20. Role of astroglial pentose phosphate pathway in ROS production under acutely increasing glucose concentrations
Y. Izawa, S. Takahashi and N. Suzuki
Department of Neurology, Keio University School of Medicine, Tokyo, Japan
Objectives: Brain function is exclusively dependent on the oxidative metabolism of glucose. Reactive oxygen species (ROS) derived from mitochondria in the neural cells play an essential role in brain aging as well as in neurodegenerative disorders. Hyperglycemia is well known to enhance ROS production, resulting in oxidative stress in vascular endothelial cells (Brownlee, Diabetes 2005;54:1615–25). Although acute hyperglycemia does not alter the rates of glucose utilization in the brain as measured using the [14C]deoxyglucose method (Orzi et al.J Cereb Blood Flow Metab 1988;8:346–56), whether the oxidative metabolism of glucose is affected remains to be elucidated (Blomqvist et al.Acta Physiol Scand 1998;163:403–15). Therefore, the effect of acutely increasing glucose concentrations on oxidative stress in different types of neural cells remains unclear.
We reported that chronic (2 weeks) exposure to a high glucose environment (22 mmol/L) suppresses the oxidative metabolism of glucose in astroglia but not in neurons (Abe et al.J Cereb Blood Flow Metab 2006;26:153–60). Moreover, even short-term exposure (24 h) to high glucose elicits a similar suppression in astroglia. In the present study, we examined the effect of acutely increasing glucose concentrations on the rates of conversion of D-[14C]glucose ([14C]glc) to 14CO2, production of ROS, and GSH formation in cultured rat neurons and astroglia.
Methods: Primary neurons and secondary astroglia were prepared from SD rats, as described previously. The rates of total glucose oxidation (y: pmol glucose/mg protein/60 min) based on the conversion from [U-14C]glc to 14CO2 over 60 mins were measured at different concentrations of D-glucose (x: 2, 10, 20 mmol/L) containing tracer doses of [U-14C]glc (0.5, 2.5, and 5 mmol/L, respectively).
The activity of the pentose phosphate pathway (PPP) was measured essentially as described by Hothersall et al. (Arch Biochem Biophys 1979;198:478–492) based on the determination of the difference in 14CO2 production from [1-14C]glucose (decarboxylated by the 6-phosphogluconate dehydrogenase-catalyzed reaction and by the Krebs cycle) and from [6-14C]glucose (decarboxylated only by the Krebs cycle).
The production of ROS, mainly H2O2, in cells was assessed using H2DCFDA and semi-quantitative fluorometric measurements (Ex/Em: 485/530 nm) and that of GSH in astroglia was assessed using monochlorobimane (Chatterjee et al.Glia 1999;27:152–161) (Ex/Em: 360/460 nm); the results were expressed as the percent-increase in the fluorescent signal over 60 mins.
Results: Increasing glucose concentrations elicited linear increases in glucose oxidation both in neurons (yn = 0.26x+9.81; R2 = 0.996) and astroglia (ya = 0.04x+0.75; R2 = 0.995). ROS production in neurons also increased (100%, 115%, 145%), while ROS production decreased somewhat in astroglia (100%, 70%, 15%). Neither mixed cultures of neurons & astroglia nor neurons grown on an astroglial cell layer showed an increase in ROS production. Increasing glucose concentrations enhanced PPP activities in astroglia, resulting in elevated CO2 production, GSH formation, and ROS reduction.
Conclusions: These results indicate that astroglial PPP exerts a protective role against oxidative stress under acutely elevated glucose environments, such as hyperglycemia in diabetic patients.
230. Astrocytes—the key sensors of O2 tension in the brain?!
T. Kulik1, S. Aronhime1 and H.R. Winn2
1Neurosurgery, Mount Sinai School of Medicine; 2Neurosurgery, Mount Sinai Hospital, New York, New York, USA
Background: Adenosine (Ado) is an endogenous purine nucleoside, whose level is markedly increased during hypoxia,1 causing an Ado 2a receptor mediated vasodilatation.2 The cellular source of Ado under hypoxic conditions remains elusive, but astrocytes have been implicated. They are a critical component of the glial-vascular unit: Interposed between neuron and cerebral blood vessels, they are strategically positioned to affect cerebral blood flow in response to neuronal activity.
Objective: We sought to determine if astrocytes produce Ado in response to changes in O2 levels.
Methods: Primary, mixed cultures were established from the cerebral hemispheres of 1–3 day old Wistar rat pups. Cultures were purified based on differential adhesion after 5–7 DIV and a high purity of astroyctes (>90%) ascertained with GFAP/DAPI immunolabeling. Astrocytes were trypsinized after 11 DIV and attached to microcarrier beads kept in spinner flasks. After 23 DIV these flasks were flushed with N2, and dissolved O2 in medium measured continuously utilizing a fluorometric technique. Samples of supernatant were acquired at predetermined time intervals and given O2 levels. Using solid phase extraction, samples were cleaned, the eluent nitrogen evaporated, reconstituted and submitted to HPLC analysis for adenine nucleotides. Compounds of interest were identified by retention time as well as wavelength spectrum, and confirmed by enzymatic peak-shifting techniques and spiking of samples.
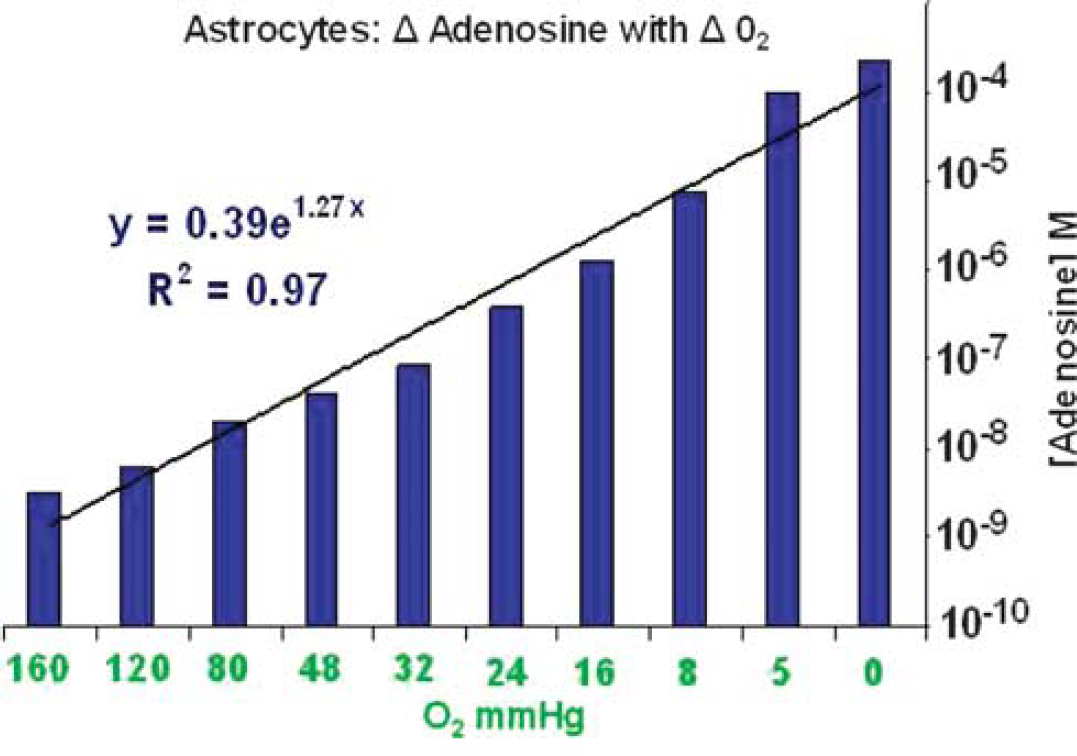
Results: Levels of Ado rose rapidly from 25.2±0.3 nmol/L at normoxia to 173.2±94.5 μmol/L at anoxia (Figure). This increase represents a >10,000 × elevation from baseline levels and was significant (P<0.005). We derived the following equation: y = 0.39e1.27x (R2 = 0.97), where y = Ado concentration (M) and × = O2 (mm Hg).
Conclusion: Astrocytes release Ado in an O2 tension dependant fashion. The rise in Ado concentration occurs rapidly. In addition, concentrations of Ado produced by astrocytes are similar to those found in the hypoxic brain1 and are in the range which evokes vasodilatation of penetrating arteries.3 We would conclude that astrocytes may be the main source for vasoactive adenosine in the hypoxic brain.
255. Astrocytes are neuroprotective against transient forebrain ischemia in CA3 hippocampus
S. Yamamoto, Y. Wang, T. Sakurai and S. Terakawa
Photon Med Res Ctr, Hamamatsu Univ Sch Med, Hamamatsu, Japan
Objective: The recent evidences suggest that the astrocytes participate in the excitatory neurotransmission. In the present study, we sought to determine the role of astrocytes in the process of delayed neuronal death (DND) following transient forebrain ischemia in rats.
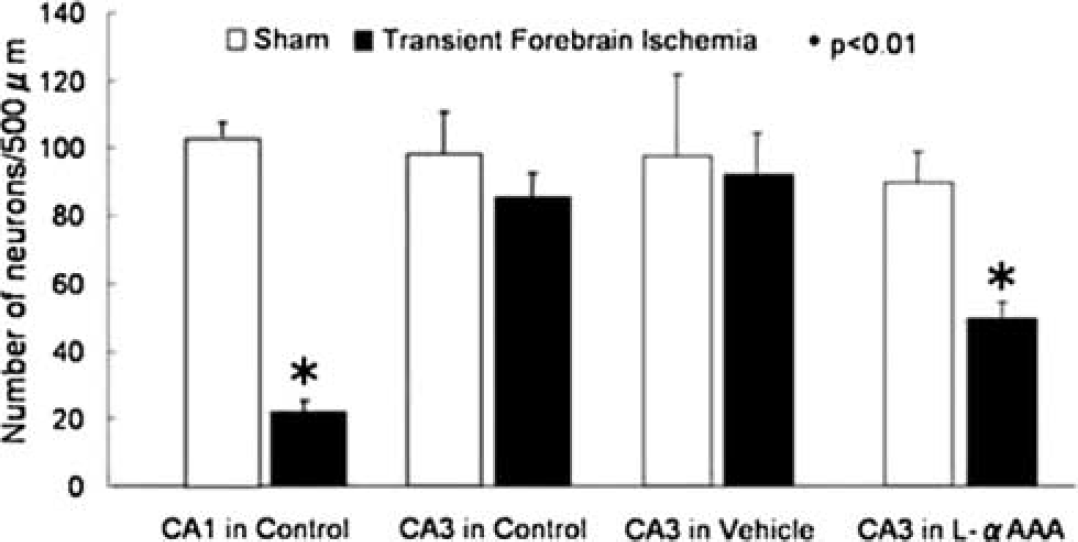
Methods: In anesthetized SD rats (300 g), the studies were performed. Three days before 4-vessel occlusion (4VO), L-α-aminoadipic acid (L-αAAA, 2 mmol/L, 10 μl), a gliotoxin, was injected by pressurized bolus into rt. CA3 hippocampus. Transient (10-min) forebrain ischemia was induced by 4VO, CBF was measured by laser-Doppler flowmetry, and intracellular Ca2+ concentration ([Ca2+]i) changes were examined by intravital fluorescence imaging using fluo3/AM, a Ca2+ indicator. To obtain the images, we employed the fiber-coupled confocal microscope, which is capable of observing confocal images inside the brain.1 In different animals, seven days after 4VO, numbers of residual neurons were counted on HE-stained slices. The results were compared among the control group (non-treated rats), the L-αAAA group (L-αAAA-injected rats), and the vehicle group (aCSF-injected rats).
Results: Three days after the injection, glial fibrillary acidic protein (GFAP) expression decreased in rt. CA3 of the L-αAAA-treated brains, indicating the reduction of astrocytes. 4VO decreased CBF (mean±s.d.%) to 25±18 in CA1 (n = 6) and to 20±11 in CA3 (n = 6) of the control group, and to 28 ± 5 in CA3 of the L-αAAA group (n = 5). They did not significantly differ among the groups. DND was remarkable, seven days after 4VO, in CA1 but not in CA3 of the non-treated brains (n = 5) (Figure 1). In rt. CA3 of the L-αAAA-treated brains (n = 4), but not of the vehicle-treated brains (n = 4), the comparable DND was noticed (Figure 1). In the control group, the [Ca2+]i increased in CA1 (n = 6) upon 4VO and returned to the baseline level after 30-min reperfusion, although in CA3 (n = 6) the increase was significantly (P<0.05) less. Interestingly, in CA3, L-αAAA (n = 5) but not vehicle (n = 5) produced similar [Ca2+]i increases to those in CA1 of the control group upon 4VO.
Effect of Gliotoxin on Delayed Neuronal Death.
Conclusion: We observed that:
L-αAAA, a gliotoxin, decreased a number of astrocytes in CA3 three days later, but did not change CBF responses upon 4VO;
L-αAAA induced DND in CA3 seven days after 4VO, while without ischemia it did not induce it; and
L-αAAA produced similar calcium responses in CA3 upon 4VO to those observed in CA1, a vulnerable region.
These results suggested that astrocytes play neuroprotective roles in CA3 through suppressing [Ca2+]i increase against ischemia/reperfusion.
Supported by the grant from MEXT #18053010 (SY).
412. Effect of S100B on cellular injury and glial activation
A. Kleindienst1, Z. Qu2, C. Stadelmann3, F. Hesse1, I. Emtmann1 and M. Buchfelder1
1Department of Neurosurgery, University Erlangen-Nuremberg, Erlangen; 2Department of Neurosurgery, 3Department of Neuropathology, University of Göttingen, Göttingen, Germany
Objective: The supposed contrasting effects of S100B, the beneficial one in acute injury like traumatic brain injury (TBI), and the detrimental one in chronic injury and neurodegeneration like Alzheimer′s disease, have been speculated to be due to a variation of the exposure to S100B. Studies in S100B transgenic mice suggest S100B to accelerate neuronal development.1 We demonstrated an exogenous S100B treatment to enhance hippocampal neurogenesis and to improve cognitive recovery following experimental TBI in the rat.2 The purpose of the present study was to elucidate temporal and spatial effects of S100B on cellular injury, microglial and astrocyte activation in the same model of acute brain injury.
Methods: Following lateral fluid percussion injury in male Sprague-Dawley rats (n = 32), we infused S100B (50 ng/h) or vehicle into the lateral ventricle for 7 days using an osmotic micro-pump. The animals were sacrificed on day 5 or 5 weeks post-injury, and 5 mm sections, 100 mm apart (bregma −3.3 to −5.6 mm) were analysed histologically. Cell death was assessed using TUNEL and hematoxylin-eosin staining, activation of astrocytes was examined applying the glial markers GFAP and S100B, microglial activation by ED1 immunostaining, and axonal damage by APP.
Results: TUNEL-positive cells were present directly beneath the lesion site in vehicle and S100B-treated animals on day 5 post-injury (238±6 and 234±24 cells/mm2, respectively, n.s.), but not after 5 weeks. The intraventricular S100B infusion did not significantly affect the early (TBI P = 0.004, TBI+S100B P = 0.036) or late (TBI P = 0.039, TBI+S100B P = 0.002) axonal injury, but resulted in an unspecific microglial activation opposite to the injury site as documented by an increased ED1 expression (TBI+S100B P = 0.001). After 5 weeks, both injury and S100B treatment resulted in an increased number of GFAP expressing cells in the corpus callosum (TBI P = 0.005, TBI+S100B P = 0.003, sham+S100B P = 0.005), while in the hippocampal granular cell layer (GCL) this effect was only present in non-injured control animals (P = 0.048). The S100B expression in the GCL was increased by a S100B treatment after 5 weeks, both in injured and non-injured animals (P = 0.017 and P<0.001).
Conclusion: In accordance to the participation of S100B in injury-induced cell proliferation, we found the S100B expression in the germinative area of the hippocampus on the lesion side to be significantly enhanced by an intraventricular infusion at 5 weeks post-injury. The S100B infusion in non-injured control rats provoked a symmetrically increased S100B expression and astrocytosis after 5 weeks. S100B did not exert an effect on early cell death or axonal injury. The significance of some delayed APP and ED1 accumulation and reactive astrocytosis has to be clarified by long-term experiments.
480. Activation of astrocytes by albumin is mediated by p38 MAPK and ERK, but not JNK, -dependent pathways
H. Ralay-Ranaivo1,2 and M. Wainwright1,2
1Department of Pediatrics, Division of Neurology, Northwestern University, Feinberg School of Medicine; 2Center for Interdisciplinary Research in Pediatric Critical Illness and Injury, Children's Memorial Research Center, Chicago, Illinois, USA
Background and aims: Albumin, which may infiltrate brain parenchyma after compromise of the blood brain barrier, has been implicated in the evolution of neurologic injury and mechanisms of epileptogenesis. Astrocytes are activated in response to neurologic insults and contribute to the mechanisms of repair, neurologic injury, or susceptibility to subsequent neurologic injury. The contribution of albumin to astrocyte activation is not well understood. This study aims to determine the effect of albumin on astrocyte injury, activation and release of inflammatory markers. Here, we tested the hypothesis that specific mitogen activated protein kinase (MAPK) pathways (p38 MAPK; extracellular signal-regulated protein kinase, ERK; and c-Jun N-terminal kinase, JNK) selectively regulate astrocyte responses to albumin.
Methods: Enriched astrocytes were cultured from neonatal rat brains and treated with 0.1 mmol/L bovine serum albumin. Astrocyte activation was quantified by serial measurements of the pro-inflammatory cytokine IL-1β, S100B, iNOS, and the nitric oxide metabolite, nitrite. Cell injury was quantified by lactate dehydrogenase (LDH) release. MAPK activation was quantified by Western blotting. The level of the regulatory chemokine fractalkine (FKN) produced by astrocytes in response to albumin was measured by ELISA. To distinguish the specific contribution of p38MAPK, ERK or the JNK pathway to each of these albumin-induced responses, cells were treated with specific MAPK inhibitors (SB203580, PD98059, and SP600125).
Purpose: To determine the specific MAPK pathways which regulate cell injury, cytokine release, chemokine release and increase in NO in astrocyte responses to albumin.
Results: After 24 h exposure, albumin induced an increase in the release of LDH, consistent with cell injury. Albumin induced a significant increase in the levels of the inflammatory markers IL-1β and nitrite released in the media as well as the level of iNOS in the cell lysates. Notably, levels of S100B levels were decreased under the same conditions, while levels of the chemokine FKN were increased by treatment with albumin. Albumin activated all MAPKs, producing increases in the levels of phosphorylated p38 MAPK, ERK, and JNK as early as 30 mins. The increase in LDH and the decrease in S100B produced by albumin were not dependent on MAPK activation. In contrast, inhibition of p38MAPK and ERK pathway, but not JNK partially attenuated the albumin-induced increases in IL-1β, nitrite and FKN.
Conclusions: Albumin activates all MAPKs in cultured astrocytes. Exposure to albumin is both cytotoxic to astrocytes and induces markers of glial activation including both IL-1β and iNOS. In response to albumin, astrocytes produce divergent response in levels of S100B (decreased) and FKN (increased). Of these responses, the increase in the pro-inflammatory cytokine IL-1β and the increase in nitrite are MAPK-dependent, mediated by p38MAPK and ERK, not JNK. These findings identify a role for specific MAPK signaling pathways in the mechanisms of astrocyte activation produced by albumin and implicate these pathways in the protective and pathologic responses to albumin in the CNS.
524. Neurometabolic origin of flavoprotein autofluorescence signal in the cerebellar cortex
W. Gao1, K. Reinert2, X. Wang1, C. Gang1 and T. Ebner1
1Neuroscience, University of Minnesota, Minneapolis, Minnesota; 2Neurology, Systemic Neuroscience Inst., University of Pittsburgh, Pittsburgh, Pennsylvania, USA
Objectives: Flavoprotein autofluorescence imaging in the cerebellar cortex in vivo revealed that stimulation of the parallel fibers evokes a beam-like optical response consisting of an initial, brief increase in fluorescence (light phase) followed by a longer duration decrease (dark phase). In addition, off-beam parasagittal bands of decreased fluorescence are evoked due to feedforward inhibition of Purkinje cells and interneurons by molecular layer inhibitory interneurons.1,2 The present experiments examined the cellular and metabolic origins of these three response components. The astrocyte-neuron lactate shuttle hypothesis3 states that during excitatory neurotransmission, the astrocytes that are activated by glutamate transporters glycolytically convert glucose to lactate and transport that lactate to neurons to fuel mitochondrial oxidative phosphorylation. We hypothesized that the light phase is neuronal and the dark phase is glial.
Methods: In the anesthetized mouse, mitochondrial flavoprotein autofluorescence1 was used to image the responses to parallel fiber stimulation in the cerebellar cortex in vivo. Pharmacological manipulations were used to selectively block glutamatergic synaptic transmission, glial activation or glycolysis. Field potential recordings were used to assess for the status of the parallel fiber-Purkinje cell circuit.
Results: Parallel fiber stimulation evokes a beam like response and intersecting inhibitory bands. First, blocking neuronal synaptic transmission using a cocktail of glutamate receptor blockers (DNQX, D-APV and LY367385) selectively suppressed both the light phase and the inhibitory bands. The dark phase was largely intact. Preventing glial activation with the glial glutamate transporter blocker, DL-TBOA, abolished the dark phase. These findings imply a neuronal origin for the light phase and a glial origin for the dark phase. Second, substituting the glycolytic products pyruvate or lactate for glucose in the bathing solution selectively abolished the dark phase and the light phase remained unchanged, demonstrating that dark phase is primarily due to glycolysis and light phase oxidative metabolism.
Conclusions: Together these results establish that the light phase evoked by parallel fiber stimulation is due to oxidative metabolism of lactate in neurons driven by excitatory glutamate transmission. Conversely, the dark phase is primarily due to increased glycolytic metabolism in glia following neuronal oxidative metabolism. The inhibitory bands are primarily neuronal and are due to energy saving. These findings provide strong in vivo support for the astrocyte-neuron lactate shuttle hypothesis.
654. Oligodendrogenesis after chornic cerebral hypoperfusion in mouse
N. Miyamoto1, Y. Tanaka1, K. Yatomi2, Y. Ueno1, R. Tanaka3, N. Hattori1 and T. Urabe1
1Department of Neurology; 2Department of Neurosurgery, Juntendo University School of Medicine, Tokyo; 3Department of Neurology, Juntendo Urayasu hospital, Chiba, Japan
Background and purpose: Cerebrovascular white matter (WM) lesions are observed in aging and stroke and constitute the core pathology of Binswanger disease, a form of subcortical vascular dementia. These WM lesions are believed to be responsible for cognitive impairment and are caused by chronic cerebral hypoperfusion. The neuropathological changes in these lesions are characterized by diffuse demyelination, the loss of the axons, and gliosis, but the process leading to these changes remains unclear. The rat model of chronic cerebral hypoperfusion is accompanied by cognitive impairment and cholinergic deficits and is used most widely. However, genetic studies are hampered because of limited accessibility to molecular technologies using knockout or transgenic animals. Within 5–7 years, a mouse model of chronic cerebral hypoperfusion was established. In this study, we induced white matter lesions in a mouse chronic cerebral hypoperfusion model, and then tested the capability of tissue regeneration in the lesioned area after ischemia using bromodeoxyuridine (BrdU) and a series of neuronal and glial markers.
Methods: Rats underwent on narrowing the bilateral CCAs with microcoils (BCAS, n = 20). 5-Bromodeoxyuridine (BrdU, 50 mg/kg) was injected for analysis of newly generated cells in white matter. Immunohistochemistry for BrdU, glutathione S transferase-pi (GST-pi), platelet derived growth factor receptor-α (PDGFR-α), and single stranded DNA (ssDNA) were analyzed at pre-operation, 7, 14 and 28 days after hypoperfusion.
Results: The white matter lesions were progressed in time dependent manner in KB staining. Newly generated BrdU-positive cells were transiently increased at 7days after ischemia then decrased. Oligo progenitor cells labeled by PDGFR-α in white matter significantly increased in time dependence after ischemia compared with pre-operation (P<0.01). However, more mature oligodendrocyte labeled by GST-pi in white matter transiently increased in early period, but decreased in later period compared with pre-operation (P<0.01). Moreover, apoptotic cells were increased in time dependent manner.
Conclusions: Our result indicated that chronic cerebral hypoperfusion transiently promoted generation of oligo progenitor cells, but white matter lesion were progressed because of decreasing mature oligodendrocyte and increasing apoptotic cell death. Moreover, the mouse, which is readily amenable to gene knockout and manipulation and has advantages in cognitive evaluation, can be a model of subcortical vascular dementia suited for pathogenetic analysis.
690. Selenoprotein S protects astrocytes against ischemia-induced apoptosis
N. Fradejas Villar, D. Pastor Herrera, S. Mora-Lee, P. Tranque and S. Calvo
Medical Sciences, University of Castilla-La Mancha, Albacete, Spain
Objectives: Astrocytes are involved in maintaining brain integrity in conditions of disease and injury1 and are thought to confer neuroprotection during brain ischemia.2 Since the molecular mechanisms involved in the astrocyte response to ischemia are not yet completely understood, our work is aimed to ascertain the relevance of the endoplasmic reticulum stress response in the astrocyte injury caused by ischemia.3 Specifically, the work presented here was devoted to analyze the functions of Selenoprotein S (SEPS1)4 in the astrocyte response to ischemia and ER-stress inducers, as this protein is involved in the retrotranslocation step of ER stress.5
Methods: Differential display technique was used to uncover the genes whose expression was modified in cultured astrocytes subjected to oxygen and glucose deprivation (OGD) as an in vitro model of ischemia. Northern-blot and qRT-PCR were used to quantify Selenoprotein S expression levels. Selenoprotein S downregulation was reached by means of a specific siRNA. Cell viability was analyzed by the MTT method and flow cytometry of Annexin V/propidium iodide stained astrocytes.
Results: Differential display analysis of astrocytes exposed to OGD uncovered the upregulation of Selenoprotein S gene during ischemia. Subsequent Northern blot and quantitative RT-PCR studies showed that OGD-induced Selenoprotein S mRNA increase was maximum after 4-h and returned to control levels following reoxygenation. Astrocyte treatment with thapsigargin or tunicamycin also induced a significant increase of Selenoprotein S expression, indicating that this gene is expressed in astrocytes in the context of the ER-stress response.
siRNA technology was used to assess astrocytic Selenoprotein S functions. This approach caused a prominent diminution of Selenoprotein S expression which had a strong deleterious effect on astrocyte viability, decreasing survival to 4-h OGD by 40%. In agreement with Selenoprotein S functions on ER-stress response, astrocyte survival to thapsigargin or tunicamycin treatment was also intensely diminished by Selenoprotein S siRNA.
In summary we can conclude that Selenoprotein S activation during ischemia has protective functions in astrocytes that might be related to the activation of retrotranslocation machinery.
767. Sex differences in response to dihydrotestosterone and flutamide in astrocyte cell death
M. Liu and P.D. Hurn
Anesthesiology and Peri-Operative Medicine, Oregon Health & Sciences University, Portland, Oregon, USA
Introduction: Male sex is an acknowledged risk factor for stroke. Testosterone (T) can be converted to 17 β-estradiol via the aromatase, or metabolized to dihydrotestosterone (DHT) via the enzyme 5a-reductase, a step that then does not permit aromatization to estradiol. We previously demonstrated that astrocytic cell death induced by oxygen-glucose deprivation (OGD) is sex-specific.1,2 However, it is not clear if sex differences in response to T and DHT in astrocyte cell death contributes to the observed sex difference in response to ischemia. In the current study, we tested the hypothesis that male (XY) and female (XX) astrocytes are respond differently to T and DHT.
Methods: Primary sex-specific cultured cortical astrocytes were prepared from 1–3-day old male and female rat pups separately and grown to confluency in steroid-free medium.1,2 Confluent monolayers (10–14 days in vitro) were incubated in anoxia chamber in glucose-, serum-free medium for 6 h (oxygen-glucose deprivation, OGD), and then returned to normoxia and glucose-containing medium for 24 h. Cell death was induced by OGD alone, or in combination with T, DHT, or Flutamide. These reagents were added 24 h before OGD, and maintained during OGD and re-oxygenation. Cell death was estimated by lactate dehydrogenase (LDH) assay.
Results: Female astrocytes are less sensitive to OGD alone, or in combination with T or DHT than male astrocyte. Androgen receptor antagonist, Flutamide, protect against T or DHT combined with OGD-induced cell death in male astrocytes but not in female astrocytes.
Conclusions: We conclude that there are sex differences in ischemic sensitivity in female and male astrocytes, and male astrocytes (but not female) metabolize testosterone to DHT via the enzyme 5 alpha reductase, resulting in androgen receptor activation and enhanced cell death after OGD.
770. Differential activation of microglia in the white matter after closed skull traumatic brain injury in the mouse
C. Venkatesan1,2, M. Chrzaszcz1,2, N. Choi1,2 and M. Wainwright1,2
1Department of Pediatrics, Division of Neurology, Northwestern University, Feinberg School of Medicine; 2Center for Interdisciplinary Research in Pediatric Critical Illness and Injury, Children's Memorial Research Center, Chicago, Illinois, USA
Background and aims: The specific mechanisms by which microglia are activated and contribute to injury and repair following traumatic brain injury (TBI) have not been elucidated. In a mouse closed skull model, we have previously shown sustained long-term activation of glia as well as neurobehavioral deficits following TBI (Lloyd et al.J Neuroinflammation 2008;5:28). The differential role of microglia in the mechanisms of injury and repair of the white matter following TBI is not known.
Methods: Adult CD1 mice were subject to midline closed skull injury or sham operation using a stereotactically guided pneumatic compression device. Animals were sacrificed 8 or 14 days after injury or sham procedure. Coronal frozen brain sections were cut at 40 micrometers on a freezing microtome and processed using standard immunohistochemical techniques. Antibody to the microglial marker IBA-1 was used to identify both resting and activated microglia. Antibody to Mac-2 was used to identify activated microglia alone. In order to evaluate the functional role of activated microglia, expression of the trophic factor Insulin-like growth factor (IGF) within microglia was also examined using an anti-IGF antibody. Patterns of labeling within the white matter underlying the impact site were analyzed qualitatively and quantitatively. Semi-quantitative analysis was performed in a blinded manner on digitized images.
Purpose: We tested the hypothesis that following TBI, there is a specific increase in the expression both of activated (Mac-2 immunoreactive) microglia, and a change in the functional properties of these activated microglia (increased IGF-expressing microglia) within the white matter.
Results: Qualitative and quantitative evaluation of IBA-1 immunureactive microglia showed no statistically significant differences between the sham and TBI groups. In contrast, there was a significant increase in the density of Mac-2 immunoreactive microglia within the white matter underlying the impact site 8 and 14 days post-injury (P<0.01 vs sham). The increase was greatest 8 days after TBI. Comparison of Mac-2 staining in the two injury groups showed that there was a significant attenuation in the density of labeled cells 14 days following injury (P<0.01 vs 8-days post injury). There was also an increase in IGF-immunoreactive microglia following TBI within the white matter.
Conclusions: These studies show that microglia have diverse phenotypes and a subset of microglia (identified by MAC-2 immunoreactivity) within the white matter are activated after TBI. This activation may involve a phenotypic switch of pre-existing microglia since there was no increase in the region examined in the density of IBA-1-immunoreactive cells following TBI. Further, the finding of an increase in the density of IGF-immunoreactive microglia supports a protective role for microglia for this microglial phenotype. Greater understanding of the mechanisms which distinguish the protective and pathologic phenotypes of activated microglia may advance the development of new therapeutic strategies to prevent the long term neurologic sequelae of TBI.
813. Loss of astrocytic glutamate transporters in Wernicke's encephalopathy; involvement of oxidative stress
A. Hazell1, D. Sheedy2, C. Wang1, D. Wang1, R. Oanea1, M. Aghourian1, S. Sun1 and J. Yong1
1Department of Medicine, University of Montreal, Montreal, QC, Canada; 2Department of Pathology, University of Sydney, Sydney, NSW, Australia
Objectives: Wernicke's encephalopathy (WE), a neurological disorder caused by thiamine deficiency (TD), is characterized by structural damage in brain regions that include the thalamus and cerebral cortex.1 The basis for these lesions is unclear but evidence suggest it may involve a disturbance of glutamatergic neurotransmission.2 We have therefore investigated levels of the astrocytic glutamate transporters EAAT1 and EAAT2 in these vulnerable areas in order to evaluate their role in the pathophysiology of this disorder.
Methods: Postmortem samples of frontal cortex were studied from patients with a neuropathological diagnosis of WE and a clinical history of consumption of greater than 80 g of absolute alcohol per day, and from control cases with a history that included consumption of less than 20 g of absolute alcohol per day. Tissue was provided by the New South Wales Tissue Resource Centre which is supported by the University of Sydney, Neuroscience Institute of Schizophrenia and Allied Disorders, National Institutes of Alcohol Abuse and Alcoholism and New South Wales Department of Health. In addition, TD rats and their pair-fed controls were prepared as previously described,3 with and without co-treatment with N-acetylcysteine (NAC), and their brains studied at the loss of righting reflex stage. WE and TD samples were examined by immunoblotting and immunohistochemical methods.
Results: Histological assessment of the frontal cortex revealed a significant loss of neurons in WE cases compared to age-matched controls, concomitant with decreases in α-internexin and synaptophysin protein content of 67% and 52% by immunoblotting. EAAT2 levels were diminished by 71% in WE, with levels of EAAT1 also reduced by 62%. Development of TD in rats caused a profound loss of EAAT1 and EAAT2 in the thalamus accompanied by decreases in other astrocytic-specific proteins including GFAP, glutamine synthetase, and the GABA transporter GAT-3, which in the thalamus is localized in these cells, while β-actin levels were unchanged. Treatment of TD rats with NAC prevented the downregulation of both EAAT1 and EAAT2 in the medial thalamus, concomitant with increased neuronal survival.
Conclusions: Our results suggest that:
loss of astrocytic glutamate transporters is associated with structural damage to the frontal cortex in patients with WE,
oxidative stress plays an important role in this process, and
TD has a profound effect on the functional integrity of astrocytes.
Based on these findings, we recommend that early treatment using a combination of thiamine replenishment AND antioxidant approaches should be an important consideration in cases of WE, particularly those in which recurrent bouts of TD are part of the clinical history and where the underlying damaging processes may therefore be having a cumulative effect.
825. Astrocytic metabolism assessed from 14C-acetate after oxygen glucose deprivation and chemical activation in vitro
T. Sasaki1, K. Kitagawa2, K. Kajimoto3, Y. Terasaki1, E. Omura-Matsuoka1, N. Ohyama1, Y. Sugiyama2, S. Okazaki2, Y. Yagita2 and J. Hatazawa4
1Division of Stroke Center, Department of Internal Medicine; 2Department of Neurology, Osaka University Graduate School of Medicine, Osaka; 3Stroke Divison, Department of Internal Medicine, National Cardiovascular Center, Suita, Osaka; 4Department of Nuclear Medicine, Osaka University Graduate School of Medicine, Osaka, Japan
Background: Increasing evidence has suggested that stroke should be integrative, and thus a concept of dynamic interaction between cells belonging to the neurovascular unit, such as endothelial cells, astrocytes and neurons, is emerging. In the neurovascular unit, astrocyte supports their maintenance of the permeability barrier. Also, considering the coupling of neuronal activation to cerebral blood vessel responses, astrocytes play key roles between these cells. Thus, whereas astrocytes play crucial roles in both normal and injured brain, the astrocytic metabolism after ischemia are not yet fully known. Exogenous acetate is preferentially metabolized by astrocytes in the CNS. Acetate uptake by astrocytes appears to be mediated by a carrier of monocarboxylate transporter (MCT). We have assessed astrocytic metabolism based on the incorporation of radiolabel 14C-acetate after in vitro ischemia and chemical stimulation by lipopolysaccharide (LPS).
Subjects and methods: Astrocytes for culture were obtained from 1 to 2 day neonatal Wister rats and grown in DMEM+10% fetal bovine serum (FBS)+antibiotics. The purity of astrocytes has been shown to be >95% glial fibrillary acidic protein positive cells. For oxygen-glucose deprivation (OGD), the culture medium was replaced with a glucose-free EBSS that had previously been saturated with 95% N2/5% CO2 and heated to 37 °C. The cultures were then put into an anaerobic incubator (pO2 < 2 mm Hg) with an atmosphere of 95% N2 and 5% CO2 (1% O2) at 37 °C for 6 hours. The cells were subjected to 6 h of OGD followed by reoxygenation. Reoxygenation was achieved by returning cell cultures to normoxic conditions (37 °C in a humidified 5% CO2 atmosphere). To elucidate the mechanism of acetate uptake, we assessed the ability of selective inhibitor of TCA cycle, fluoroacetate, or MCT 1, 2 or 4 siRNAi. On the other hand, the cultures were exposed for 24 h to 0.5 to 2.0 μg/ml LPS to study the incorporation of 14C-acetate under chemical activated conditions of astrocyte.
Results: Following OGD, acetate uptake significantly increased at 3 to 24 h after reoxygenation than basal control levels (Figure). Fluoroacetate produced significant inhibition at 10 mmol/L. In addition, MCT-1 siRNAi, but not MCT-2 or MCT-4 siRNAi, significantly decreased the acetate uptake. We also observed the increase of acetate uptake following LPS treatment.
Conclusion: These findings indicate that astrocytic function dynamically changes following ischemia and chemical activation. The images of 14C-acetate may be particularly useful in the elucidation of mechanism of human stroke and neurodegenerative disease.
964. Spatio-temporal changes of apolipoprotein E (ApoE) in the rat brain after experimental stroke. Enriched housing condition attenuates ApoE expression
K. Ruscher, E. Johannesson, M. Rickhag and T. Wieloch
Laboratory for Experimental Brain Research, Lund University, Lund, Sweden
Objectives: Apolipoprotein E (ApoE), a cholesterol transporter and an immunomodulator, is brain protective after stroke and implicated in brain repair. The present study was designed to assess the involvement of ApoE in the restoration of brain function after experimental stroke, by employing animal housing conditions that differentially improve recovery after brain injury.1
Methods: The temporal and cellular pattern of ApoE expression was analyzed from different brain regions from rats subjected to 2 h of transient occlusion middle cerebral artery (tMCAO). Permanent MCAO was induced in spontaneous hypertensive rats, housed in either standard or enriched housing conditions (starting 2 days after pMCAO), and the tissue levels and cellular cellular distribution of ApoE determined.
Results: ApoE levels increased in the injured hemisphere over a 30 days recovery period, with the exception of a proximal narrow peri-infarct rim, conspicuously devoid of ApoE at 4 to 7 days after tMCAO. In contrast to the ischemic core, this region showed no accumulation of CD11b+ microglial cells but presented a normal distribution of NeuN+ neurons indicative of a proximal non-injured peri-infarct zone. In this region, we further observed only a limited amount of GFAP+ astrocytes suggesting an inhibition of astrocyte proliferation in this zone. Still, in the peri-infarct rim, ramified S100β+ reactive astrocytes with high expression of ApoE are present. Further, enriched housing following pMCAO caused an approximately 50% decrease in ApoE levels compared to standard housing conditions, particularly in the peri-infarct region and in ApoE/S100β+ reactive astrocytes.
Conclusions: The reduced levels of ApoE and reduced number of ApoE/S100β+ reactive astrocytes in animals housed in an ee implies that enriched housing conditions might attenuate the inflammatory response after stroke. Our study also support the notion of a proximal regenerative zone in the peri-infarct zone after stroke, with low levels of ApoE.
Grant support: The EU 7th workprogram through the European Stroke Network, Swedish Research Council, Pia Ståhls Foundation, The Swedish Brain Fund, the Kungliga Fysiografiska Sällskapet i Lund, and the Greta and Johan Kocks stiftelser.
996. Impact of cell therapies on post-ischemic astrogliosis
D.-C. Wagner1, A. Kranz1,2, U.-M. Riegelsberger1,2, C. Poesel1, F. Emmrich1,2,3 and J. Boltze1,2,3
1Neurorepair Research Group, Fraunhofer Institute for Cell Therapy and Immunology; 2Institute for Clinical Immunology and Transfusion Medicine; 3Translational Centre for Regenerative Medicine, University of Leipzig, Leipzig, Germany
Objectives: Astrogliosis is a common phenomenon following brain injury. However, the actual role of astrocytes within the complex spatio-temporal events after ischemic stroke is still not elucidated satisfactorily. On the one hand, multitude of evidence displayed a negative influence of activated astrocytes such as the release of proinflammatory cytokines, an increase of ischemic damage by the astrocytic gap-junction system and the formation of an anti-regenerative glial scar. Nevertheless, several aspects also support the hypothesis of a beneficial role of astrocytes after brain damage. The ablation of reactive astrocytes after traumatic brain injury resulted in the augmentation of CNS damage and inflammatory response. Hence, astrocytes may be an interesting target for modulating therapeutic approaches such as cell therapies. A recent study showed increased astrocytic survival by BMSC after hypoxic conditions. The aim of this study is the investigation of a potential interrelation between cell therapies and a modulation of astrocytic characteristics.
Methods: We examined the astrocytic reaction to permanent middle cerebral artery occlusion (pMCAO) dependent on cell therapy in two different experiments. Firstly, we investigated brain specimen of an effective long-term cell transplantation experiment. Sixteen male spontaneously hypertensive rats (SHR) were subjected to pMCAO and randomly assigned to therapy (intravenous administration of 2 × 10E6 human placenta cells; n = 8) and control group (intravenous application of vehicle solution; n = 8). Efficacy of cell treatment was examined weekly by behavioral tests and MR investigations until Day 60. Secondly, we performed a short-term investigation of astrocyte-associated markers within a 48 h time window following stroke. Twenty-seven SHRs were subjected to pMCAO and received either 8x10E6 human umbilical cord blood cells (HUCBC) per kilogram bodyweight (n = 9) or vehicle solution (n = 9) intravenously. The animals were sacrificed 30 h, 36 h and 48 h after stroke onset. Additional control subjects were sacrificed without pMCAO (n = 3) and 6 h and 24 h after pMCAO (n = 6), respectively. Messenger RNA expression of IL-1β, nestin, GFAP, and glutamate transporter-1 (GLT-1) was quantified by real time PCR within two areas in the cortical lesion border or corresponding contralateral domains.
Results: The long-term investigation of astrocytic reactions to brain ischemia and cell therapy showed a significant increase of GFAP+ cells in the infarct border of cell treated subjects at Day 60 after stroke onset. This observation was associated with a reduced infarct volume and a decline of functional deficits. The short-term investigation showed a fast increase of IL-1β followed by increased values of GFAP and nestin and a decline of GLT-1 mRNA expression. The development of IL-1β, GLT-1 and nestin appeared homogenously while there was a strong trend towards less GFAP expression in control animals compared to cell treated subjects at 36 h and 48 h.
Conclusions: Efficacy of cell therapies after stroke is, at least partly, mediated by a modulation of astrocytic survival or activation. The mentioned developments proceed within a comparatively short time upon stroke onset and might therefore determine a limited window for experimental therapies aiming to modulate these processes. However, an increase of the group size and further immunohistological examinations are under evaluation to confirm these preliminary results.
References
1.
WinnHRRubioRBerneRM. Brain adenosine concentration during hypoxia in rats. Am J Physiol1981;241:H235–42.
2.
MiekisiakGKulikTKusanoYChenJ-FWinnHR: Cerebral blood flow response in adenosine 2a receptor knockout mice during transient hypoxic hypoxia. J Cereb Blood Flow Metab2008;28(10):1656–64.
3.
NgaiACCoyneEFMenoJR. Receptor subtypes mediating adenosine-induced dilation of cerebral arterioles. Am J Physiol Heart Circ Physiol2001;280:H2329–N35.
4.
SakuraiTYamamotoSMiyakawaA. Fiber-coupled confocal microscope for real time imaging of cellular signals in vivo. Proc of SPIE2006;6088:608803–1–608803–6.
5.
ShapiroLAMarksAWhitaker-AzmitiaPM. Increased clusterin expression in old but not young adult S100B transgenic mice: Evidence of neuropathological aging in a model of Down Syndrome. Brain Res2004;1010:17–21.
6.
KleindienstAMcGinnMJHarveyHBColelloRJHammRJBullockMR. Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. J Neurotrauma2005;22:645–55.
7.
ReinertKCDunbarRLGaoWChenGEbnerTJ. Flavoprotein autofluorescence imaging of neuronal activation in the cerebellar cortex in vivo. J Neurophysiol2004;92:199–211.
8.
GaoWChenGReinertKCEbnerTJ. Cerebellar cortical molecular layer inhibition is organized in parasagittal zones. J Neurosci2006;26:8377–87.
9.
PellerinLMagistrettiPJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA1994;91(22):10625–9.
10.
NedergaardMDirnaglU. Role of glial cells in cerebral ischemia. Glia2005;50:281–6.
11.
LiLLundkvistAAnderssonDWilhelmssonUNagaiNPardoACNodinCStahlbergAApricoKLarssonKYabeTMoonsLFotheringhamADaviesICarmelietPSchwartzJPPeknaMKubistaMBlomstrandFMaragakisNNilssonMPeknyM. Protective role of reactive astrocytes in brain ischemia. J Cereb Blood Flow Metab2008;28:468–81.
12.
BenavidesAPastorDSantosPTranquePCalvoS. CHOP plays a pivotal role in the astrocyte death induced by oxygen and glucose deprivation. Glia2005;52:261–75.
13.
KryukovGregory V.CastellanoSergiNovoselovSergey V.LobanovAlexey V.ZehtabOmidGuigóRodericGladyshevVadim N.. Characterization of mammalian selenoproteomes. Science2003;300(5624):1439–43.
14.
YeYShibataYYunCRonDRapoportTA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature2004;429:841–7.
15.
LiuMHurnPDRoselliCE and AlakyedNJ. Role Of P450 aromatase in sex-specific astrocytic cell death. Journal of Cerebral Blood Flow & Metabolism. 2007;27:135–41.
16.
LiuMOyarzabalEAYangRMurphyMJHurnPD. A novel method for assessing sex specific and genotype specific response to injury in astrocyte culture. Journal of Neuroscience Methods. 2008;171:214–7.
17.
VictorMAdamsRDCollinsGH. The Wernicke-Korsakoff Syndrome and Related Neurologic Disorders due to Alcoholism and Malnutrition. F.A. Davies, Philadelphia, 1989.