
Abstract
Riluzole is believed to modulate glutamatergic function by reducing glutamate release and facilitating astroglial uptake. We measured 13C labeling in metabolites in prefrontal cortex and hippocampus during a 10 mins infusion of [1-13C]glucose in urethane anesthetized rats treated with riluzole (21 days, 4 mg/kg per day, i.p.) or saline. Total and 13C concentrations of metabolites were determined in extracts using 1H-[13C] NMR spectroscopy. In prefrontal cortex (P < 0.05) and hippocampus (P < 0.05) riluzole increased 13C labeling over saline in glutamate-C4 (to 112% and 130%), GABA-C2 (to 142% and 171%), and glutamine-C4 (to 118% and 233%) without affecting total metabolite levels (P > 0.2). Our findings indicate that contrary to expectation chronic riluzole enhanced glucose oxidative metabolism and glutamate/glutamine cycling.
Introduction
Glutamate (Glu) and GABA (γ-aminobutyric acid) are the major excitatory and inhibitory neurotransmitters in the brain. These amino-acid neurotransmitters play vital roles in the regulation of several important CNS processes, and have been linked to the pathogenesis and pathophysiology of several neurodegenerative disorders. Riluzole (2-amino-6-trifluoromethoxy benzothiazole), a neuroprotective agent with antiglutamatergic actions, is effective in prolonging the median survival in patients with amyotrophic lateral sclerosis (Miller et al, 2007).
Although riluzole's mechanism of action is not completely understood, its antiglutamatergic pharmacological properties are believed to include inhibition of Glu release through inactivation of voltage-dependent ion channels (Doble, 1996; Huang et al, 1997; Urbani and Belluzzi, 2000), effects on the expression and signal transduction through Glu receptors (Doble, 1996; Du et al, 2007), and facilitation of astrocytic Glu uptake (Frizzo et al, 2004). Recent clinical studies also suggest riluzole possesses antidepressant and anxiolytic-like effects in patients with depression and anxiety disorders (Pittenger et al, 2008), thereby suggesting that riluzole has effects on regions involved in emotion regulation. To better understand the potential mechanism of riluzole's action, we sought to determine the drug's effects on neuronal Glu metabolism and the Glu/glutamine (Gln) cycle in the prefrontal cortex and hippocampus.
Nuclear magnetic resonance (NMR) spectroscopy used in conjunction with 13C-labeled substrates can provide dynamic information on neuronal and glial metabolism and neuron-glial trafficking of Glu and GABA in the Glu/GABA-Gln cycle (Chowdhury et al, 2007b). [1-13C]Glucose is oxidized to a quantitatively greater extent in neurons than glia, labeling the large and predominately neuronal Glupool. 13C-labeled Glu and GABA released from active neurons is taken up and converted to Gln in nearby astroglia, the latter being a precursor for their neuronal resynthesis. The rates of 13C incorporation into amino-acid pools of neurons (Glu and GABA) and astroglia (Gln) using [1-13C]glucose with NMR detection in vivo or ex vivo can provide unique information on neuronal and glial oxidative metabolism and substrate trafficking (Rothman et al, 2003; Hertz et al, 2007).
In this study we examined the effects of chronic (21 days) riluzole treatment using 13C labeling from [1-13C]glucose infused intravenously on the metabolism of the major brain amino acids in prefrontal cortex and hippocampus of anesthetized rats. Our findings suggest that riluzole enhances overall Glu metabolism.
Materials and methods
All experiments were conducted under protocols approved by the Yale IACUC. Two groups of male Sprague—Dawley rats (300 to 350 g) were studied: (1) saline-injected controls, n = 5; (2) riluzole (4 mg/kg per day, i.p, Sigma-Aldrich, St Louis, MO, USA, n = 5). Saline and riluzole were administered once daily for 21 days. We chose this dose based on neuroprotection and previous studies, for example, Risterucci et al (2006) and behavioral effects observed in our laboratory (Chowdhury et al, 2008) suggesting this dose targeted behavioral effects of interest.
As part of a larger study to assess riluzole's antidepressant effects, rats in both groups received a behavioral assessment (sucrose preference test) on day 21. Rats were exposed to a palatable sucrose solution (1%; Sigma) for 48 h followed by 4 h of water deprivation and a 1h exposure to two identical bottles—one filled with sucrose solution and the other with water. Sucrose and water consumption by each animal was determined by measuring the change in volume of fluid consumed. Sucrose preference was defined as the ratio of the volume of sucrose versus water consumed during the 1 h test.
One day after (day 22) the last exposure to riluzole or saline, rats were anesthetized with urethane (1.5 g/kg, i.p.) and a tail vein cannulated. The core body temperature was monitored and maintained near 37°C with a heating pad connected to a temperature-regulated, recirculating water bath. Thirty minutes after injection of urethane, a solution of [1-13C]glucose (99 atom%; Cambridge Isotopes, Andover, MA, USA) dissolved in water (0.75 mol per 200 g body wt. per min) was infused for 10 mins ( Chowdhury et al, 2007b ). Immediately after the infusions rats were euthanized using directed pulsed microwave irradiation (5 kW, Model TMW 6402C, Muromachi Microwave Fixation System) to the head, arresting brain metabolism in < 1 sec, allowing brain tissue removal from prefrontal cortex and hippocampus. Blood was sampled from the heart following microwave irradiation, centrifuged, and the plasma removed. Blood plasma was frozen in liquid N2 and stored at −80°C for subsequent analysis. Cortical and hippocampal tissue extracts were prepared as described previously ( Chowdhury et al, 2007a ) using ethanol and the supernatants removed and lyophilized. Lyophilized samples were resuspended in D2O and H2O (90:10 v/v) for NMR analysis at 11.7 T (1H resonance frequency of 500.13 MHz) using a Bruker AVANCE spectrometer (Bruker Instruments). Fully relaxed 1H-[13C] NMR spectra were acquired as two subspectra—one involving broadband 13C inversion pulses applied in alternate scan blocks, whereas 13C-decoupling was applied in both. Subtraction of the scans obtained with 13C inversion (12C—13C) from those without inversion (12C +13C) gave the difference spectrum (2 × 13C), containing only 13C coupled 1H resonances at twice the true intensity. The total carbon isotope composition was given by the 12C+13C subspectrum. The 13C atom percentage enrichment was calculated as the ratio, 13C/(12C + 13C) × 100, followed by subtraction of 1.1% to remove 13C arising from natural abundance ( Chowdhury et al, 2007a ). Absolute concentrations of metabolites were determined relative to the [2-13C]glycine, added during tissue extraction. The isotopic 13C enrichments of Glu-C4, GABA-C2, and Gln-C4, were calculated from the ratio of the areas of these resonances in the 1H-[13C] NMR difference spectrum (2 × 13C only) and the nonedited spectrum (12C+13C). Plasma glucose concentrations were determined using a Beckman Glucose Analyzer II (Model 6517; Beckman Instruments Inc., Brea, CA, USA). Plasma glucose was determined using 1H NMR under fully relaxed pulsing conditions (repetition time, 20 secs) without 13C decoupling. The 13C fractional enrichment of glucose-C1 was determined from the H1a glucose resonance at 5.2 p.p.m. as the ratio of the 13C satellites to the apparent triplet using the downfield 13C peak (and multiplied by 2) to avoid distortion from the water suppression (Chowdhury et al, 2007a). Formate was added to the sample as a concentration standard.
Statistical Analysis
The statistical significance of differences in metabolite concentrations and 13C enrichments between control and riluzole-treated rats were assessed by two-tailed, Student's t-test. Differences in mean values were considered significant for P < 0.05. All values are presented as mean ± s.d.
Results
Concentration and 13C Enrichment of Plasma Glucose
The plasma concentrations and percentage 13C enrichments at the end of the 10 mins infusions were similar in both groups of animals (control: 21.1 ± 1.3 mmol/L and 36.1 ±3.4% versus riluzole: 19.7 ± 1.1 mmol/L and 37.5 ±3.3%; P < 0.2).
Effects of Riluzole on Levels of Amino Acids and Metabolites in Prefrontal Cortex and Hippocampus
The total concentrations of amino acids and metabolites were determined in the prefrontal cortex and hippocampus of [1-13C]glucose-infused control and riluzole-treated rats (Table 1). Total concentration (μmol/g) of Glu, Gln, and aspartate were higher (P < 0.01) in prefrontal cortex than hippocampus in both groups, whereas GABA and alanine levels were lower (P < 0.05). However, within the same brain region no significant differences were found between control and riluzole-treated rats (P < = 0.4; Table 1).
Concentration (μmol/g) of amino acids and metabolites from [1-13C]glucose (10 mins) in prefrontal cortex and hippocampus
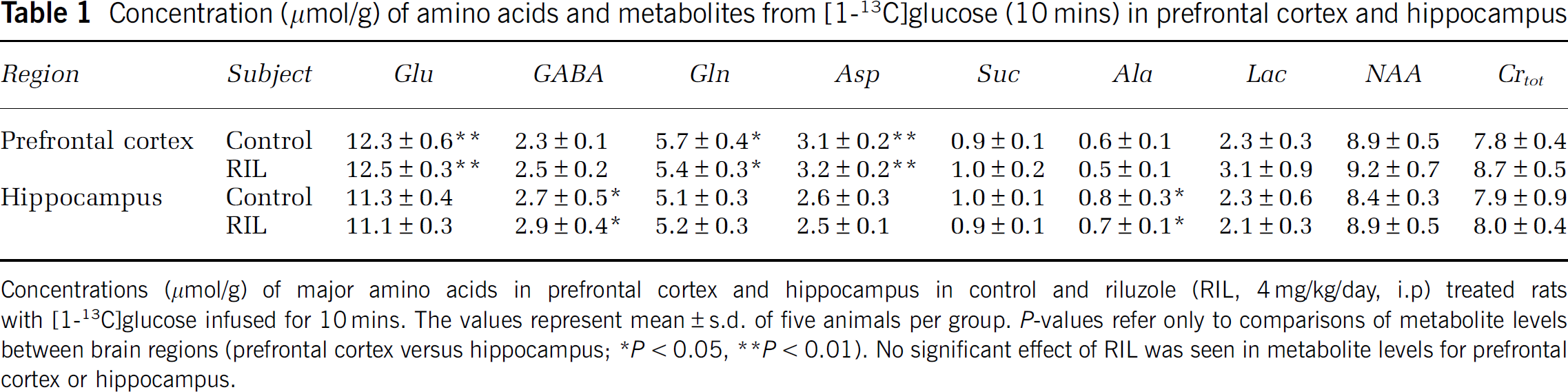
Concentrations (μmol/g) of major amino acids in prefrontal cortex and hippocampus in control and riluzole (RIL, 4 mg/kg/day, i.p) treated rats with [1-13C]glucose infused for 10 mins. The values represent mean ± s.d. of five animals per group. P-values refer only to comparisons of metabolite levels between brain regions (prefrontal cortex versus hippocampus;
P < 0.05
P < 0.01). No significant effect of RIL was seen in metabolite levels for prefrontal cortex or hippocampus.
Effects of Riluzole on Amino Acids 13C Labeling from [1-13C]Glucose in Prefrontal Cortex and Hippocampus
Figure 1 depicts the percentage 13C enrichments and concentrations of Glu-C4, GABA-C2, and Gln-C4 in the control and riluzole-treated rats infused with [1-13C]glucose. Riluzole-treated rats displayed greater 13C labeling in the respective amino acids compared with controls (P < 0.05) in terms of 13C enrichment (%) or 13C concentration (μmol/g) in prefrontal cortex and hippocampus (Figure 1B).

(
Effects of Riluzole on Behavior
No effects of riluzole were seen in the preference of sucrose over water volume in rats treated with riluzole compared with saline (ratio of sucrose to water volume consumed over 1 h: riluzole, 3.1 ± 0.4 versus saline, 3.7 ± 0.5; n = 5 per group; P > 0.4).
Discussion
The results of this study demonstrate that chronic treatment with riluzole induces increases in 13C-glucose metabolism in the prefrontal cortex and hippocampus. These effects were evidenced by increases in 13C incorporation into Glu-C4, Gln-C4, and GABA-C2 following 21 days of riluzole treatment. The increased rate of incorporation of 13C label into the major amino acids, indicative of an enhancement of oxidative metabolism by riluzole, was unexpected considering the presumed effect of riluzole in reducing Glu release in vitro. The majority of brain Glu is present in neurons and its turnover from [1-13C]glucose reflects the rate of oxidative metabolism mainly in the neuronal TCA cycle and not the rate of net Glu synthesis (see Figure 1A). The enhancement by riluzole of Glu-C4 labeling, in the presence of an unchanged total Glu concentration (12C+13C), indicated that TCA cycle flux had increased. Riluzole-induced enhancement of Gln-C4 labeling is also consistent with increased Glu/Gln cycling between neurons and astroglia, because ∼ 70 to 80% of Gln-C4 13C labeling from [1-13C]glucose is derived through this pathway (Oz et al, 2004; Sibson et al, 2001). Thus, the increase in 13C labeling is consistent with enhanced glutamatergic metabolism rather than with decreased Glu release.
We did not measure the complete time course of 13C labeling in the current study, but only a single early time point (10 mins), which our previous studies (Chowdhury et al, 2007b) indicate is well within the linear portion of the turnover curves. The blood glucose infusion protocol raises plasma glucose to a near constant level within the first 1 to 2 mins; thus, the 13C label accumulated in brain Glu, GABA, and Gln at 10 mins, after normalization by the plasma percentage 13C enrichment over that interval for each animal, reflects the associated metabolic fluxes. Oxidation of [1-13C]glucose in the glial TCA cycle will also result in labeling of Gln-C4, although the extent is less than that for neurons, comprising ∼ 15 to 25% of the total reflecting neurons and glia (Oz et al, 2004; Sibson et al, 2001).
A limitation of this study was the potential interfering effects of anesthesia. We can rule out the possibility that the effects of riluzole on metabolism seen in this study were indirectly related through an effect of riluzole on the depth of anesthesia, because we saw no evidence that riluzole altered the level of anesthesia in the animals. Another limitation in our study was that animals were hyperglycemic, which is known to depress cerebral blood flow in rats (Duckrow, 1995). With the exception of specialized subcortical nuclei involved in glucosensing and energy homeostasis, for example, hypothalamus and areas of the basal ganglia, glucose utilization in cortex and most other brain regions (including prefrontal cortex and hippocampus) is unaltered by hyperglycemia (Orzi et al, 1988; Duckrow and Bryan, 1987). As brain glucose transport is not limiting for glucose utilization for plasma levels > 2 mmol/L, and riluzole's effect was to increase (and not decrease) glucose metabolism, it is unlikely that hyperglycemia indirectly led to riluzole's metabolic effect. Potential suppression by riluzole of a hyperglycemia-induced reduction of glucose metabolism is contradicted by the studies cited above showing that glucose utilization is unaltered by hyperglycemia in the regions studied.
Riluzole is currently the only medication indicated for the treatment of amyotrophic lateral sclerosis by the US Food and Drug Administration. Although it is widely believed that decreased glutamatergic excitotoxicity is related to the drug's mode of action in delaying the progression of this disorder, there is no clear consensus regarding the exact pharmacological properties that are responsible for the effects on glutamatergic neurotransmission. More recent hypotheses support two pharmacological properties as playing critical roles in generating the drug's antiglutamatergic effects (Doble, 1996). In vitro, riluzole reduces Glu release by interaction with voltage-dependent ion channels (Wang et al, 2004). This has been posited as a likely mechanism through which the drug decreases glutamatergic excitotoxicity associated with neuronal damage (Huang et al, 1997; Urbani and Belluzzi, 2000). However, several recent studies also demonstrate riluzole's ability to facilitate Glu clearance from the extrasynaptic space (Fumagalli et al, 2008; Frizzo et al, 2004), thus suggesting a second potentially important mechanism of decreasing the excitotoxic effects of Glu.
Riluzole has been shown to restore levels of N-acetylaspartate, a marker of neuronal viability, in motor cortex of patients with amyotrophic lateral sclerosis; increases in N-acetylaspartate within as little as 1 day of treatment (Kalra et al, 1998) suggests relatively rapid metabolic effects. An increase in N-acetylaspartate was also observed in hippocampus of individuals with generalized anxiety disorder at 8 weeks of riluzole therapy (Mathew et al, 2008). Whether the increase in N-acetylaspartate reflects a direct effect of riluzole on neuronal metabolism or is a consequence of reduced excitotoxicity is not clear. Our finding that chronic riluzole treatment increased 13C labeling from glucose in physiologically normal brain suggests that riluzole may have enhancing effects on mitochondrial function beyond those related to the suppression of excitotoxicity. It is also possible the increases in 13C incorporation into Glu-C4, Gln-C4, and GABA-C2 following riluzole treatment could be due to direct effects of the drug on mitochondrial function. Previous studies have demonstrated enhanced mitochondrial function following riluzole administration (Mu et al, 2000).
The idea that riluzole's neuroprotective effect is mediated, at least in part, by enhancing Glu uptake from the extrasynaptic space is consistent with the model of neurotoxicity proposed by Hardingham (2006). This model proposes that Ca2+ influx through excessive extrasynaptic NMDA (N-methyl-D-aspartate) receptor activity leads to increased release of neuronal nitric oxide synthase and mitochondrial dysfunction. Stimulation of synaptic NMDA receptors, in contrast, promotes cellular signaling cascades that enhance neuronal survival. Thus, the ratio of extrasynaptic to synaptic NMDA receptor stimulation is a main determinant in processes of neurotoxicity and neuroprotection. Considering our present findings, riluzole led to increased Gln-C4 labeling from 13C-glucose, suggesting increased Glu/Gln cycling with more Glu being released and cycled through this pathway. As long as Glu removal keeps pace with release, extrasynaptic Glu levels would not be expected to rise. If, as found in several previous studies (Frizzo et al, 2004; Fumagalli et al, 2008), riluzole is acting to enhance Glu clearance from the extrasynaptic space, then it could be expected that chronic administration of this drug would result in decreased excitation of extrasynaptic NMDA receptors while allowing for an increase in synaptic stimulation. Synaptic NMDA receptor stimulation has been previously shown to have prosurvival effects on the brain (Hardingham, 2006).
A common model of glutamatergic disturbance has been purported for several neurological and psychiatric disorders. Interestingly, riluzole has recently been shown to have clinical efficacy across several neuropsychiatric disorders, perhaps reversing or preventing common pathophysiological effects of Glu. The findings of this study suggest that chronic riluzole enhances both neuronal oxidation of glucose and Glu/Gln cycling between neurons and glia.
Footnotes
Acknowledgements
We thank Dr James CK Lai and Dr Fahmeed Hyder for helpful discussions, and Dr Prajna P Siddiqui for her assistance in the preparation of brain tissue extracts.