
Abstract
The aim of this study was to investigate the relationship between aspirin resistance, ischaemic stroke subtype, stroke severity, and inflammatory cytokines. Aspirin resistance was assessed by thrombelastography in 45 people with ischaemic stroke and 25 controls. Plasma interleukin (IL)-6 was measured. Stroke severity was assessed using the modified Rankin scale and National Institute of Health Stroke Score within 72 h of stroke. Aspirin resistance was more common in the stroke than the control group (67% versus 40%, P=0.028), and within the stroke group the aspirin-resistant group had a higher Rankin score (4.0 versus 2.0, P=0.013). Aspirin resistance was greater in lacunar than embolic strokes (platelet activation 79% versus 59%, P=0.020). The stroke aspirin-resistant group had higher levels of IL-6 than the stroke aspirin-sensitive group (2.4±1 versus 1.8±0.9 ng/mL, P=0.037). Using multivariate analysis, we examined the interrelationships between aspirin resistance, IL-6, and stroke severity. These analyses showed that IL-6 was independently associated with stroke severity as the outcome (B=3.738, P=0.036), and aspirin resistance was independently associated with IL-6 (B=0.765, P=0.005) as the outcome. In conclusion, aspirin resistance is related to stroke severity and aspirin resistance is more common in lacunar strokes than embolic strokes.
Introduction
Large population studies have shown that aspirin reduces the incidence of both primary and secondary (Antithrombotic Trialists' Collaboration, 2002) cardio- and cerebrovascular disease by approximately 25%. However, aspirin fails to prevent myocardial infarctions and strokes in a large proportion of people (Mueller et al, 1997; Eikelboom et al, 2002) and the phenomenon of aspirin resistance may contribute to this problem. Aspirin resistance may contribute to failure of aspirin therapy in the secondary prevention of cerebrovascular events in as many as 30% to 40% of patients (Grundmann et al, 2003). Previous studies have shown that aspirin resistance occurs in 5% to 65% of people with ischaemic strokes, as reviewed by Schror et al (2006). Aspirin resistance was also associated with an increased risk of death, myocardial infarction, or stroke compared with patients who were aspirin sensitive (24% versus 10%, hazard ratio (HR) 3.12, 95% confidence interval) 1.10 to 8.90, P=0.03) (Gum et al, 2003).
Ischaemic stroke is associated with a proinflammatory profile. Inflammation in the acute post-stroke period may hinder recovery and people who have infections after stroke have a poorer functional outcome (Aslanyan et al, 2004; Kwan and Hand, 2007). Mechanistically, inflammatory cytokines such as interleukin (IL)-6, tumor necrosis factor α (TNFα), and IL-1β have been shown to influence neural damage (Carlson et al, 1999), vascular endothelial function (Burke-Gaffney and Keenan, 1993), and platelet activation (Lumadue et al, 1996).
To date, no reports have been published linking inflammation with aspirin resistance. In addition, it is not clear whether aspirin resistance is related to the subclass of ischaemic stroke or with the severity of stroke.
The aim of this study was to investigate whether aspirin resistance is related to the subclass of ischaemic stroke, the severity of stroke, and inflammation in the acute stroke period.
Materials and methods
Participants
This study complied with the Declaration of Helsinki, the locally appointed ethics committee approved the research protocol, and informed consent was obtained from the subjects (or their guardians).
Forty-five volunteers who had an ischaemic stroke in the previous 72 h were recruited. These subjects were all taking low-dose aspirin (75 mg). A total of 37% of the stroke group had taken aspirin only in hospital whereas 57% of the stroke group had been taking aspirin regularly at home as well as when in hospital before blood samples were taken. Exclusion criteria included occurrence of any of the following in the last 6 months: deep vein thrombosis, pulmonary embolism, myocardial infarction, stroke (not including their current event), transient ischaemic attack, surgery, or cancer. Occurrence of stroke was confirmed by computed tomography scan. Stroke outcome (degree of disability) was assessed using the modified Rankin scale (van Swieten et al, 1988) and the National Institute of Health Stroke Score (NIHSS) scale (Brott et al, 1989) within 72 h of stroke. Stroke subtypes were categorized according to the Oxfordshire Community Stroke Project classification system, identifying by clinical features (Bamford et al, 1991). Strokes were grouped as total anterior circulation stroke (TACS), partial anterior circulation stroke (PACS), lacunar stroke (LACS) and posterior circulation stroke (POCS), or undeterminable (Bamford et al, 1991). Information was collected on previous thrombotic events that occurred more than 6 months ago (termed ‘previous’ in Table 1). Atrial fibrillation was defined according to a diagnosis recorded in medical notes. Information on prescribed medications, including aspirin, was collected from pharmacy charts in medical notes whereas the details of aspirin taken at home were taken from medical notes made on admission. Diabetes was self-reported or noted from medical notes. Blood pressure values were the most recent readings in the previous 24 h using a sphygmometer. Body fat was calculated from the means of triplicate biceps and triceps skin fold measurements when standing, sitting, or lying. We have previously shown in the elderly that there were no differences in measurements when sitting or lying compared with that when standing (Horsfield et al, 2005).
Characteristics of control and stroke groups

BMI, body mass index; CVD, cardiovascular disease (MI or angina or other recorded outcome); TIA, transient ischemic attack; DVT, deep vein thrombosis; PE, is pulmonary embolism.
P-values represent analysis of variance or χ2-tests between groups.
Defined more fully in Materials and methods.
For comparison, 25 healthy controls of a similar age who were currently taking aspirin (75 mg for at least 1 month) were recruited by advertising in the local community. Exclusion criteria for the controls were the same as for the stroke group except that they were excluded if they had ever had a stroke; however, they were not excluded if they had a suspected previous transient ischaemic attack. Blood pressure and body fat were measured as for the stroke group. Information was collected on previous thrombotic events that occurred more than 6 months ago. Information on prescribed medications was self-reported but they were specifically asked whether they took aspirin regularly and whether they had taken aspirin in the past 24 h. Diabetes was self-reported. Other conditions in Table 1 for the control and stroke groups included multiple sclerosis, asbestosis, emphysema, arthritis, polycythaemia, chronic obstructive pulmonary disease, and hiatus hernia.
Procedures
Within the first 72 h of having a stroke, venous blood was collected for measurement of IL-6, TNFα, and IL-1β (R&D Systems, Abingdon, UK) by enzyme-linked immunosorbent assay. Aspirin resistance was measured using PlateletMapping Assays (Haemoscope, USA) on the Thrombelastograph Haemostasis Analyser (Haemoscope, Niles, USA, supplied by Medicell, London, UK). The Thrombelastograph Haemostasis Analyser detects clot formation kinetics and strength as changes in the torque of a rotating sample cup via a stationary pin. The contribution of platelets to the clot strength directly influences pin movement and is referred to as maximum amplitude (MA). The heparinized blood sample is split into three cups: one containing activator (MA results from fibrin only), one containing activator plus arachidonic acid (MA results from conversion of arachidonic acid to thromboxane A2 in the presence of cyclooxygenase and thromboxane A2 then stimulates platelet activation) and one containing heparinase and kaolin (measures total platelet function based on thrombin activation because the effect of heparin has been reversed). As aspirin inhibits cyclooxygenase function, the difference between MA when stimulated by arachidonic acid and the MA in the heparinase/kaolin-treated sample determines the degree of resistance to aspirin in terms of the percentage inhibition or activation. The MA related to fibrin formation is also taken into account during this calculation, which is carried out automatically by the software. Aspirin resistance was defined as more than 50% platelet activation after stimulation with arachidonic acid (Tantry et al, 2005; Agarwal et al, 2006; Gwozdziewicz et al, 2006).
Statistical Analysis
Data that were not normally distributed were converted to a normal distribution by logarithmic transformation. Data represent mean±s.d. (or median±s.d. and range for Rankin score). Differences in frequencies between groups were examined using χ2-tests (Fisher's exact test, P-values are shown). Differences in measurements across the stroke subgroups were analysed by analysis of variance. Differences in measurements between groups were assessed by unpaired t-tests. Multivariate regression models were used to investigate factors associated with IL-6 and stroke severity. P<0.05 was considered statistically significant.
Results
Aspirin Resistance is More Prevalent in People Who have had a Stroke than Controls and is Associated with More Severe Strokes
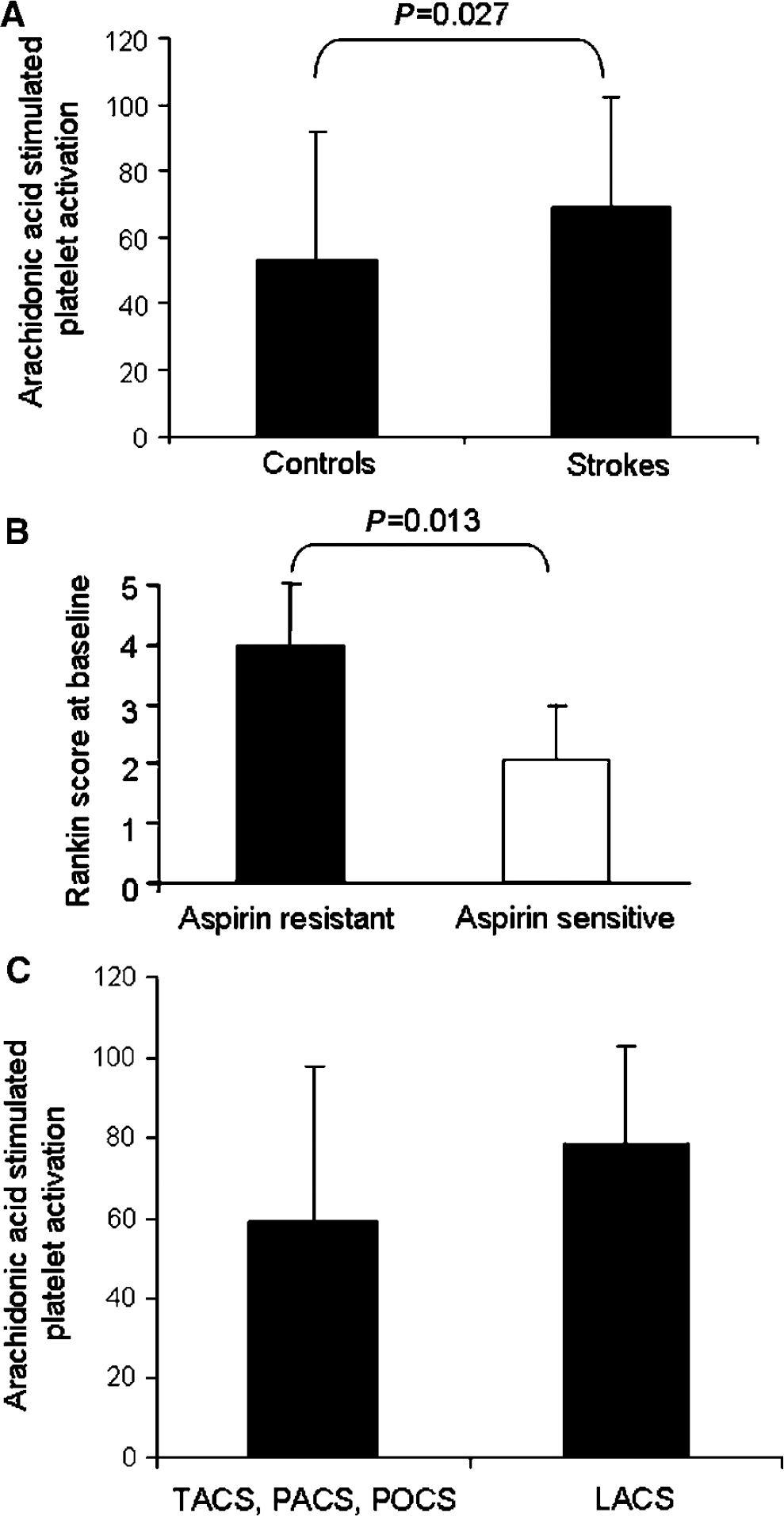
The proportion of people with aspirin resistance was significantly higher in the stroke than the control group (67% versus 40%, P=0.028). The stroke group had significantly more arachidonic acid-stimulated platelet activation than the control group (69%±33% versus 53%±39%, P=0.027; Figure 1A). Therefore, aspirin had less of an inhibitory effect on platelet activation in the stroke group than the control group. The aspirin-resistant group had more severe strokes than the aspirin-sensitive group using the Rankin score (4.0±1.15, range 1 to 5 versus 2.0±1.12, range 1 to 4, P=0.013; Figure 1B) with a similar trend (although nonsignificant) for the NIHSS score (7.6±7.2 versus 4.9±5.8, P=0.122). The baseline characteristics were similar between the control group who are aspirin sensitive, the control group who are aspirin resistant, the stroke group who are aspirin sensitive, and the stroke group who are aspirin resistant except for systolic blood pressure (Table 1).
(
Aspirin had Less of an Inhibitory Effect on Platelet Activation in People with Lacunar Strokes than Strokes of an Embolic Origin
Likely embolic strokes (TACS, PACS, and POCS, n=23) had more inhibition by aspirin than intrinsic arteriosclerotic strokes (LACS, n=16) (59%±39% versus 79%±24% platelet aggregation, P=0.020; Figure 1C). There was a trend for higher arachidonic acid-stimulated platelet activation in those taking aspirin across different stroke subgroups. This was because of higher platelet activation in the LACS (78.9%±24.0%) versus POCS (32.9%±28.2%, P=0.004), LACS versus TACS (61.6%±44.7%, P=0.067), and LACS versus PACS (64.4%±40.0%, P=0.079) subgroups. There was no significant difference in the proportion of people with aspirin resistance across stroke subgroups although there was a tendency for more people to have aspirin resistance in the LACS and PACS groups (45% LACS, 35% PACS, 10% TACS, and 5% of POCS strokes had aspirin resistance, χ2=0.771). There were no differences in age, body mass index, blood pressure, smoking status, or medication between embolic and intrinsic arteriosclerotic strokes. Lacunar stroke had significantly smaller infarct volumes than embolic strokes (53±8 versus 167±144 units, P=0.036), but there were no statistical differences in stroke severity between embolic and LACS groups, regardless of whether NIHSS or Rankin score was used. There were no differences in any of the above parameters between aspirin-resistant and aspirin-sensitive groups in either the embolic or LACS group.
Interleukin-6 is Higher in Aspirin-Resistant Stroke Patients and is Associated with Embolic Strokes and Stroke Severity
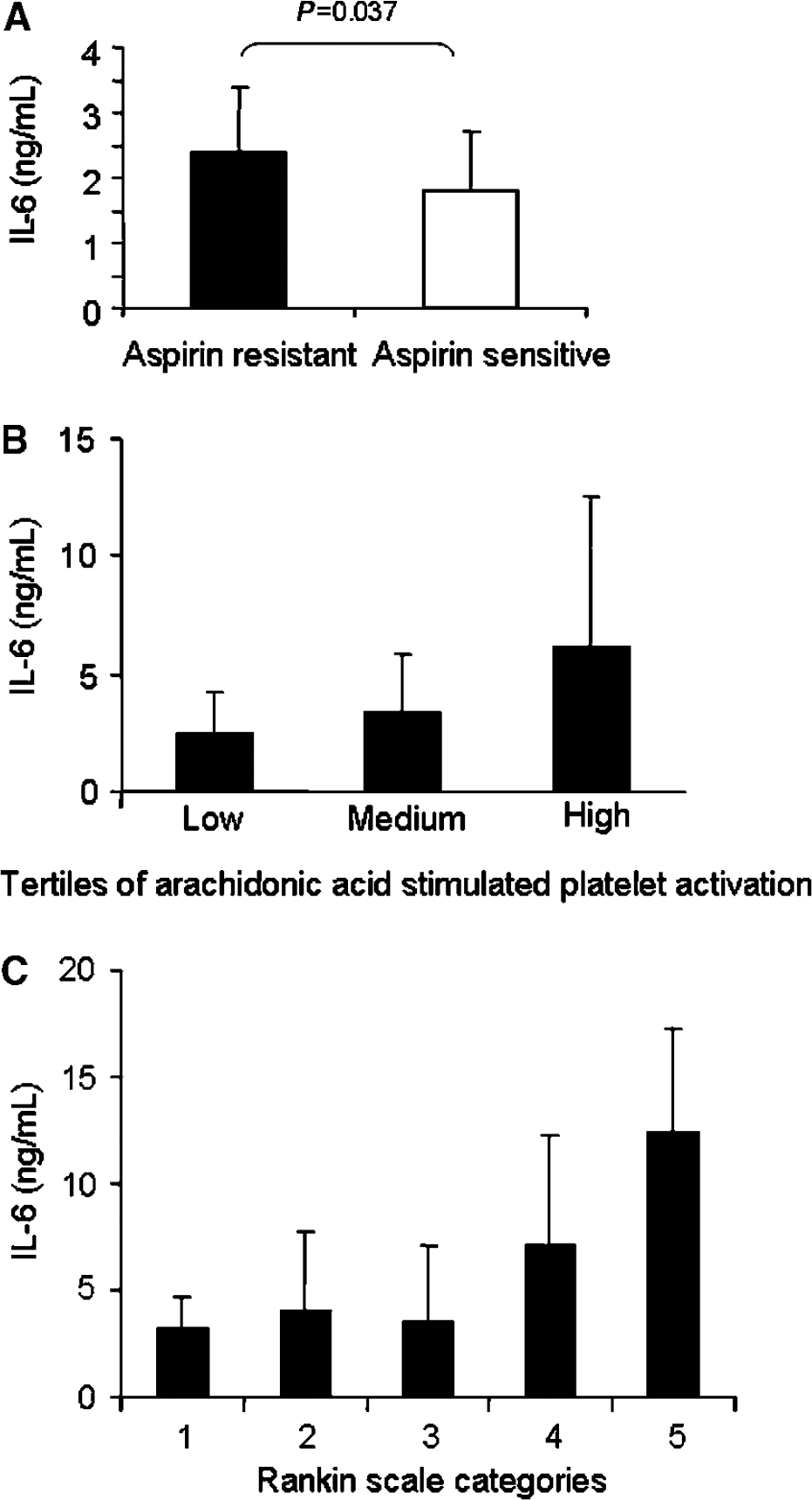
Those with aspirin resistance in the stroke group had higher levels of IL-6 than those from the stroke group who were aspirin sensitive (2.4±1 versus 1.8±0.9 ng/mL, P=0.037; Figure 2A), but there was no statistically significant difference in IL-6 between aspirin-resistant and aspirin-sensitive controls (1.7±0.8 versus 1.5±0.3 ng/mL, P=0.451). The stroke group had significantly higher IL-6 than the control group (2.2±0.9 versus 1.6±0.6 ng/mL, P=0.001). There was a significant trend for increasing IL-6 levels across tertiles of arachidonic acid-stimulated platelet activation (P=0.034; Figure 2B) in the stroke group, corroborating the suggestion that IL-6 is highest in those people who have aspirin resistance. The levels of IL-6 were also increased across Rankin score (P=0.017; Figure 2C) and NIHSS score (R=0.713, P<0.001), suggesting that plasma IL-6 was highest in the most severe strokes. There were no significant differences in plasma TNFα and IL-1β concentrations between aspirin-resistant and aspirin-sensitive groups (for the control group, IL-1β was 3.5±1.1 ng/mL in the aspirin-resistant group versus 3±1 ng/mL in the aspirin-sensitive group, P=0.384; for the stroke group, IL-1β was 3.2±0.7 versus 3.3±0.7 ng/mL, P=0.830; for the control group, TNFα was 3.9±1.6 versus 3.8±4 ng/mL, P=0.703; for the stroke group, TNFα was 3.9±0.9 versus 3.5±1.2 ng/mL, P=0.298).
(
Interleukin-6 was higher in embolic strokes than in LACS (2.4±1 versus 1.8±0.7 ng/mL, P=0.032). This may appear paradoxical when platelet activation is highest in LACS and high plasma IL-6 is associated with high levels of platelet activation. However, within each type of stroke, there was always a trend for IL-6 levels to be highest in the aspirin-resistant patients: for embolic strokes, IL-6 in the aspirin-sensitive group was 2.7±1.0 versus 2.1±0.9 ng/mL (P=0.100); for LACS, IL-6 was 4.1±2.9 versus 2.1±1.4 ng/mL (P=0.092).
Interleukin-6 is Independently Associated with Stroke Severity, and Aspirin Resistance is Independently Associated with Interleukin-6
Multivariate analysis was used to determine which factors were most important for determining stroke severity, as stroke severity has the biggest impact on quality of life after stroke. The NIHSS score was entered as the dependent variable whereas aspirin resistance (or platelet activation), embolic/LACS categorization, IL-6, infarct volume, age, smoking status, systolic blood pressure, and atrial fibrillation were entered as independent variables because they had shown differences between groups as described above or have been suggested to be important in the literature. Only plasma IL-6 independently associated with stroke severity. Interleukin-6 was associated with 57% of the variation in stroke severity (B=3.738, P=0.036, 95% confidence interval 0.282 to 7.194). When the modified Rankin score was used as the dependent variable instead of NIHSS score, there was a strong trend for IL-6 to still be the only independent variable (P=0.079).
As IL-6 was independently associated with stroke severity, we used multivariate analysis to determine independent associations with plasma IL-6. The same independent variables (minus stroke severity score) were entered with IL-6 as the dependent variable. Aspirin resistance was associated with 21.6% of the variation in plasma IL-6 (B=0.765, P=0.005, 95% confidence interval −0.021 to 0.926). When stroke severity (either NIHSS or Rankin) was included as an independent variable, aspirin resistance did not remain in the model for IL-6.
Discussion
We have shown for the first time that aspirin resistance is more common in lacunar strokes than PACS, POCS, and TACS. We have also shown for the first time that aspirin resistance, stroke severity, and inflammation are interrelated.
We have shown a higher prevalence of aspirin resistance and lack of inhibition of platelet activation by arachidonic acid in the stroke group than the control group. Both the values for the control and stroke groups are consistent with the prevalence of aspirin resistance in control and stroke groups obtained using other methods, although there is variation in the published literature (Buchanan and Brister, 1995; Knoepp and Laposata, 2005; Mason et al, 2005; Takahashi et al, 2006; Schror et al, 2006; Macchi et al, 2006). We defined aspirin resistance as greater than 50% platelet activity, in agreement with previous work (Tantry et al, 2005) showing a good correlation between the aggregometry and the Thrombelastograph Haemostasis Analyser (r=0.85, P<0.001). A 50% cutoff is also used clinically (Gwozdziewicz et al, 2006). Given that we are using the same equipment and reagents, it is likely that the proportion of people with aspirin resistance is not an overestimation. Discrepancies between studies may be because of timing, as it is plausible that platelet activation during and immediately after a stroke is thrombin-driven and therefore unaffected by aspirin, whereas platelet activation a few days after stroke may be driven by other activation pathways, for example by collagen, that are affected by aspirin. It may also be that platelet stimulation is so overwhelming at the time of stroke that aspirin has little effect.
Our data also suggest that there might be less of an effect of aspirin in LACS. Interestingly, TACS, PACS, and POCS are often embolic strokes originating in larger vessels whereas LACS is often because of intrinsic atherosclerotic disease of the smaller vessels (Bamford et al, 1991). It is possible that lack of inhibition of platelet activation by aspirin leads to formation of microthrombi or platelet—fibrin complexes that totally occlude blood flow through a vessel already narrowed by intrinsic disease in lacunar stroke.
One of the strengths of this study is that 94% of the stroke group were administered aspirin in hospital and therefore compliance was guaranteed. Of these volunteers, 37% had taken aspirin only in hospital whereas 57% had been taking aspirin regularly at home as well as when in hospital before blood samples were taken. Given that there was no difference in the proportion of aspirin resistance between these two groups, it is likely that the effect of taking different numbers of doses of aspirin does not affect the results.
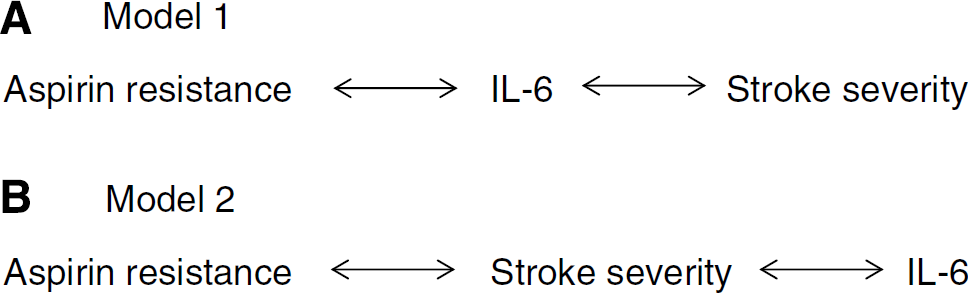
Data from this study suggest that there is interaction between aspirin resistance, IL-6, and stroke severity. The multivariate analysis suggests two potential models between these factors: aspirin resistance → IL-6 → stroke severity (model 1) and aspirin resistance → stroke severity → IL-6 (model 2).
Model 1
Figure 3A shows that aspirin resistance may be associated with stroke severity through IL-6. This is consistent with Figures 2A and 2B, which show that IL-6 is higher in people with aspirin resistance, and with Figure 2C, which shows that IL-6 is highest in those with the most severe strokes. There is biologic evidence to support this hypothesis. Aspirin resistance is associated with IL-6: Lack of inhibition of platelet activation may lead to increased arachidonic acid turnover, thromboxane, platelet-activating factor 4, and leukotriene biosynthesis and/or release. It has been shown that leukotrienes stimulate IL-6 production by phagocytes (Stankova and Rola-Pleszczynski, 1992; Gum et al, 2001), platelet-activating factor 4 stimulates IL-6 production in endothelial cells (Lacasse et al, 1997; Gum et al, 2001), and prostaglandins regulate IL-6 production in a variety of cell types (Rola-Pleszczynski et al, 1993; Gum et al, 2001). Platelets release CD40L, which increases inflammatory cytokine release from monocytes and endothelial cells, including IL-6 (Kiener et al, 1995). Aspirin is known to reduce plasma IL-6 in a variety of patient groups, and it is thought that this may occur by inhibition of the transcription factor nuclear factor-κB. Therefore, people who are insensitive to aspirin may not achieve a reduction in IL-6. The release of vasoactive molecules associated with oxidative stress and inflammation, such as isoprostanes (Kranzhofer and Ruef, 2006), may also link inflammation with aspirin resistance through priming of platelets. IL-6 is associated with stroke severity: Experimental stroke is associated with rapid and widespread activation of inflammation, including IL-6, in multiple tissues (Offner et al, 2006). In animal models, IL-6 appears to have neuroprotective effects when present at high levels in the brain, but is associated with poor outcome when present at high levels in the circulation. It is this latter effect that we have addressed in our research. Interleukin-6 controls the expression of other acute-phase proteins and is therefore important in amplifying inflammation. It also initiates tissue repair mechanisms, although it is not yet clear how systemically raised IL-6 is involved in the repair of neural tissues.
Schematic diagrams of models linking aspirin resistance, stroke severity, and IL-6. (
Model 2
Figure 3B shows the second possible model to explain the data generated in this study. This model is consistent with an association between aspirin resistance and stroke severity (Figure 1B) and between stroke severity and IL-6 (Figure 2C). As with the first model, there are possible biologic explanations for each step in the model. Aspirin resistance is associated with stroke severity: It is possible that insensitivity to aspirin leads to recurrent production of microthrombi that are not visualized by computed tomography scanning but which lead to maintenance of occlusion of flow of blood for longer periods. It is clear from trials with recombinant tissue plasminogen activator that there is short window where it is most beneficial, suggesting that the shorter the time the occlusion occurs, the less severe the stroke. Stroke severity is associated with IL-6: As discussed above, it is well known that circulating IL-6 is elevated after a stroke, although it is not clear why this is so. So, from the two models generated from the results of this study, it is possible that IL-6 is upregulated by either the inflammatory response to stroke or by aspirin resistance. The elucidation of which model is correct merits further investigation, and it is possible that both models contribute to this phenomenon.
Interleukin-6 was higher in embolic strokes than LACS, despite LACS having the highest levels of platelet activation and the relationship between plasma IL-6 and platelet activation. However, within each type of stroke, IL-6 is highest in aspirin-resistant patients, suggesting that the relationship between IL-6 and aspirin resistance/platelet activation still holds true. It has been shown previously that IL-6 is higher in patients with large infarcts rather than lacunar strokes (Beamer et al, 1995) and is related to neurologic deterioration (Castellanos et al, 2002). A relationship between IL-6 polymorphisms and lacunar strokes has also been shown previously (Revilla et al, 2002; Chamorro et al, 2005). Interestingly, it has also been suggested that the role of IL-6 might be vascular bed specific, especially in lacunar stroke (Salobir and Sabovic, 2004), suggesting that IL-6 might have differential effects in different vessels relevant to aetiological subtypes of stroke.
Given that aspirin is standard treatment after an ischaemic stroke, it is important to understand more about aspirin resistance in stroke. Aspirin irreversibly inhibits the activity of cyclooxygenase 1. By inhibiting the production of prostaglandins from arachidonic acid, aspirin impacts upon platelet activation and thrombus formation, but also probably on inflammation, endothelium, neutrophils, and antioxidant effects (Steer et al, 1997; Husain et al, 1998; Altman et al, 2004). It is possible that there are multiple reasons why aspirin does not always effectively inhibit platelet function in vitro and in vivo. Sanderson et al (2005) suggested that problems with adherence (less likely in our study because of aspirin ingestion in hospital in most cases), dosage (as adherence), comorbid conditions (not associated with aspirin resistance in our study but the numbers are small precluding a firm conclusion), and drug interactions (as for comorbid conditions) were important in causing aspirin resistance. The same authors concluded that aspirin resistance in vitro was associated with an increase in the risk for vascular events (Sanderson et al, 2005). An alternative classification of aspirin resistance is drug-related mechanisms (pharmacokinetics and pharmacodynamics, insufficient bioavailability because of drug interactions, impaired sensitivity of cyclooxygenase 1, gene polymorphisms) and disease-related mechanisms (platelet hyperactivity, changes in collagen receptor and platelet sensitivity by isoprostanes) (Schror et al, 2006). It is possible that the disease-related mechanisms are particularly important in our study, as acute stroke is associated with platelet activation, oxidative stress, and inflammation. Finally, it is unclear what effect endothelial cells and other circulating cells have on aspirin resistance (Szczeklik et al, 2005). There is evidence that aspirin resistance may contribute to the incidence of stroke (Eikelboom et al, 2002; Grundmann et al, 2003) and recurrence of stroke (Grotemeyer, 1991; Grotemeyer et al, 1993) but this is the first study to examine the effect of aspirin resistance on stroke severity. Therefore, the clinical effects and mechanisms of aspirin resistance have not yet been fully determined. What is clear is that there are plausible mechanistic reasons why aspirin resistance is clinically important in stroke and that identification of aspirin resistance and treatment by alternative platelet-inhibitory reagents should be explored further.
If we could better understand the factors linking aspirin resistance, inflammation, and severity of stroke, it may be possible to tailor treatments for the individual to aid recovery. It may be possible in the future to identify a subgroup of people with ischaemic stroke who are likely to have more severe strokes and who are characterized by aspirin resistance. These individuals may benefit from higher doses of aspirin after acute stroke, as it has been suggested that increasing the aspirin dosage may overcome aspirin resistance (Stankova and Rola-Pleszczynski, 1992; Williams et al, 2005), or from combination therapy targeting cyclooxygenase-independent pathways. The link between aspirin resistance and LACS also deserves further investigation, as it is important to identify patients most likely to benefit from antiplatelet therapy.
Footnotes
Acknowledgements
We gratefully acknowledge Research into Ageing for funding NAE's fellowship. We thank Kim Laxton and Anne Linge for assistance with collection and analysis of samples, Dr Tim Bryant and Dr Mary Gawne-Cain for calculating infarct volumes, Dr Giles Durward and Dr Pamela Crawford for help in recruitment, and Dr Sarah Wild for critically reading the manuscript. We are grateful to the Wellcome Trust for providing excellent research facilities and nursing support. We are grateful to Medicell/Haemoscope for kits, analysers, and technical assistance.
The authors declare no conflicts of interest. Neither funding source influenced study design, collection, analysis, and interpretation of data, writing of the report, or the decision to submit the paper for publication.