 4 h, indicating that IL-1β production was not because of microglial contamination. Immunofluorescence revealed a few IL-1β/GFAP-positive astrocytes in GFP/GFAP and CX3CR1 +/− cultures, but more IL-1β/GFAP-positive cells were detected in CD1 astrocyte cultures 4 h after OGD (Figures 7D and 7E). A population of IL-1β-expressing cells was not positive to GFAP but was labeled with glutamine synthetase, an alternative marker for astrocytic cells. 24 h after OGD, cell lysates contained more IL-1β compared with 4 h in both GFP/GFAP and CX3CR1 +/− astroglial cultures (75 and 103 pg/mL, respectively).
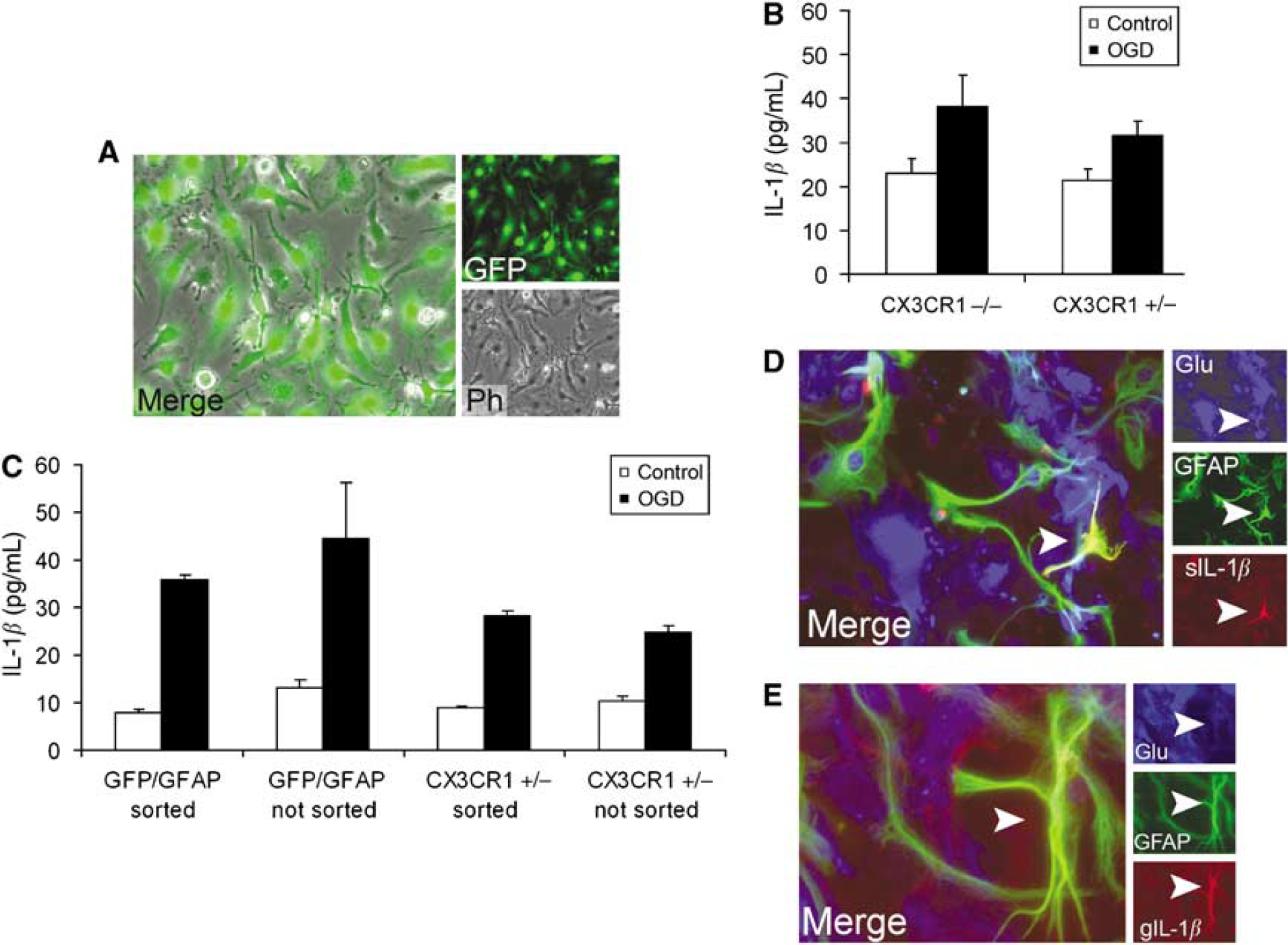
Interleukin-1β expression in vitro after OGD in microglia-enriched cultures and astrocytes. (
Quantitative Real-Time PCR Measurement Reveals Elevated Levels of IL-1Ra and TNFα mRNA after MCAo in CX3CR1+/− Mice
A total of 24 h after MCAo, IL-1Ra and TNFα mRNA expression was increased in brain homogenates of CX3CR1 +/− mice compared with CX3CR1−/−animals both in the ipsilateral striatum and cortex (Figures 8A–8D). Irrespective of the CX3CR1 genotype, significant correlation was observed between IL-1β and IL-1Ra mRNA levels (P < 0.001). The level of TNFα mRNA was also elevated in mice expressing higher amount of IL-1β.
Elevation of IL-1Ra and TNFα mRNAs was observed on the contralateral hemisphere after MCAo, but it was only 2% to 10% of that seen in homogenates of the ipsilateral hemisphere. The levels of IL-1Ra and TNFα mRNAs were higher in CX3CR1 +/− mice on the contralateral hemisphere, compared with CX3CR1−/− animals 24 h after MCAo (Figures 8E–8H).
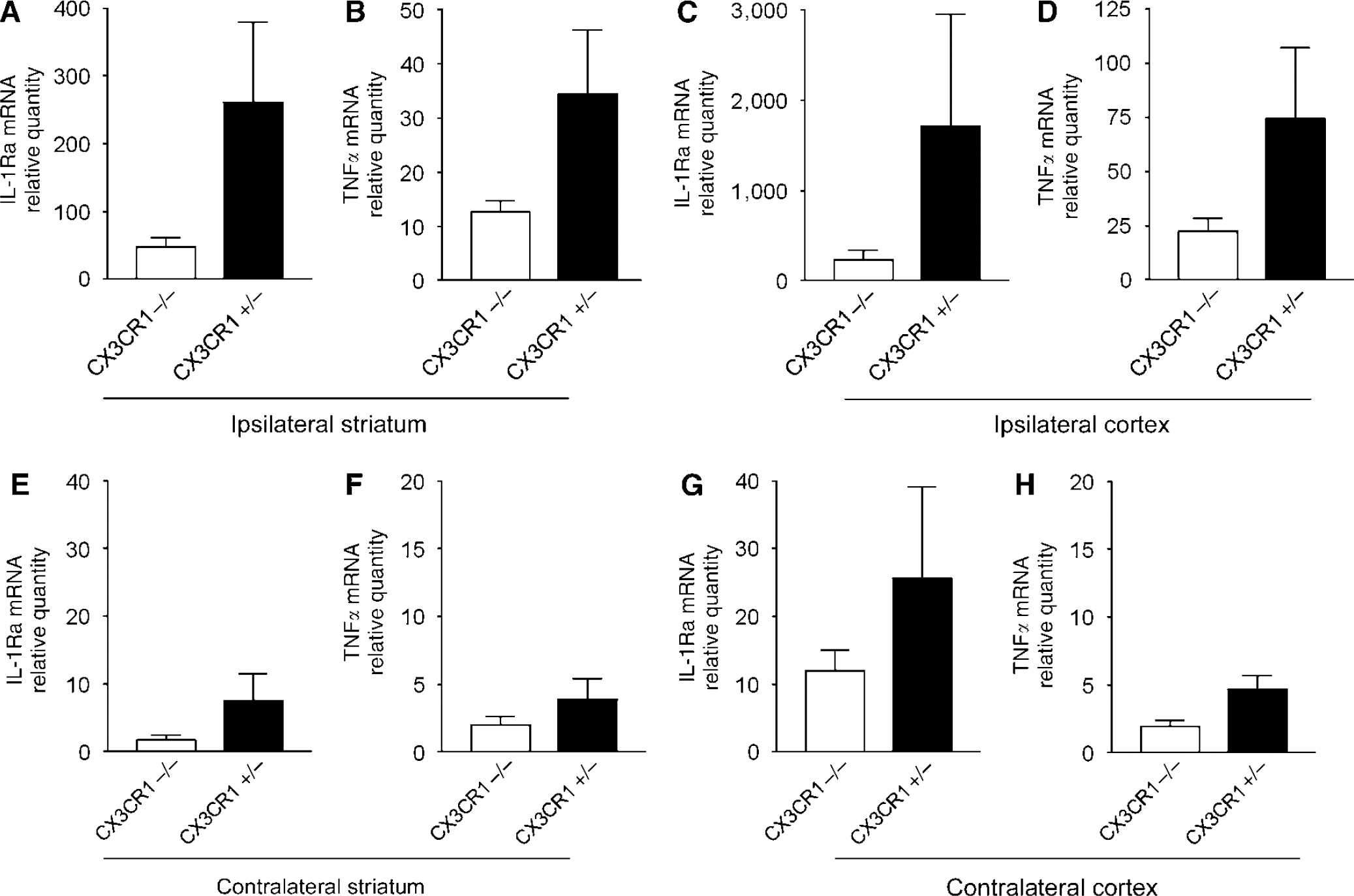
CX3CR1 +/− mice express higher amount of IL-1 Ra and TNFα mRNA after MCAo than CX3CR1−/− animals. Quantitative real-time PCR measurement reveals elevated amount of IL-1 Ra and TNFα mRNA after MCAo. Graphs showing the relative quantity of mRNAs in the same brain homogenates of the ipsilateral (left) striatum (
Discussion
We observed reduced brain damage, a lower number of apoptotic cells, CD45-positive infiltrating leukocytes, and highly blunted IL-1β expression after MCAo in mice lacking the CX3CR1 (fractalkine) receptor. These results indicate that CX3CR1 is involved in the pathophysiology of cerebral ischemia in the current mouse model and the absence of this receptor results in better general recovery and reduced inflammation in the brain.
One possible explanation for our findings is that fractalkine—CX3CR1 signaling modulates microglial acute and/or late response to ischemic challenge. Microglial activation after MCAo and phagocytosis of Annexin V-positive cells were observed in both genotypes, indicating that CX3CR1 deficiency does not profoundly affect microglial movement and detection of apoptotic signals.
Absence of fractalkine receptor in CX3CR1−/−mice was reported to cause cell-autonomous microglial neurotoxicity after systemic LPS treatment in a toxic model of Parkinson's disease and a transgenic model of amyotrophic lateral sclerosis (Cardona et al, 2006). Furthermore, CX3CR1−/− microglia were shown to accumulate and promote retinal degeneration (Combadiere et al, 2007). In contrast, we observed significantly smaller ischemic damage/neuronal apoptosis after MCAo in CX3CR1−/− mice than in mice having CX3CR1, suggesting that loss of CX3CR1 confers neuroprotection in ischemia. Supporting this finding, fractalkine knockout mice were similarly shown to be less susceptible to cerebral ischemia—reperfusion injury (Soriano et al, 2002), which indicates that loss of CX3CR1–fractalkine signaling mediates these changes. In addition, CX3CR1 inhibition is protective against ischemic acute renal failure in mice (Oh et al, 2008). Therefore, it seems that lack of CX3CR1 has different consequences under different pathologic conditions. One plausible mechanism could be the altered IL-1β secretion by microglia/macrophages in the absence of CX3CR1. After activation by LPS, microglia derived from CX3CR1−/− mice expressed more IL-1β, and adoptive transfer of these cells resulted in increased neuronal death, which was blocked by IL-1Ra (Cardona et al, 2006). In contrast, we observed blunted IL-1β mRNA and protein expression in CX3CR1−/− mice 24 h after MCAo, which correlated with smaller infarcts, whereas no difference was seen in the number of IL-1β-positive microglia and in in vitro IL-1β production by microglia-enriched cultures after OGD. It is likely, therefore, that CX3CR1−/− microglia are not neurotoxic in stroke and/or microglia are not directly involved in worsening the outcome after MCAo. Experiments that usually show the toxic potential of microglia in vitro often use stimuli eliciting defense-oriented reactions, such as LPS (Hanisch and Kettenmann, 2007), which results in production of a high amount of proinflammatory cytokines. This situation is not identical to other challenges, such as hypoxia and ischemia. It has previously been reported in several studies that microglia have beneficial as well as detrimental effects in cerebral ischemia, which indicate a highly complex microglial acute/late function in damage formation, inflammation, and repair. It is not clear whether loss of CX3CR1 might have led to reduced microglial inflammatory function after MCAo or decrease in microglial-derived inflammatory molecules directly contributed to the formation of smaller ischemic damage. Therefore, it cannot be completely excluded that the reduced level of inflammatory cytokines and lower number of infiltrating leukocytes in KO mice after MCAo were (at least in part) because of the smaller ischemic damage itself. Inflammation is observed in the brain after several types of pathologic insults. However, it has also been reported that inflammatory mediators can profoundly exacerbate ischemic damage in the MCAo model (McColl et al, 2007).
It is possible that lack of CX3CR1 modifies microglial response in response to MCAo, which, in turn, could blunt the release of inflammatory molecules from other cells too, resulting in a better outcome in CX3CR1−/− mice. It was reported recently that IL-1β induces neuronal death through an astrocyte-dependent mechanism (Thornton et al, 2006). In a prion model of chronic neurodegeneration and inflammation, CX3CL1 immunoreactivity is upregulated in astrocytes and CX3CR1 expression is elevated on microglia (Hughes et al, 2002). Our results might also indicate that fractalkine—CX3CR1 signaling could modulate synthesis, cleavage, and/or release of IL-1β directly. It has recently been shown that fractalkine exerts a dose-dependent differential regulation of cytokine secretion from macrophages. Relatively low concentrations of fractalkine suppressed LPS-induced TNFα secretion, but higher concentrations of fractalkine, which may represent a local inflammatory condition, was not immunosuppressive; instead, upregulation of IL-23 was seen (Mizutani et al, 2007). Our data indicate that fractalkine receptor deficiency may not increase microglial neurotoxicity or IL-1β release from microglia after transient MCAo in the current model.
We found that astrocytes (and in less part neurons) can represent a significant source of IL-1β in vivo, after MCAo, and in vitro after OGD at 4 to 24 h in both models. Furthermore, the appearance of these cells near the ependyma and meninges (and possibly neuronal IL-1β-expression) would, in part, explain elevated IL-1β peptide levels measured in the contralateral hemisphere after MCAo. It has been previously reported that astrocytes express IL-1β 2 to 7 days after MCAo in the rat (Pearson et al, 1999), and that hypoxia-inducible factor-1 mediates transcriptional activation of IL-1β in astrocyte cultures (Zhang et al, 2006). Interestingly, endogenous caspase-1 activation and bioactive IL-1β production are prerequisites for iNOS induction and excessive NO formation in brain-derived endothelia and astrocytes after challenge with proinflammatory cytokines (Juttler et al, 2007). Recent data indicate the potential cytotoxicity of astrocytes in vitro and in vivo in neurodegenerative diseases (Hashioka et al, 2008; Thornton et al, 2006). However, it requires further investigation whether IL-1β is released early from astrocytes in response to hypoxia or needs contribution of other cells/stimuli in vivo. Because cytokines, including IL-1β, can be released into the extracellular space and into the cerebrospinal fluid from the cells of origin, brain homogenates might have contained more IL-1β peptide than the amount indicated by IL-1β-immunopositive cells. In addition, the detection of elevated amount of IL-1β and TNF mRNAs in heterozygote mice with real-time PCR, even on the contralateral side, indicates local expression of these cytokines. Moreover, IL-1β in situ hybridization data were compatible with results of IL-1β immunohistochemistry regarding the number and location of IL-1β-expressing microglia and astrocytes.
It is not clear how fractalkine—CX3CR1 signaling could affect late central inflammatory events and how peripheral components contribute to the formation of ischemic damage. A total of 12 h after transient MCAo in rat, fractalkine expression was lost from the ischemic striatum, but fractalkine immunoreactivity was strongly increased in morphologically intact cortical neurons of the ischemic penumbra 24 and 48 h after ischemia as well as in endothelial cells 48 h and 7 days after ischemia (Tarozzo et al, 2002). We observed significantly less CD45 +/GFP- leukocytes in the ipsilateral hemisphere in CX3CR1−/− mice 72 h after MCAo. CX3CR1−/− mice were reported to have a significant reduction in macrophage recruitment to the vessel wall and decreased atherosclerotic lesion formation (Lesnik et al, 2003). Others found that the CX3CR1highCCR−2Gr− subset of murine blood monocytes is characterized by CX3CR1-dependent recruitment to noninflamed tissues and that a short-lived CX3CR1lowCCR2−Gr1− cell population is actively recruited to inflamed tissue (Geissmann et al, 2003). We observed a lower number of GFP +/CD45+ microglia/macrophages in the ipsilateral cerebral cortex of CX3CR1 +/− mice at 72 h, possibly as a result of more serious ischemia and decreased microglial proliferation (Denes et al, 2007) and/or recruitment. Our results indicate that a reduction in damage formation observed relatively early in CX3CR1−/− mice, and large differences seen in IL-1β expression in the brain at 24 h, may be explained by central effects rather than by the result of reduced macrophage infiltration and/or peripheral monocytic IL-1β secretion in the current model.
Fractalkine has been shown to serve as a potent chemoattractant for natural killer cells, CD8+ T cells (Imai et al, 1997), and mast cells (Papadopoulos et al, 2000). Neutrophils can be recruited to the brain independently of fractalkine by various substances released under pathologic conditions. For example, cytokine-induced neutrophil chemoattractant-1 is expressed by astrocytes under inflammatory conditions (Koyama et al, 2007). In addition, hypoxic challenge increases the expression of fractalkine and intercellular adhesion molecule-1 on human endothelial cells and promotes neutrophil adhesion through activation of the Jak-Stat5 pathway (Yang et al, 2007). The fact that significantly more CD45 +/GFP- (i.e., CX3CR1-negative) leukocytes were recruited to the brain in heterozygote mice than in CX3CR1−/− mice indicates that the fractalkine receptor may not be directly involved in recruitment and adhesion of these cells after MCAo. In addition, loss of central CX3CR1–fractalkine signaling in CX3CR1−/− mice could result in reduced amount of other brain-derived chemokines and adhesion molecules too. The lower number of leukocytes in the brain and reduced IL-1β, TNFα, and IL-1Ra expression in CX3CR1−/− mice together indicate a lower level of central inflammation after MCAo.
Taken together, we identify CX3CR1 as an important contributor to cerebral ischemia and postischemic inflammation. Through modulation of microglial and/or astrocytic function and IL-1β expression, CX3CR1 might be an important target in clinical approaches of treatment of ischemic stroke.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.