
Abstract
Indirect evidence from laboratory studies suggests that mitochondrial energy metabolism is impaired in progressive supranuclear palsy (PSP), but brain energy metabolism has not yet been studied directly in vivo in a comprehensive manner in patients. We have used combined phosphorus and proton magnetic resonance spectroscopy to measure adenosine-triphosphate (ATP), adenosine-diphosphate (ADP), phosphorylated creatine, unphosphorylated creatine, inorganic phosphate and lactate in the basal ganglia and the frontal and occipital lobes of clinically probable patients (N= 21; PSP stages II to III) and healthy controls (N= 9). In the basal ganglia, which are severely affected creatine in PSP patients, the concentrations of high-energy phosphates (= ATP + phosphorylated creatine) and inorganic phosphate, but not low-energy phosphates (=ADP+ unphosphorylated creatine), were decreased. The decrease probably does not reflect neuronal death, as the neuronal marker N-acetylaspartate was not yet significantly reduced in the early-stage patients examined. The frontal lobe, also prone to neurodegeneration in PSP, showed similar alterations, whereas the occipital lobe, typically unaffected, showed less pronounced alterations. The levels of lactate, a product of anaerobic glycolysis, were elevated in 35% of the patients. The observed changes in the levels of cerebral energy metabolites in PSP are consistent with a functionally relevant impairment of oxidative phosphorylation.
Keywords
Introduction
Progressive supranuclear palsy (PSP) is a sporadic neurodegenerative disorder characterized by an akinetic-rigid syndrome with prominent postural instability, oculomotor deficits and neuropsychological deficits in frontal lobe-related functions (Litvan et al, 1996). Neuropathologically, PSP is characterized by neuronal loss and somatodendritic aggregation of the microtubule-associated protein tau, predominantly in the basal ganglia, brain stem nuclei and the frontal lobes; the occipital lobes and the cerebellum are relatively spared (Hauw et al, 1994). The median survival time is 5 to 10 years (Golbe and Ohman-Strickland, 2007). To date, there are no effective symptomatic or neuroprotective treatments available (van Balken and Litvan, 2006). Therefore, a detailed understanding of the pathogenesis underlying PSP is urgently needed to allow the development of novel rational therapies.
Although the etiology of PSP is unknown, several lines of evidence point to an impairment of mitochondrial energy production: (1) Cerebral glucose metabolism is reduced, as shown by [18F]-2-fluoro-2-deoxy-
Experimental findings also suggest that mitochondrial failure might play a crucial role in the pathophysiology of PSP. A PSP-like tauopathy endemic in the island of Guadeloupe has been linked to the consumption of fruit and infusions of the leaves of Annona muricata (Lannuzel et al, 2007), which contain the complex I inhibitor, annonacin, that induces a PSP-like pattern of neurodegeneration in the rat brain (Champy et al, 2004) and tau pathology in cultured neurons (Escobar-Khondiker et al, 2007). Similar results have been obtained with rotenone, another natural complex I inhibitor (Höglinger et al, 2003, 2005).
The main function of the mitochondrial respiratory chain is to maintain adequate concentrations of high-energy phosphates (HEP), particularly ATP, in cells. It is important to note, however, that brain mitochondria can tolerate a 60% decrease in complex I activity, without a significant decrease in ATP production (Davey et al, 1997). In the experimental models using annonacin and rotenone, a significant decrease in ATP levels was required to induce tau pathology; mild complex I inhibition without ATP depletion had no effect (Höglinger et al, 2005; Escobar-Khondiker et al, 2007). To date, however, there has been no direct demonstration in vivo regarding the bioenergetic alterations identified in PSP patients lead to significant changes in brain ATP levels.
High-energy metabolites (ATP and phosphorylated creatine (pCre)) and inorganic phosphate (Pi) can be quantified in vivo by phosphorus magnetic resonance spectroscopy (31P-MRS). Lactate, an indicator of anaerobic glycolysis, and N-acetylaspartate (NAA), a marker of neuronal integrity, can be quantified by proton [1H]MRS. Combining 31P- and 1H-MRS additionally allows the quantification of the low-energy metabolites, ADP and unphosphorylated creatine (uCre). Therefore, we have combined 31P- and 1H-MRS to measure these metabolites in differentially vulnerable brain regions in PSP patients and in healthy controls.
Materials and methods
This study was approved by the Ethics Committee of Marburg University and is registered online (http://clinicaltrials.gov/, identifier NCT00328874).
Participants
Patients examined in the Department of Neurology of the University of Marburg were qualified for participation in the study if they had clinically probable PSP (Litvan et al, 1996). This criterion was chosen to obtain the highest degree of diagnostic certainty possible in vivo in patients who were not more advanced than stage III (Golbe, 1997), thus avoiding a bias in the measured levels of energy metabolites because of pronounced neuronal loss.
The healthy volunteers were free of neurologic, systemic or psychiatric diseases, including alcohol or substance abuse, as verified by a detailed evaluation of their medical histories and by a comprehensive physical examination. Furthermore, participants were included only if they were willing and able to give written informed consent to participate in the study.
We excluded a variety of conditions that might influence the levels of cerebral energy metabolites. Exclusion criteria for all participants were age > 50 or > 85 years; Parkinson's syndromes other than PSP; dementia (Mini-Mental State Examination (MMSE) score ⩽24); a history of epilepsy, structural brain disease, brain surgery, electroconvulsive therapy, stroke and arterial hypertension (systolic > 180 or diastolic > 110mmHg); systemic disorders affecting the metabolism or function of the brain (e.g., diabetes mellitus); other serious illnesses; participation in drug studies or the use of drugs that modify mitochondrial activity (e.g., coenzyme Q10, statins) or antioxidants (e.g., vitamin E, C) within the last 60 days; the use of drugs interfering with catecholamine metabolism (e.g., reserpine, amphetamines, MAO-A inhibitors, methylphenidate and cinnarizine) within the last 30 days; an unstable dosage of CNS-active drugs (e.g., antiparkinsonian drugs, anxiolytics, hypnotics, tranquillizers and antidepressants) within the last 30 days.
Clinical Evaluation
Motor functions were assessed with the Unified Parkinson's Disease Rating Scale (UPDRS), part III, the PSP staging system (Golbe, 1997) and the PSP rating scale (PSP-RS) (Golbe and Ohman-Strickland, 2007). Activities of daily living were scored with the UPDRS, part II. Global cognitive function was evaluated with the MMSE. Neuropsychiatric symptoms were evaluated with the Frontal Assessment Battery (FAB) (Dubois et al, 2000) and the Montgomery-Åsberg Depression rating scale (MADRS).
1H-MRS
A 3-T system (Magnetom Trio; Siemens, Erlangen, Germany) with a double-tuned 1H/31P volume head coil (Rapid Biomedical, Würzburg, Germany) was used for 1H-MRS, with a two-dimensional chemical shift imaging double echo (CSI) sequence with PRESS volume selection. A 1.5-cm axial slice, including the basal ganglia, was recorded. Data were acquired with circular-phase encoding using a 16 × 16 matrix, which was extrapolated before Fourier transformation to 32 × 32 matrix. The field of view was 240mm2, the repetition time 1,500 msecs and the echo time 30 msecs. For quantitative analysis, spectra were fitted in the frequency domain by a linear combination of a set of model spectra using the LCModel software (Provencher, 1993). Cramer–Rao lower bounds for NAA, choline (Cho) and creatine (Cre) were typically between 5% and 10%. Data with the Cramer–Rao lower bounds of more than 20% were discarded. Corrections for 1H-T1 and 1H-T2 relaxation were applied (Hattingen et al, 2007).
Phosphorus Magnetic Resonance Spectroscopy
Phosphorus magnetic resonance spectroscopy data were acquired using a three-dimensional CSI phase-encoding scheme applying a pulse-free induction decay sequence with WALTZ4 proton decoupling. A 300 × 300 × 200mm3 three-dimensional CSI slab was recorded with angulation and offset identical to the 1H-slice. Circular-phase encoding was employed with a weighted acquisition scheme on a 10 × 10 × 8 matrix extrapolated to 16 × 16 × 8, resulting in a series of axial slices with nominal 25mm thickness and 18.75 × 18.75mm2 in plane resolution (flip angle 60°, repetition time 2000 msecs and echo time 2.3 msecs). Before spatial Fourier transformation, the data set was multiplied with a linear increasing phase in the foot to head direction. Phase increments were adjusted to cause a shift of 12.5mm toward the feet, resulting in a perfect alignment of slice 5 from the 31P data with the 1H-slice. Spectra were analyzed in the time domain with jMRUI software employing the non-linear least square-fitting algorithm AMARES (Vanhamme et al, 1997). The time domain model function was composed of 14 exponentially decaying sinusoids in the frequency domain. Six of those, which had identical damping, corresponded to peaks assigned to pCre, phosphoethanolamine, phosphocholine, glycero-phosphocholine, glycero-phosphoethanolamine and Pi. pCre was adjusted to 0 p.p.m. and, except for Pi, constraints for the chemical shifts of the other signals were applied as a fixed difference with respect to the position of pCre. ATP was represented by seven exponentially damped sinusoids, defining each multiplet by the respective number of peaks with identical damping and adequate amplitude ratios. The coupling constant was fixed at 18 Hz. One signal with a fixed chemical shift of 2.24 p.p.m. and maximum line width of 50 Hz was used to account for potential macromolecule signals in the phosphodiester region. Corrections were performed for 60° flip angle and partial saturation using T1 of 2.4 secs for pCre, 1.1 secs for Pi and 1.0 secs for ATP, resulting in respective intensity correction terms of 1.60, 1.27 and 1.24. The relaxation times are within the error margins of published data measured at 1.5 T (Buchli et al, 1994) and 4 T (Hetherington et al, 2001).
External Calibration
Signal intensities were quantified in terms of absolute concentrations by the phantom replacement method (Michaelis et al, 1993). The phantom solution, which was used as external calibration reference, contained 20 mmol/l creatine and 20 mmol/l phosphate. In contrast to the in vivo sequence for 31P data, a two-dimensional CSI sequence with full 16 × 16 matrix and 240mm2 field of view was performed using a 90° flip angle and a repetition time of 40 secs.
Regions of Interest
Figure 1A shows the axial plane of a T1-weighted magnetic resonance image from a control brain. The grid indicates the resolution of each modality, whereas the location of the templates defines the regions of interest (ROIs) in the ‘basal ganglia’, ‘frontal lobe’ and ‘occipital lobe’ for 1H- and 31P-MRS. As the data from the left and right ‘basal ganglia’ and ‘frontal lobe’ ROIs did not differ significantly from each other, only the averaged values are given. The ROI for the ‘occipital lobe’ crossed the midline and thus yielded only one set of data.
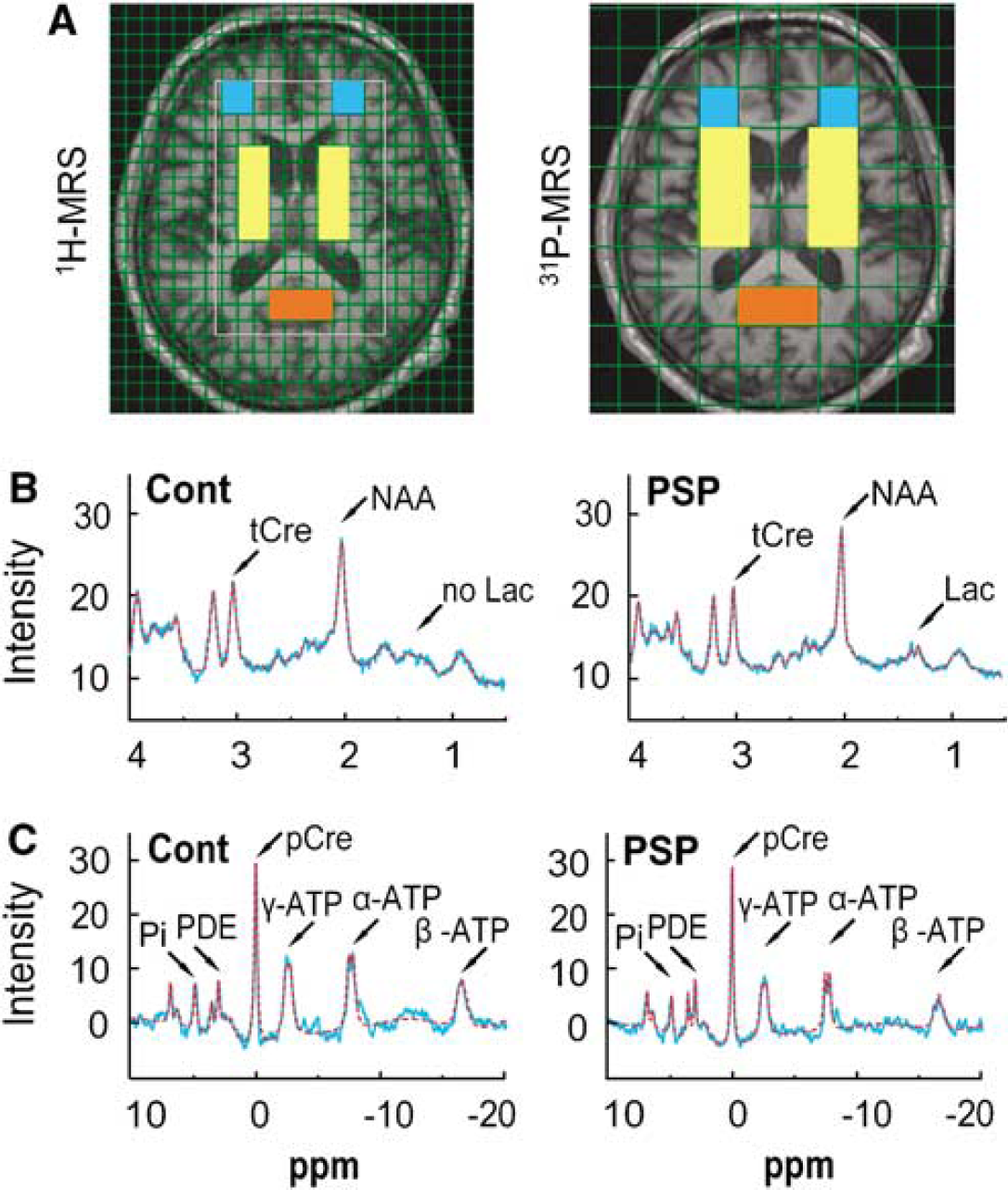
Magnetic resonance spectroscopy (MRS). (
MRS Data Acquisition
The signals for tCre, NAA and lactate were obtained from the 1H spectra (Figure 1B). Those for Pi, pCre and ATP were from the 31P spectra (Figure 1C).
Lactate: In healthy control brains, lactate is not detectable by 1H-MRS, using the data acquisition scheme described by Isobe et al (2007). However, anaerobic glycolysis (i.e., an excess of glycolysis over oxidative phosphorylation) can lead to elevated and, therefore, detectable lactate levels (Prichard et al, 1991). The LCModel software provides a value for the lactate concentration and an estimation of its accuracy is given (% s.d.). Each complete CSI data set was screened for lactate s.d. values > 20%. The presence of a clear positive doublet signal at 1.3 p.p.m. in the spectra was then verified. Voxels fulfilling these criteria were considered to have elevated lactate levels (see Figure 1B).
ATP: The 31P-MRS spectra for ATP have three signals corresponding to the three phosphorous nuclei of the nucleotide (Figure 1C). α-ATP contains contributions from reduced nicotinamide adenine dinucleotide (NADH) and α-ADP; γ-ATP contains contributions from β-ADP, whereas the β-ATP peak area is proportional to the total cellular ATP concentration (Iles et al, 1985). We therefore used the β-ATP value as representative of total ATP levels (Hu et al, 2000; Pietz et al, 2003; Rango et al, 2006).
The following parameters were calculated from the measured values:
ADP= [(tCre–pCre) *β–ATP]/[pCre × H × KCK]
with KCK = 1.66 × 109 mol/L−1 and H= 10−pH mol/L (Pietz et al, 2003);
uCre = tCre–pCre;
HEP= β-ATP + pCre (Rango et al, 2006);
LEP =ADP+ uCre (Rango et al, 2006);
Phosphorylation potential (PP) =ATP/(ADP × Pi) (Pietz et al, 2003).
pH: Tissue pH was estimated from the chemical shift difference between Pi and pCre according to the formula
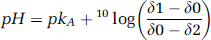
where δ1 = 3.27, δ2 = 5.63, pkA = 6.75, and δ0 equals a chemical shift difference between the Pi and pCre signals (Petroff et al, 1985).
Statistical Analysis
As data on cytosolic ADP levels measured by 1H-MRS in the occipital lobes of PSP patients compared with those of healthy controls are available (Martinelli et al, 2000), ADP was used as the main outcome parameter for sample size calculation. With a 2:1—the patient/control ratio, 16:8 available individuals are needed to obtain 90% power to detect a significant difference (P > 0.05) of 7.5 µmol/L with a standard deviation of 5 µmol/L in a two-sided Wilcoxon–Mann–Whitney test. In view of possible dropouts, the planned sample size was 20 PSP patients and 10 healthy controls. The confirmatory analysis of the primary end point was performed on data of all patients and controls for which ADP in the basal ganglia could be calculated using the above-mentioned test. The analysis of all secondary data is descriptive. Data are shown as mean±s.d. The two-sided Wilcoxon–Mann–Whitney test was used to compare metric variables and the two-sided Fisher exact test for categorical variables. P-values > 0.05 were considered to be statistically significant. No adjustment for multiple testing was performed. Statistical analyses were performed with the SAS statistical package (version 9.1).
Results
Study Population
A total of 67 patients were screened for eligibility, of which 46 were excluded: 21 for clinically possible but not clinically probable PSP, 21 for Parkinson's syndrome other than PSP, 14 for a PSP stage > III, 14 for dementia, 5 for structural brain disease, 2 for insufficiently treated arterial hypertension, 2 for a systemic disease affecting the brain and 2 for another serious illness. The total number of excluded patients was less than the sum of the individual items, as some patients fulfilled more than one of the exclusion criteria. Twenty-one patients met the inclusion and none of the exclusion criteria and participated in the study.
The clinical characteristics of the analyzed participants are shown in Table 1.
Clinical characteristics of the participants
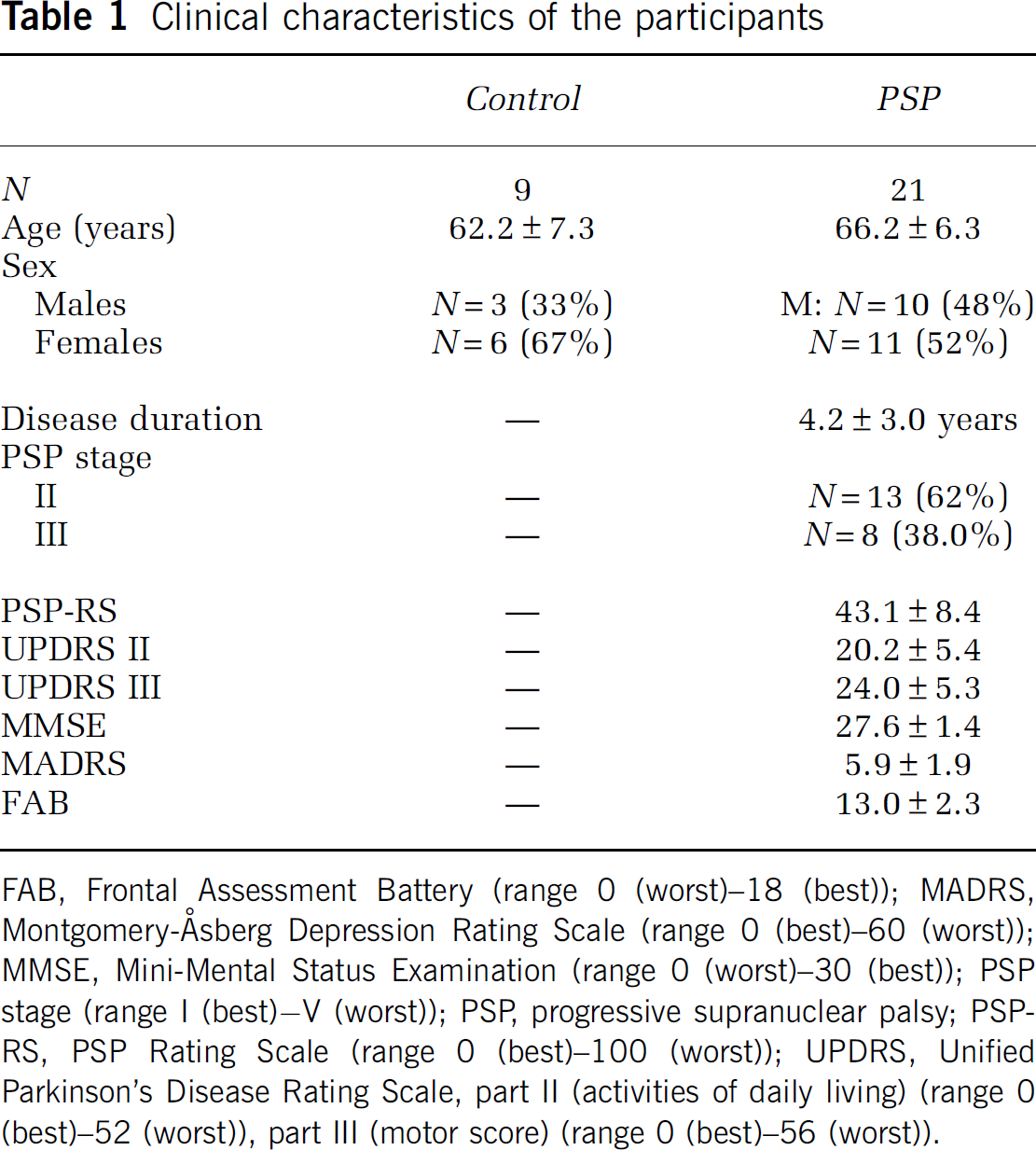
FAB, Frontal Assessment Battery (range 0 (worst)−18 (best)); MADRS, Montgomery-Åsberg Depression Rating Scale (range 0 (best)−60 (worst)); MMSE, Mini-Mental Status Examination (range 0 (worst)−30 (best)); PSP stage (range I (best)–V (worst)); PSP, progressive supranuclear palsy; PSP-RS, PSP Rating Scale (range 0 (best)−100 (worst)); UPDRS, Unified Parkinson's Disease Rating Scale, part II (activities of daily living) (range 0 (best)−52 (worst)), part III (motor score) (range 0 (best)−56 (worst)).
N-Acetylaspartate
We found no significant differences in NAA, tCre or the NAA/tCre ratio in patients and controls in any of the ROIs analyzed (Figure 1B, Table 2), suggesting that the patients studied, who were in early stages of the disease, had only mild neuronal disintegrity in the analyzed ROIs.
Cerebral metabolites measured by magnetic resonance spectroscopy
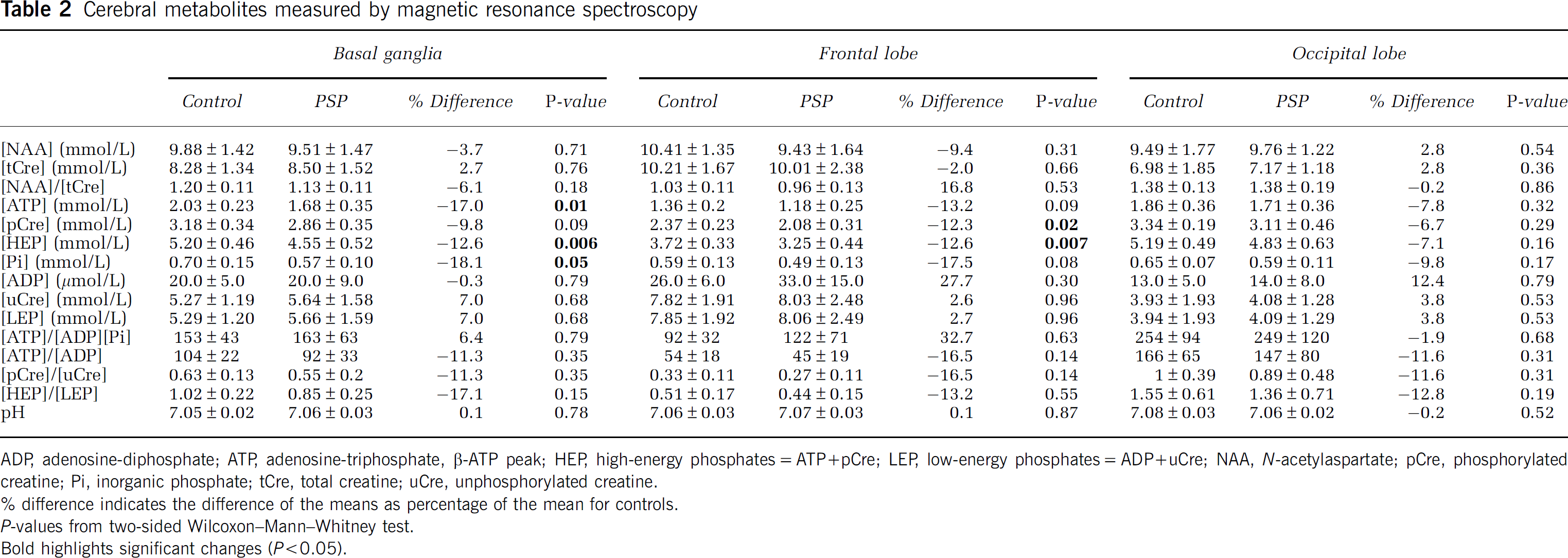
ADP, adenosine-diphosphate; ATP, adenosine-triphosphate, β-ATP peak; HEP, high-energy phosphates=ATP+pCre; LEP, low-energy phosphates=ADP+uCre; NAA, N-acetylaspartate; pCre, phosphorylated creatine; Pi, inorganic phosphate; tCre, total creatine; uCre, unphosphorylated creatine.
difference indicates the difference of the means as percentage of the mean for controls.
P-values from two-sided Wilcoxon–Mann–Whitney test.
Bold highlights significant changes (P>0.05).
Energy Metabolites in the Basal Ganglia
In the ‘basal ganglia,’ ROI (averaged value of the left and the right hemisphere), there was a significant reduction in the concentration of ATP (P = 0.01) in PSP patients compared with that in controls; pCre showed a consistent trend (P = 0.09); the sum of the HEPs (=ATP + pCre) was also significantly lower in patients (P = 0.006) (Figure 1C, Table 2).
When the analysis of the ‘basal ganglia’ ROI was carried out separately for the left and right hemispheres, the HEP concentration was also significantly lower in patients on both sides (left: −12.1%, P = 0.03; right: −13.1%, P = 0.02); the reduction in pCre reached significance on the left side (–10.5%, P = 0.05); and the reduction in ATP reached significance on the right side (–19.1%, P = 0.02).
Free phosphate (Pi) levels were also decreased in the averaged ‘basal ganglia’ ROI of patients (P = 0.05) (Figure 1C, Table 2).
In contrast, there were no significant differences in the LEPs, ADP and uCre, and the sum of both (LEPs) in the averaged ‘basal ganglia’ ROI of patients and controls (Table 2).
The ratios of the HEPs to their low-energy counterparts (PP =ATP/(ADP × Pi), ATP/ADP, pCre/uCre and HEP/LEP), which are considered to represent the cellular energy load that drives all energy-dependent biochemical processes (Iles et al, 1985; Pietz et al, 2003), were not altered in the ‘basal ganglia’ ROI of the PSP patients (Table 2). Neither were the HEP/Pi (Rango et al, 2006), ATP/Pi (Hu et al, 2000) and pCre/Pi ratios (Penn et al, 1995) (data not shown).
Energy Metabolites in Other Brain Areas
In the averaged ‘frontal lobe’ ROI of patients, changes in the concentrations of energy metabolites were similar to those in the ‘basal ganglia’ ROI: there were significantly lower concentrations of HEP (P = 0.007) and pCre (P = 0.02); there was a consistent trend for ATP (P = 0.09) and Pi (P = 0.08); and the LEPs (ADP, uCre, LEP) were unaltered (Table 2).
In the averaged ‘occipital lobe’ ROI, none of the parameters measured differed significantly in patients and controls (Table 2).
Lactate
In healthy control brains, lactate is not detectable by 1H-MRS, using the data acquisition scheme described earlier (Isobe et al, 2007). Consistently, we found no lactate peaks in the 1H-MRS data of any of healthy control subjects. In contrast, lactate peaks suggestive of increased anaerobic glycolysis (Prichard et al, 1991) were found in 7 of 20 (35%) PSP patients. These lactate peaks were typically found in a paraventricular location in the ‘basal ganglia’ ROIs (Figure 1B).
pH
The intracellular pH was not significantly different in any of the ROIs of the patients compared with those of the controls (Table 2).
Dependence of MRS Outcomes on Age and Sex
In view of slight, nonsignificant imbalances in the age and sex distributions of patients and healthy controls (Table 1), we calculated robust regressions of the MRS outcomes in patient status adjusted for age and sex as sensitivity analyses. The results were in line with the nonadjusted analyses (data not shown).
Clinical Correlations
We calculated the Spearman rank correlations between the concentrations of the HEPs (ATP, pCre, HEP) and the PP in the averaged ‘basal ganglia’ ROI and the scales measuring motor dysfunction (PSP-RS total score and UPDRS III). No significant correlations were observed (data not shown). We also found no significant rank correlations of the concentrations between the HEPs (ATP, pCre, HEP) and the PP in the frontal lobes with the FAB scale measuring frontal lobe dysfunction (not shown).
Differentiation of Patients From Controls
In our data set, a preliminarily assumed threshold of ⩽4.95 mmol/L for HEP would have distinguished PSP patients from controls free of neurologic symptoms with a sensitivity of 85% and a specificity of 71%.
Discussion
Phosphorus magnetic resonance spectroscopy is an excellent technique for monitoring cerebral energy metabolism in vivo, as ATP, pCre and Pi can be identified and quantified by 31P resonance peaks. The concentrations of HEP and pH can also be derived from the 31P spectrum. However, to quantify NAA, tCre and Lac, 1H-MRS must be used. Furthermore, the concentrations of ADP, uCre, LEP and the PP can be only derived without further a priori assumptions from joint 31P- with 1H-MRS measurements. Therefore, we used combined 31P- and 1H-MRS on a 3-T high-field system, calibrated both modalities to external standards to quantify absolute metabolite concentrations rather than using raw signal intensities and applied this technique in three ROIs in differentially vulnerable brain areas in 21 early-stage PSP patients compared with those in 9 healthy controls. Thus, we provide here the first comprehensive representation of brain energy metabolism in PSP patients in vivo.
Reduced cerebral glucose metabolism has already been shown in vivo in PSP patients using 18F-2-fluoro-2-deoxy-
N-acetylaspartate is an amino acid that is present in high concentrations almost exclusively in neurons. It contributes to the most prominent signal in 1H-MRS of the adult human brain. Reduced regional NAA concentrations have been reported in conditions that are characterized by neuronal cell or axon loss (Rango et al, 2007). In addition, experimental data suggest a linkage between reduced NAA levels and compromised neuronal metabolism (Moffett et al, 2007). On the basis of these data, the quantification of the NAA peak is presently considered as the single most sensitive parameter in magnetic resonance spectroscopy to indicate neuronal loss or disintegrity (Moffett et al, 2007; Rango et al, 2007). Many studies normalize NAA values with respect to tCre levels used as an internal standard, as tCre levels are thought to be relatively constant across the brain and do not change in most pathologies. The NAA/tCre and the NAA/Cho ratios have been reported to be reduced in PSP (Federico et al, 1997, 1998; Tedeschi et al, 1997; Abe et al, 2000). In our sample of mildly affected early-stage PSP patients, the decrease in the high-energy metabolites appeared to exceed rather than to reflect neuronal disintegrity, as it was observed without concomitant reductions in the NAA/tCre ratio. Partial volume effects might mimic a decrease in high-energy metabolite concentrations as a result of brain atrophy when comparing patients with controls. However, as concentrations of HEPs (ATP, pCre) were significantly reduced in patients without concomitant significant reductions in parenchymal reference metabolites (NAA, tCre and NAA/tCre) and with a nonsignificant tendency of increased concentrations of LEPs (ADP, uCre) in most ROIs, this effect appears to be specific and not merely an artifact resulting from brain atrophy. These observations support the idea that mitochondrial dysfunction is indeed implicated as an upstream event in the pathophysiology of PSP rather than being a secondary epiphenomenon.
An important question arising from our observations concerns the nature of the biochemical deficit at the origin of the metabolic alterations. The decrease in ATP might be explained by either an increase in ATP hydrolysis or a decrease in ATP production through glycolysis and oxidative phosphorylation. In the first case, one would expect an increase in glucose metabolism, but the inverse is seen in the brains of PSP patients (Foster et al, 1988). Therefore, our results support the hypothesis of a primary impairment of mitochondrial ATP production in PSP (Figure 2). The increase in lactate levels in a proportion of PSP patients, which was not observed in controls, is indicative of an excess of anaerobic glycolysis over aerobic oxidative phosphorylation (Prichard et al, 1991), suggesting that ATP generation through anaerobic glycolysis is increased to compensate for the reduction in ATP generation caused by defective oxidative phosphorylation. The reduction of pCre, the second high-energy metabolite that contributes to rapid ATP production in situations of high-energy demand, is also compatible with an impairment of oxidative phosphorylation that prevents the regeneration of this metabolite. Our earlier published observation that oral administration of coenzyme Q10, a physiologic cofactor of complex I of the mitochondrial respiratory chain, improves not only clinical disability scores but also the increased ATP/ADP, pCre/uCre and HEP/LEP ratios measured by the same 31P-MRS and 1H-MRS method, in the same PSP patients studied here (Stamelou et al, 2008), which provides further support for the concept of a functionally relevant impairment of oxidative phosphorylation in PSP.

Schematic representation of the basic pathways of energy metabolism and the alterations observed in PSP patients in this study. (
It is important to note that brain mitochondria can tolerate a 60% decrease in complex I activity, without a significant decrease in ATP production (Davey et al, 1997). Inversely, the presence of a small significant ATP loss strongly suggests the presence of a much more severe inhibition of the respiratory chain. The reduction in high-energy metabolites in PSP was detected at rest, whereas in Parkinson's disease, another neurodegenerative disorder with presumed mitochondrial complex I dysfunction (Schapira, 2006), similar metabolic alterations were only observed after functional activation (Rango et al, 2006). This may suggest that the mitochondrial dysfunction in PSP is more severe than in Parkinson's disease. Therefore, we are tempted to conclude that the small, yet significant alterations in the cerebral high-energy metabolites determined in this study provide important insights into the pathophysiology of PSP.
It is interesting that the decrease in high-energy metabolites observed in the PSP patients was not accompanied by an increase in their low-energy counterparts, as it occurs during acute ischemia (Hems and Brosnan, 1970) or in Leber's hereditary optic neuropathy (Lodi et al, 2000). However, there is solid in vitro and in vivo experimental evidence that mammalian cells, in which oxidative phosphorylation is impaired, have the potential to downregulate the free cytosolic fraction of uCre, ADP and Pi, by (1) increasing the protein-bound fraction of uCre in the cytoplasm and (2) by the compartmentalization of ADP and Pi in the inner mitochondrial membrane, which would render them immobile and thus invisible by MRS (Iles et al, 1985; Balaban, 1990; Brown, 1992; Iotti et al, 1996). Such a compensatory counterregulation might be the biochemical basis for the changes in cerebral energy metabolites in PSP observed in this study. Such a regulation would also appear biologically meaningful, as it would, first, prevent the inactivation of ATPases by abnormally high cytosolic concentrations of ADP and Pi. Second, increased translocation of ADP and Pi to the inner mitochondrial membrane would stimulate the rate of oxidative phosphorylation. Third, downregulation of cytosolic LEPs in a metabolic state, in which HEPs are chronically reduced would stabilize the phophorylation potential PP (Iles et al, 1985; Balaban, 1990; Brown, 1992) (Figure 2).
Indeed, the fact that the PP [=ATP/(ADP × Pi)] and the other ratios of high-energy metabolites when compared with their low-energy counterparts (ATP/ADP, pCre/uCre and HEP/LEP), which determine the energy level driving all cellular energy-dependent processes (Iles et al, 1985; Pietz et al, 2003; Schapira, 2006), was not significantly altered in the patients suggests that the affected cells can still compensate, at rest, for the decrease in high-energy metabolites (Iles et al, 1985; Pietz et al, 2003; Schapira, 2006). This may mean that the neurons in the affected brain areas in the early-stage PSP studied were not dysfunctional at rest. This perspective is consistent with the observation that the severity of the clinical symptoms was not correlated with the concentrations of HEPs or the PP in PSP patients. Further, the observed metabolic deficit, particularly the ATP depletion, may render the affected neurons more prone to neurodegeneration and somatodendritic tau accumulation, as we have shown experimentally (Höglinger et al, 2005; Escobar-Khondiker et al, 2007).
In summary, our data provide the first in vivo evidence of a significant depletion in high-energy metabolites at rest in vulnerable brain areas of early-stage PSP patients without significant alterations in the NAA/tCre ratio. The observed pattern of metabolic alterations is more compatible with a primary decrease in oxidative phosphorylation than with a secondary consequence of neurodegeneration. This suggests that mitochondrial dysfunction is indeed crucially implicated in the pathophysiology of PSP, not a consequence thereof. Interestingly, the difference between the concentrations of HEPs in the ‘basal ganglia’ ROI of early-stage PSP patients and healthy controls was such that these metabolites might even prove to be helpful diagnostic markers. This should be examined further in independent samples.
Footnotes
Acknowledgements
We thank O Arias-Carrión for skilful help with the graphics.
The authors declare no financial interest related to the materials and methods presented here.