1079. Upregulation of blood-brain barrier drug efflux pumps by the constitutive-androstane receptor (CAR, NR1I3): a xenobiotic-activated nuclear receptor
D.S. Miller and X. Wang
Laboratory of Pharmacology, NIH/NIEHS, Research Triangle Park, North Carolina, USA
Objectives: p-Glycoprotein and breast cancer-related polypeptide (BCRP), are ATP-driven drug efflux pumps expressed at the blood-brain barrier. They are major obstacles to CNS pharmacotherapy, limiting delivery of drugs used to treat brain cancer, neuroAIDS and epilepsy.1 In peripheral barrier and excretory tissues, several ligand-activated nuclear receptors, e.g., pregnane-X receptor (PXR) and CAR, are master regulators of drug metabolizing enzymes and drug efflux transporters. We previously found that PXR was expressed in rat and mouse brain capillaries and that receptor activation in vitro and in vivo substantially increased p-glycoprotein transport activity and protein expression.2,3 Here we demonstrate CAR-mediated upregulation of p-glycoprotein and BCRP in brain capillaries.
Methods: p-Glycoprotein and BCRP transport activity was measured in isolated brain capillaries from rats and mice using fluorescent substrates (cyclosporine A and prazosin derivatives, respectively), confocal microscopy and quantitative image analysis; transporter protein expression was measured in western blots of brain capillary plasma membranes.2,3
Results: CAR protein was immunolocalized to the rat brain capillary endothelium; RT-PCR detected CAR mRNA in capillary extracts. Exposure of isolated rat capillaries to the CAR ligand, phenobarbital, roughly doubled protein expression and transport activity of both transporters. These increases were abolished by nanomolar concentrations of the protein phosphatase 2A (PP2A) inhibitor, okadaic acid, a finding consistent with the role of that phosphatase in CAR activation. In mouse brain capillaries, the specific murine CAR ligand, TCPOBOP, increased transport mediated by p-glycoprotein and BCRP, again by a PP2A-dependent mechanism. These increases were not seen in capillaries from CAR-null mice. Thus, CAR upregulated p-glycoprotein and BCRP expression and activity. Finally, dosing mice with TCPOBOP increased transport mediated by p-glycoprotein and BCRP, as well as p-glycoprotein and BCRP protein expression, all measured in isolated brain capillaries ex vivo.
Conclusions: CAR is a xenobiotic responsive transcription factor that is activated by a number of ligands, including, bilirubin, and a variety of foreign compounds, steroid hormones, and prescription drugs. The present in vitro and in vivo experiments with rats and mice show that CAR ligands upregulate expression of two key drug efflux pumps at the blood-brain barrier. Increases in transporter expression were comparable to that seen previously with PXR upregulation of p-glycoprotein. That level of transporter induction was previously associated with 70% reduction in the efficacy of the p-glycoprotein substrate, methadone, in an antinociception assay.3
177. Nadph oxidase from circulating inflammatory cells exacerbates injury in experimental stroke
X. Tang1,2, Z. Zheng1, R. Giffard2 and M. Yenari1
1Neurology, UCSF SF VAMC, San Francisco; 2Anesthesia, Stanford University School of Medicine, Stanford, California, USA
NADPH oxidase (Nox2) is a major enzyme system which generates superoxide generation in inflammatory cells, but has recently been found in non inflammatory cells such as endothelial cells and neurons. Here we show that Nox2 contributes to experimental stroke, especially in circulating inflammatory cells. Experimental stroke was produced in mice by 2 h transient middle cerebral artery occlusion (tMCAO) with an intraluminal suture model, followed by 22 h reperfusion. Three different paradigms were studied:
Mice treated with the Nox2 inhibitor, apocynin (Apo, 2.5 mg/kg IV 30 min prior to reperfusion) or vehicle (Veh),
Nox2 deficient (X-CGD, deficient in the gp91 subunit) versus wildtype (Wt) mice, and
to determine whether Nox2 in circulating cells versus brain resident cells contribute to ischemic injury, bone marrow chimeras were generated by transplanting bone marrow from Wt or X-CGD into X-CGD or Wt, respectively.
Brains were assessed for infarct volume, hemorrhage, in situ O•− detection, as well double labeling for O•− in neurons (NeuN), endothelial cells (CD31) and microglia (CD11b). Brain tissue within peri-infarct regions was sampled for Western blots. Infarct size was reduced whether Nox2 was pharmacologically inhibited (by 37% versus vehicle, P<0.05) or genetically absent (by 54% versus Wt, P<0.001). This was also associated with reduced incidences of cerebral hemorrhage (17% versus 58%, Apo versus Veh; 14% versus 58%, X-CGD versus Wt). After ischemia, most of the O•− was generated by neurons, some microglia, and rare endothelial cells. O•− was markedly reduced by Apo treatment and in X-CGD mice in all cell types (1% versus 448%, Apo versus Veh; 11% versus 448%, X-CGD versus Wt). Apo treatment and X-CGD mice showed decreased MMP9 (40% versus 86%, Apo versus Veh; 50% versus 86%, X-CGD versus Wt) and decreased loss of ZO-1(182% versus 27%, Apo versus Veh; 48% versus 27%, X-CGD versus Wt). Infarcts in Wt mice who received Nox2 deficient marrow (40.1±6.7 mm3) were decreased significantly compared to either the Wt mice who received Wt marrow (100.4±9.9 mm3, P<0.01) or X-CGD mice who received Wt marrow (74.5±6.5 mm3, P<0.05). Furthermore, mice receiving Nox2 deficient marrow had smaller infarcts than X-CGD deficient mice transplanted with Wt marrow. We conclude that either pharmacologic or genetic inhibition of Nox2 leads to reduced brain injury and hemorrhage, and is correlated to decreased O•− and MMP9 expression and prevents the loss of ZO-1. Nox2 originating from the circulating inflammatory cells contributes more to exacerbating experimental stroke than that of the brain resident cells.
990. Blood brain barrier endothelial cells are directly involved in ischemic preconditioning
R. Gesuete1, E.R. Zanier1, F. Orsini1, L. Garzetti1, M. Deli2 and M.G. De Simoni1
1Mario Negri Institute, Milan, Italy; 2Biological Research Centre, Szeged, Hungary
Objective: Ischemic PreConditioning (IPC) is a phenomenon whereby a sub-threshold ischemic insult activates neuroprotective pathways.1 Although the concept of preconditioning has been widely studied, little attention has focused on the effects of preconditioning on cerebrovascular unit. Endothelial cell dysfunction may play an important role in the pathophysiology of cerebral ischemia. Disruption of the endothelial cells that form the blood-brain barrier (BBB) may lead to edema formation that can exacerbate brain injury. In this study we addressed the involvement of the cerebrovascular unit in IPC using an in vitro model of BBB.
Methods: Primary mouse Brain Microvessel Endothelial Cells (BMECs) co-cultured with primary mouse mixed glial cells were used to model BBB. In this condition the endothelial cells form a differentiated monolayer that retains the endothelial markers and the same properties of in vivo cerebral endothelium.2 Short Oxygen-Glucose Deprivation (1h-OGD) was used as preconditioning stimulus (PC) and administered 24 h before severe 5h-OGD. Twenty-four and 48 h after the severe OGD we evaluated the effect of IPC on the trans-endothelium electrical resistance (TEER), endothelial permeability coefficient (Pe) for paracellular (NaFluorescein) and transcellular (Albumin) transport and the presence of tight junction proteins.
Results: In basal conditions the endothelial cells showed the typical features of functional cerebral endothelium, i.e. high TEER values (⩾100 Ωcm2), low Pe for NaFluorescein (0.53±0.02 × 10−3 cm/min) and Albumin (0.02±0.005 × 10−3 cm/min) values and expression of the tight junction protein ZO-1.
Twenty-four hours after severe OGD the endothelial cells showed a dramatic reduction in TEER values (73.1±2.2 Ωcm2), a significant increase in Pe values for both NaFluorescein (1.8±0.35 × 10−3 cm/min) and Albumin (0.29±0.07 × 10−3 cm/min) and a loss of the ZO-1 continuous staining pattern. The administration of 1h-OGD 24 h before a severe OGD significantly ameliorated all the parameters observed. In particular 24 h after severe OGD the preconditioned cells did not show significant changes in TEER values (111.2±5.8 Ωcm2) and Pe values for both NaFluorescein (0.98±0.11 × 10−3 cm/min) and Albumin (0.12±0.01 × 10−3 cm/min) compared to control condition. Similarly, the immunostaining for ZO-1 showed that the pattern of the staining for this tight junction protein was not affected in preconditioned cells compared to control cells.
Conclusions: The present study shows for the first time that IPC significantly attenuates OGD-induced brain endothelial dysfunction. We observed that a short exposure to OGD acts as an IPC stimulus preventing the reduction of TEER values, the loss of paracelluar and transcelluar Pe selectivity and the disruption of the ZO-1 organization induced by OGD. These data indicate that the BBB endothelial cells can be directly preconditioned and that the cerebrovascular unit may have an important role in IPC neuroprotection.
1028. Assessment of cerebral blood flow and blood-to-brain transfer constant for GD-DTPA in ischemic brain tissue in diabetic and normal rats
R. Knight1, T. Nagaraja2, K. Keenan2, J. Xu2, K. Karki3, P. Whitton3 and A. Ergul4
1Neurology NMR Research, Henry Ford Health System Hospital; 2Anesthesiology; 3Neurology NMR Research, Henry Ford Hospital, Detroit, MI; 4Physiology, Medical College of Georgia, Augusta, Georgia, USA
Introduction: Diabetes increases the risk of cerebrovascular disease four- to six-fold and produces marked remodeling of cerebral vessels.1 To evaluate the effects of diabetes in stroke, transient focal cerebral ischemia was induced in Wistar and Goto Kakizake (GK) rats by middle cerebral artery occlusion (MCAO). The GK strain is analogous to Wistars, but the animals develop type 2 diabetes spontaneously at 4 to 6 weeks of age. This study uses magnetic resonance imaging (MRI) measures of cerebral blood flow (CBF) and blood-brain barrier permeability to assess the effects of diabetes on stroke outcome.
Methods: Transient ischemia was induced in male Wistar (n = 6) and GK rats (n = 7) by intraluminal suture MCAO and reperfusion by withdrawal of the occluding filament after 3 h. Blood glucose and glycosylated hemoglobin (A1c) levels were measured in all GK animals, whereas blood glucose was measured in 3 Wistar rats. MRI data included T2-weighted imaging, CBF and BBB permeability assessments and were performed at 7 Tesla. Quantitative assessment of ischemia induced CBF changes was performed at baseline, during ischemia and acute reperfusion and at 24 h. BBB permeability changes were assessed at approximately 2 and 24 h after reperfusion using Look-Locker based T1-weighted imaging to generate estimates of the blood-to-brain transfer constant (Ki) for Gd-DTPA via Patlak plot methodology.2 Ischemia damaged regions of interest (ROIs) were identified by thresholding T2 values, whereas ROIs with BBB opening were indentied based on an F-test statistic. After MRI, the rats were sacrificed and brain tissues stained with 2,3,5-Triphenyltetrazolium chloride (TTC).
Results: Average blood glucose levels for the Wistar and GK groups were 145±12 and 255±12, respectively. A1c values were 5.5±0.4 for the GK group. Average baseline CBF values (Figure 1) in the GK group were significantly lower than in the Wistar group. Ischemic region CBF values for the GK animals decreased significantly during ischemia and then rebounded slightly toward baseline levels during acute reperfusion and at 24 h. CBF values fell to roughly the same level during ischemia in the Wistar group. Contralateral ROI CBF became significantly elevated compared to baseline levels in the GK group during ischemia and early reperfusion, but not in Wistars. Acute BBB disruption was detected in the preoptic area and/or striatum in all rats by Gd-DTPA enhanced MRI. Measured Ki values were significantly elevated relative to corresponding contralateral brain regions with intact BBB function, but did not differ significantly between Wistar and GK groups. The average size of the BBB damaged ROI, however, was marginally larger in the Wistar group (154±72) than in the GK group (89±84) during acute reperfusion (P = 0.08) and significantly larger (236±134 versus 122±96, respectively) at 24 h (P = 0.05). Likewise, the size of the ischemia-damaged region was larger in Wistars than in GK rats as assessed from the TTC stained sections.
Conclusions: The differences noted in CBF and BBB permeability between control and diabetic rats suggest that diabetes may play an important role in influencing outcome in stroke.
107. Microglia contribute to sepsis induced injury to bbb components via the NF-κB, JAK-STAT and map kinase pathways
R. Kacimi1, R. Giffard2 and M. Yenari3
1Neurology Department, University of California San Francisco and Stanford University, San Francisco; 2Department of Anesthesia, Stanford University, Stanford; 3Neurology Department, University of California San Francisco, San Francisco, California, USA
Cerebral inflammation can lead to stress-induced cell death during sepsis and exacerbates injury during ischemia and stroke. Microgliosis or microglia activation and subsequent inflammatory mediator release can promote neurovascular dysfunction in the injured brain. The present study was undertaken to evaluate whether iNOS signaling modulation is a prosurvival strategy in endotoxin-activated microglia. We used 3 cell types (microglia BV2, bEND.3 endothelial cell and primary astrocytes) studied either alone or in co-culture to assess whether BV2 cells potentiates injury to microvascular bEND.3. LPS was highly effective in stimulating NO production in BV2 cells. LPS also induced de novo synthesis of iNOS protein. Furthermore, viability assays showed that LPS induced cell death under these conditions. Moreover, NO and the peroxynitrite donor SIN-1 induced dose dependent NO accumulation and mimiked LPS induced nitrosative stress. While oxidative stress (OGD or H2O2) increased bEND.3 permeability and induced cell death, LPS had no effect on eNOS/NO or viability in bEND.3 microvascular endothelial cells alone. Interestingly, in co-culture with BV2 and bEND.3 cells, microglia induced monolayer disruption after LPS exposure. Pretreatment with NOS inhibitors (L-NMMA and aminoguanidine, P<0.01), ROS inhibitors (apocynin and allopurinol, P<0.01), and minocycline (P<0.01) to prevent microglia activation, prevented bEND.3 injury in co-culture. We further assessed the signaling implicated in iNOS/NO activation. Inhibitors of NF-κB and JAK-STAT (PDTC and AG490) respectively, abrogated NO accumulation. While Worthmanin, PD9805, SB203850 (PI3K, ERK, and p38 kinase inhibitors) respectively did not prevent NO accumulation by LPS in BV2 cells. SP600125, a C-jun N-terminal kinase (JNKs/SAPKs) inhibitor, however, partially prevented NO accumulation. Conversely, NF-kB, JAK/STAT, JNKs/SAPKs pathways blockade were shown to be protective against LPS induced cell death (60 to 70% viable cell versus 20% in LPS treated cell, P<0.01). Our data show that differential signaling pathways mediates endotoxin- dependent activation of inducible nitric oxide synthase involving transcription factor NF-κB and JAK-STAT. Our data confort the importance of microglia iNOS in inflammation induced injury. This possibility is emphasized by the observation that its modulation prevents the underlying oxidative/nitrosative stress induced injury in brain murine microglia BV2 cells. We also show that activated microglia -induced blood barrier disruption and microvascular endothelial cell death in co-culture model. In a similar fashion as NOS inhibitors (that inhibits NOS activity) ROS inhibitors (apocynin, allopurinol), JAK-STAT and NF-κB and JNK signaling pathways appears to be potent negative regulator of iNOS expression and/or NOS activity and show to be useful anti-inflammatory strategy to confer cytoprotection to maintain the integrity of BBB vascular unit. Our observations also shed light on new pathways that may be potential therapeutic targets to modify pro-inflammatory responses leading to increased damage of the BBB and its subsequent neuronal damage during sepsis, cerebral ischemia and stroke.
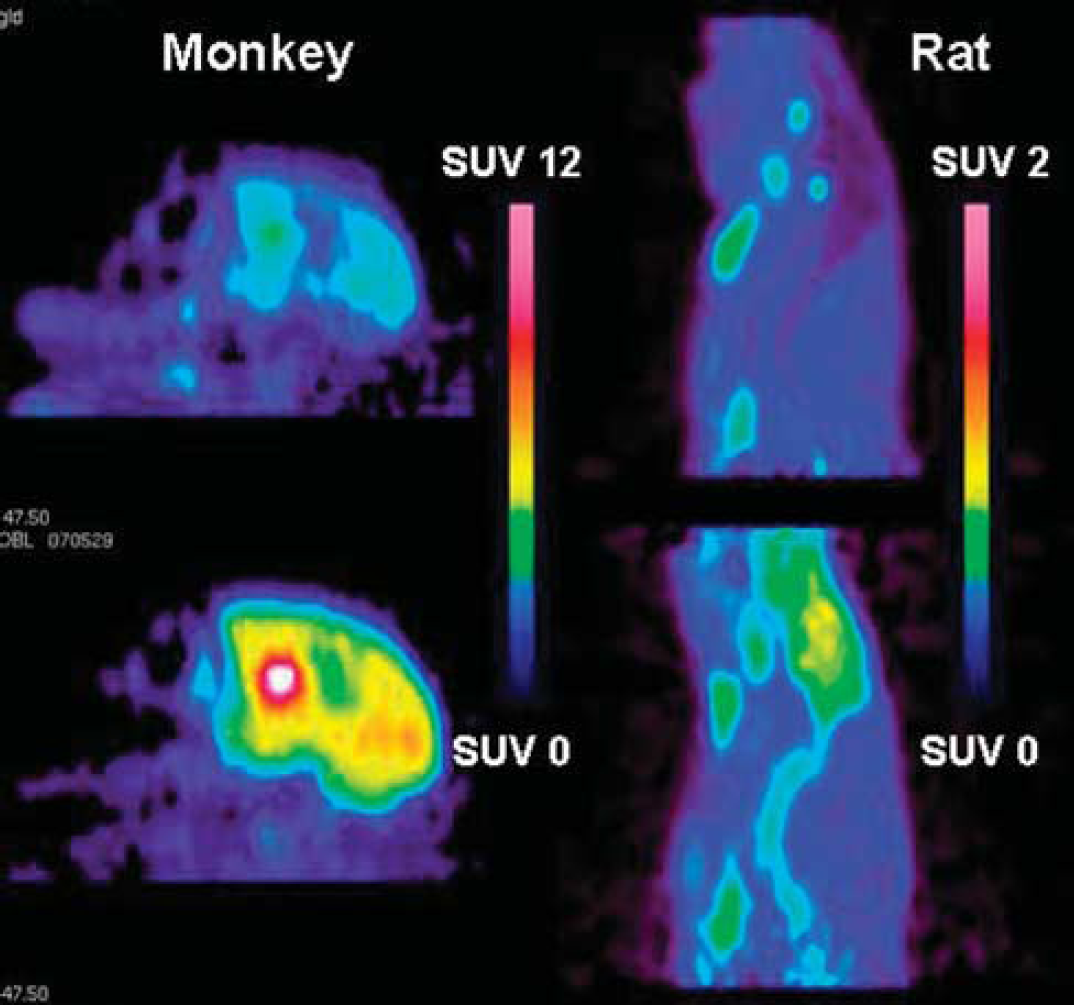
226. Species differences in brain uptake of three pet radioligands
S. Syvänen1, Ö. Lindhe2, M. Palner3, B.R. Kornum3, O. Rahman4, B. Långström4, G.M. Knudsen3 and M. Hammarlund-Udenaes5
1Division of Pharmacology, LACDR, Leiden University, Leiden, The Netherlands; 2Uppsala Imanet, GE Healthcare, Uppsala, Sweden; 3Neurobiology Research Unit, University Hospital Rigshospitalet, Copenhagen, Denmark; 4Uppsala Applied Science Lab, GEMS PET Systems, GE Healthcare; 5Department of Pharmaceutical Biosciences, Uppsala University, Uppsala, Sweden
Objectives: This Positron Emission Tomography (PET) study was designed to compare interspecies differences in brain uptake of three radiolabelled P-glycoprotein (P-gp) substrates; [11C]verapamil, a calcium channel blocker which has been used extensively in PET1–3; [11C]GR205171, a NK1-receptor antagonist; and [18F]altanserin, a 5HT2A-receptor antagonist.
Methods: The radioligands were prepared as previously reported4–6 and administered as a fast bolus to all species at the start of the emission scan. Brain radioligand concentrations, expressed as standardized uptake values (SUV), and brain-to-plasma SUV ratios were compared; [11C]verapamil in rats, guinea pigs and monkeys; [11C]GR205171 in rats, guinea pigs, monkeys and humans; and [18F]altanserin in rats, minipigs and humans. The effect of P-gp inhibition was investigated by administering cyclosporin A (CsA) 30 mins prior to the radioligand bolus injection. Blood samples were withdrawn and analyzed using HPLC to determine the concentration and metabolism of unbound radioligand in plasma.
Results: Pronounced species differences were found in the brain SUV and brain-to-plasma SUV ratio of [11C]verapamil, [11C]GR205171 and [18F]altanserin with higher brain distribution in humans, monkeys and minipigs than in rats and guinea pigs. For example, the brain-to-plasma SUV ratio of [11C]GR205171 was almost 9-fold higher in humans compared to rats. The species differences were still present after P-gp inhibition although the increase in brain concentrations after P-gp inhibition was somewhat greater in rats than in the other species. Differences in plasma concentrations, plasma protein binding and metabolism did not explain the species-related differences.
Conclusions: The findings are important for the interpretation of drug delivery to the brain when comparing or extrapolating preclinical to human data. Compounds found to be P-gp substrates in rodents are likely to be substrates also in higher species, but sufficient blood-brain barrier permeability may be retained in humans to allow the compound to act on intracerebral targets.
[11C]GR205171 brain uptake.
References
1.
MillerDSBauerBHartzAM. Modulation of P-glycoprotein at the blood-brain barrier: Opportunities to improve central nervous system pharmacotherapy. Pharmacol Rev2008;60:196–209.
2.
BauerBHartzAMFrickerGMillerDS. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol Pharmacol2004;66:413–29.
3.
BauerBYangXHartzAMOlsonERZhaoRKalvassJCPollackGMMillerDS. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol Pharmacol2006;70:1212–19.
4.
DirnaglUMeiselA. Endogenous neuroprotection: Mitochondria as gateways to cerebral preconditioning?Neuropharmacology2008;55:334–44.
5.
DeliMAAbrahámCSKataokaYNiwaM. Permeability studies on in vitro blood-brain barrier models: Physiology, pathology, and pharmacology. Cell Mol Neurobiol2005;25:59–127.