
Abstract
In this study, we examined whether rosiglitazone, a peroxisome proliferator-activated receptor gamma (PPARγ) agonist, is neuroprotective in focal ischemic brain injury, and whether rosiglitazone can enhance the protective action of tissue plasminogen activator (tPA), an agent used clinically for thrombolytic therapy. Rats were subjected to ischemic brain injury by embolizing preformed clots into the middle cerebral artery (MCA). Treatment with rosiglitazone reduced infarction and improved functional recovery; it also enhanced the neuroprotective action of tPA and lengthened the time window for initiating tPA treatment. Occlusion of MCA resulted in a loss of collagen type IV, a major structural protein of the microvascular basal lamina, and tPA treatment worsened this loss. Rosiglitazone treatment prevented the reduction of collagen type IV in the ischemic injured brain by inhibiting the activation of matrix metallopeptidase-9 (MMP-9). In addition, rosiglitazone treatment reduced inflammatory reactions in the ischemic injured brain. Rosiglitazone either alone or in combination with tPA is an effective agent in the reduction of ischemic brain injury. The reduction of microvascular damage and inflammation contributes to the beneficial actions of rosiglitazone.
Introduction
Inflammatory reactions contribute considerably to the pathogenesis of ischemic brain injury (Jean et al, 1998; Wang and Shuaib, 2002). These inflammatory reactions are characterized by a sequential series of processes, including the release of pro-inflammatory cytokines, increased expression of endothelial adhesion molecules and chemotactic factors, activation of microglia and macrophages, and infiltration of leukocytes. The inflammatory reactions exacerbate brain injury by increasing secondary expansion of ischemic infarction. Therefore, interventions aimed at suppressing postischemic inflammatory reactions are an emerging therapeutic strategy for the treatment of ischemic brain injury.
Peroxisome proliferator-activated receptor gamma (PPARγ) is a nuclear membrane-associated transcription factor (Collino et al, 2006; Sundararajan and Landreth, 2004). PPARγ was originally reported to be highly expressed in adipocytes and to have an important function in their differentiation, as well as in lipid biosynthesis and glucose homeostasis (Lemberger et al, 1996; Vamecq and Latruffe, 1999). Later studies suggest that PPARγ exhibits antiinflammatory properties by negatively regulating the expression of various pro-inflammatory molecules including interleukin-1β (IL-1β), IL-6, and tumor-necrosis factor-α (TNF-α) (Sundararajan and Landreth, 2004; Sundararajan et al, 2005). Evidence accumulated also shows the expression of PPARγ in the central nervous system (Inestrosa et al, 2005; Moreno et al, 2004). Therefore, it has also been suggested that PPARγ agonists act as potent antiinflammatory agents in the brain. Indeed, PPARγ agonists have shown promising beneficial effects in several animal models of central nervous system disorders in which an inflammatory component is strongly implicated, such as experimental autoimmune encephalomyelitis, Parkinson's disease, Alzheimer's disease, and ischemic brain injury (Sundararajan et al, 2005; Inestrosa et al, 2005; Breidert et al, 2002; Feinstein et al, 2002; Lo et al, 2002). In addition, in vitro studies have shown that PPARγ agonists protect cultured cerebellar granule cells from apoptotic cell death (Heneka et al, 1999).
Thrombolytic therapy with tissue plasminogen activator (tPA) has demonstrated efficacy in the treatment of acute stroke patients (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995; Hacke et al, 1995; Kwiatkowski et al, 1999). Unfortunately, tPA treatment is associated with increasing incidence of cerebral hemorrhage, and this risk increases with progressive delays in treatment initiation. Furthermore, the incidence of cerebral hemorrhage is directly related to the increased dosage of the thrombolytic agent. Inflammation associated with postischemic reperfusion also limits the efficacy of thrombolytic therapy in acute stroke (Jean et al, 1998). Therefore, only a small fraction of stroke patients who present to the hospital within a few hours of stroke onset are eligible for the treatment. Consequently, it would be desirable to improve the efficacy and safety of stroke therapy and to increase the time window after stroke during which thrombolytic therapy is effective, thereby more patients become eligible for receiving this treatment.
In this study, we evaluate the effect of rosiglitazone, a PPARγ agonist, in an embolic model of focal ischemic brain injury. We also studied whether rosiglitazone can enhance the protective actions of tPA. In addition, we investigated whether the protective actions of rosiglitazone are mediated through the protection of cerebral microvessels and restriction of inflammatory reactions.
Materials and methods
Focal Embolic Model of Stroke
Male Wistar rats, weighting 300 to 350 g (Charles River, St Constant, Canada), were used. Focal cerebral ischemia was induced by embolizing a preformed clot into the middle cerebral artery (MCA) as reported earlier (Wang et al, 2001). Surgery was performed in rats anesthetized with isoflurane in a mix of O2 and NO2. Infarct volume was quantified in 2% 2,3,5-triphenyltetrazolium chloride (TTC)-stained brain sections at 48 h after the MCA occlusion (Shuaib et al, 2002; Wang et al, 2001). Brain swelling was also measured in the TTC-stained brain sections and calculated using a formula: swelling (edema)=(the volume of the right hemisphere−the volume of the left hemisphere)/the volume of the left hemisphere (Shuaib et al, 2002). Neurologic deficits were recorded at 1 and 24 h after the MCA occlusion using a modified Bederson's scoring system (Wang et al, 2001). Three series of experiments were performed to examine whether treatment with rosiglitazone could reduce ischemic brain injury. In the first series, animals were randomly assigned to one of three groups. In the control group (n=9), rats received saline. In the second group (n=10), rats received rosiglitazone 2 mg/kg per day. In the third group (n=10), rats received rosiglitazone 10 mg/kg per day. Treatments were started 2 weeks before the MCA occlusion, and continued until the end of each experiment. Rosiglitazone was dissolved in saline and administered by gastric gavage, twice per day for these and also subsequent experiments. The second series consisted of four groups. In the control group (n=9), animals received saline. The other three groups of animals received rosiglitazone treatment (2 mg/kg), which was administered at 1, 3, or 6 h after the MCA occlusion (n=10 per group). The third series consisted of five groups (n=10 per group). In the control group, animals received saline. In the second group, animals received tPA (5 mg/kg). In the third group, animals received tPA (5 mg/kg) plus rosiglitazone (2 mg/kg). In the fourth group, animal received tPA (10 mg/kg). In the fifth group, the animals received tPA (10 mg/kg) plus rosiglitazone (2 mg/kg). The treatments of rosiglitazone were the same as described in the first series, and tPA was infused intravenously at 3 h after the MCA occlusion. The doses of tPA were chosen based on the findings from earlier studies, which show that the fibrinolytic effects of tPA is one-tenth as effective in rat when compared with human (Korninger and Collen, 1981; Zhang et al, 2003).
Detection of Perfusion Deficits
Perfusion deficits were analyzed using an Evans blue staining procedure (Wang et al, 2001; Noor et al, 2005). The perfused microvessels in the brain sections were visualized under fluorescent microscopy in nine brain sections taken serially and 1 mm apart starting at 3.70 mm anterior to bregma. Areas of perfusion deficits in the brain were traced, calculated, and expressed as a percentage from the ipsilateral hemisphere. The time point for the sample collection was determined based on results of our earlier studies (Noor et al, 2005; Shabanzadeh et al, 2008). There were five groups in this study. In one group (n=5), the rats were killed 5 min after the MCA occlusion. The rats in all other groups were killed 8 h after the MCA occlusion and each group consisted of nine rats. In these groups, the rats received one of the following treatments: saline, tPA alone, rosiglitazone alone, rosiglitazone plus tPA. The administration of rosiglitazone (2 mg/kg) was started at 2 weeks before the MCA occlusion as described above, and tPA (10 mg/kg) was infused intravenously 3 h after the MCA occlusion.
Detection of Collagen Type IV
Twenty-four hours after the MCA occlusion, the rats were killed and brains were removed. After sectioning, the brain sections were incubated with goat anticollagen type IV (SouthernBiotech, Richmond, VA, USA), and then fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Sigma, Atlanta, GA, USA), as described earlier (Kelly et al, 2006; Song et al, 2005). The FITC-labeled microvessels were identified under fluorescent microscope and photographed. No specific staining could be observed when first or secondary antibody was omitted, indicating detection of collagen type IV is not compromised by nonspecific background information. The numbers of microvessels per area (vascular density) and the areas of the microvessels (vascular fraction) were analyzed with Improvision Image Anaylsis System (Coventry, UK). Percentage changes of the microvessel number and area between the ipsilateral and contralateral hemispheres to the occluded MCA were then calculated. Four brain sections were collected serially from each rat, staring at 0.7 mm anterior to bregma with an interval of 1 mm. Five areas (three in the cortex and two in the striatum) were sampled from each brain section (Figure 3F). There were five groups in this study and each group consisted of three rats. In one group, the rats were killed 5 min after the MCA occlusion. The rats in all other groups were killed 24 h after the MCA occlusion. In these groups, the rats received one of the following treatments: saline, tPA alone, rosiglitazone alone, rosiglitazone plus tPA. The administration of rosiglitazone (2 mg/kg) was started at 2 weeks before the MCA occlusion as described above, and tPA (10 mg/kg) was infused intravenously 3 h after the MCA occlusion. The changes of collagen type IV were also examined in cell lysates with Western blot analyses in the brain samples collected for the gel zymography study, as described in the following sections.
Gel Zymography
Gelatin gel zymography was used to determine the matrix metallopeptidase-9 (MMP-9) activities (Kelly et al, 2006). In brief, brain samples were homogenized in lysis buffer and centrifuged. Protein concentration was determined using Bio-Rad (Hercules, CA, USA) protein assay reagent; equal amounts of proteins were loaded onto 8% acrylamide gel containing 0.1% gelatin as a substrate. Purified human MMP-9 (R&D system, Minneapolis, MN, USA) was also included as a standard control. On the completion of protein separation, the gels were incubated in Triton-X 100 and then in incubation buffer at 37°C until bands appeared. Gels were then stained with Coomassie blue and destained accordingly. MMP activation appeared as transparent bands on blue background. Images of the gels were captured by scanning on an HP ScanJet flatbed scanner (Boise, ID, USA) and analyzed with NIH image software. In these experiments, we first examined the MMP-9 activities in the brain when the rats were killed at different time points after the MCA occlusion. Then, we examined the MMP-9 activities in the brain after the treatment with rosiglitazone alone or in combination with tPA. The dosage of rosiglitazone was 2 mg/kg, and tPA was 10 mg/kg. The rosiglitazone treatment was started 2 weeks before the MCA occlusion and tPA was infused intravenously at 3 h after the MCA occlusion, as described above. The rats were killed at 24 h after the MCA occlusion. Each group consisted of five rats.
Western Blot
Western blot analyses were performed as described in the earlier studies (Song et al, 2005). Brain tissues were homogenized in cold lysis buffer and supernatants were collected. Nuclear extracts were prepared using a protocol from Active Motif (Carlsbad, CA, USA). The cell lysates (50 μg), as well as nuclear fractions (10 μg) from the subcellular fractionation, were subjected to SDS–polyacrylamide electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked in Tris-buffered saline with 0.05% Tween-20 containing 5% milk and then blotted overnight with primary antibodies. The membranes were washed and incubated with horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA). The membranes were washed and developed by chemiluminescence. Densitometric analyses of protein levels were performed with NIH image software. Rabbit antibodies used in this study included PPARγ and NF-kB p65 (Santa Cruz Biotech, Santa Cruz, CA, USA). There were eight rats per group. The treatments started 2 weeks before the MCA occlusion, as described above. Actin and Sp1 levels were used as loading controls for cytosolic and nuclear protein expression (Santa Cruz Biotech), respectively.
Immunoassay for Cytokines
The cytokines levels, IL-1β, and TNF-α, were measured with enzyme-linked immuno-sorbent assay (ELISA) kits (Biosource, Camarillo, CA, USA), according to the manufacture's instruction as described earlier (Wang et al, 1997). The levels of the cytokines in the brain were expressed as pictogram per milligram total proteins in the supernatant. For these studies, the treatments started 2 weeks before the MCA occlusion, as described above. There were four groups. Two groups of rats received saline treatment and were killed at 8 h (n=7) or 24 h (n=8) after the MCA occlusion. The two other groups received rosiglitazone treatment and were killed at 8 h (n=8) or 24 h (n=7) after the MCA occlusion.
Statistical Analysis
The data of infarct volume, brain edema, immunohistochemistry, densitometry, and ELISA were analyzed with one-way ANOVA followed by Tukey test when comparing more than two groups and analyzed with Student's t-test when comparing between the two groups. The neurologic scores were analyzed with Kruskal–Wallis test when comparing more than two groups, and analyzed with Mann–Whitney test when comparing between two groups. The rates of hemorrhage occurrence after different treatments were compared with χ2- test. Differences were considered significant when P<0.05.
Results
Rosiglitazone Protected Against Ischemic Brain Injury
Hematological parameters measured in the controls and the animals receiving treatment with rosiglitazone are shown in Table 1. Treatment with rosiglitazone did not result in significant changes in the parameters measured when compared with the appropriate control group.
Hematological parameters a
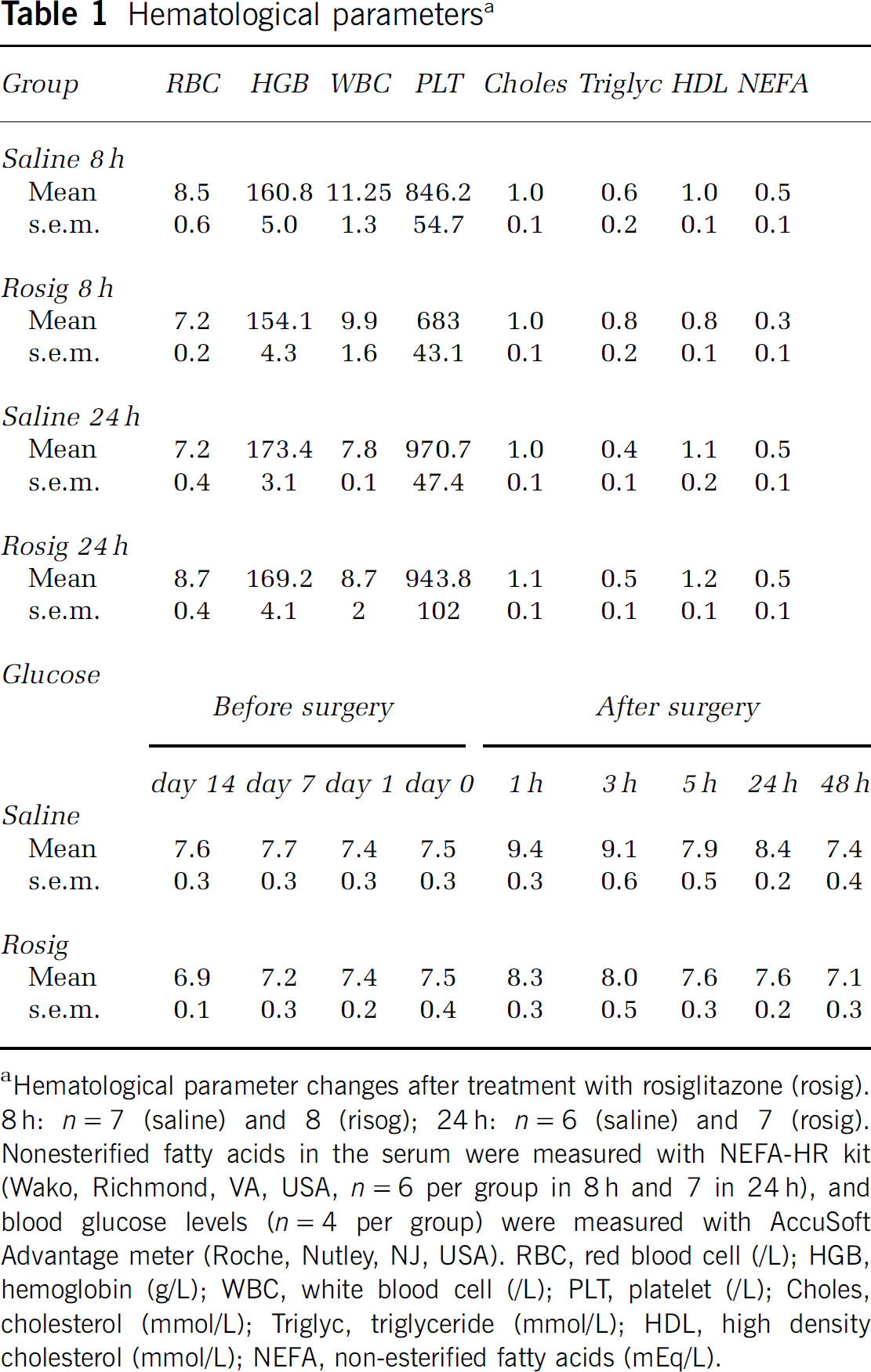
Hematological parameter changes after treatment with rosiglitazone (rosig). 8 h: n=7 (saline) and 8 (risog); 24 h: n=6 (saline) and 7 (rosig). Nonesterified fatty acids in the serum were measured with NEFA-HR kit (Wako, Richmond, VA, USA, n=6 per group in 8 h and 7 in 24 h), and blood glucose levels (n=4 per group) were measured with AccuSoft Advantage meter (Roche, Nutley, NJ, USA). RBC, red blood cell (/L); HGB, hemoglobin (g/L); WBC, white blood cell (/L); PLT, platelet (/L); Choles, cholesterol (mmol/L); Triglyc, triglyceride (mmol/L); HDL, high density cholesterol (mmol/L); NEFA, non-esterified fatty acids (mEq/L).
Dose–response study
Initially, we examined the protective effects of various doses of rosiglitazone on ischemic brain injury (Figures 1A and 1B). Administration of rosiglitazone started 2 weeks before and was continued after the MCA occlusion until the end of each experiment. Compared with the saline group, infarct volume was reduced significantly after rosiglitazone treatment with 2 and 10 mg/kg per day, respectively (P<0.05). The treatment with rosiglitazone, 2 or 10 mg/kg per day, also significantly reduced brain swelling (P<0.05). Brain hemorrhage was observed in two animals from each group. Treatment with rosiglitazone also improved functional recovery, as measured by neurologic deficit scores, Table 2. As there were no significant differences on infarct volume or neurologic deficits after treatment with rosiglitazone 2 and 10 mg/kg, the lower dose was chosen for the subsequent studies.
Neurological deficits a
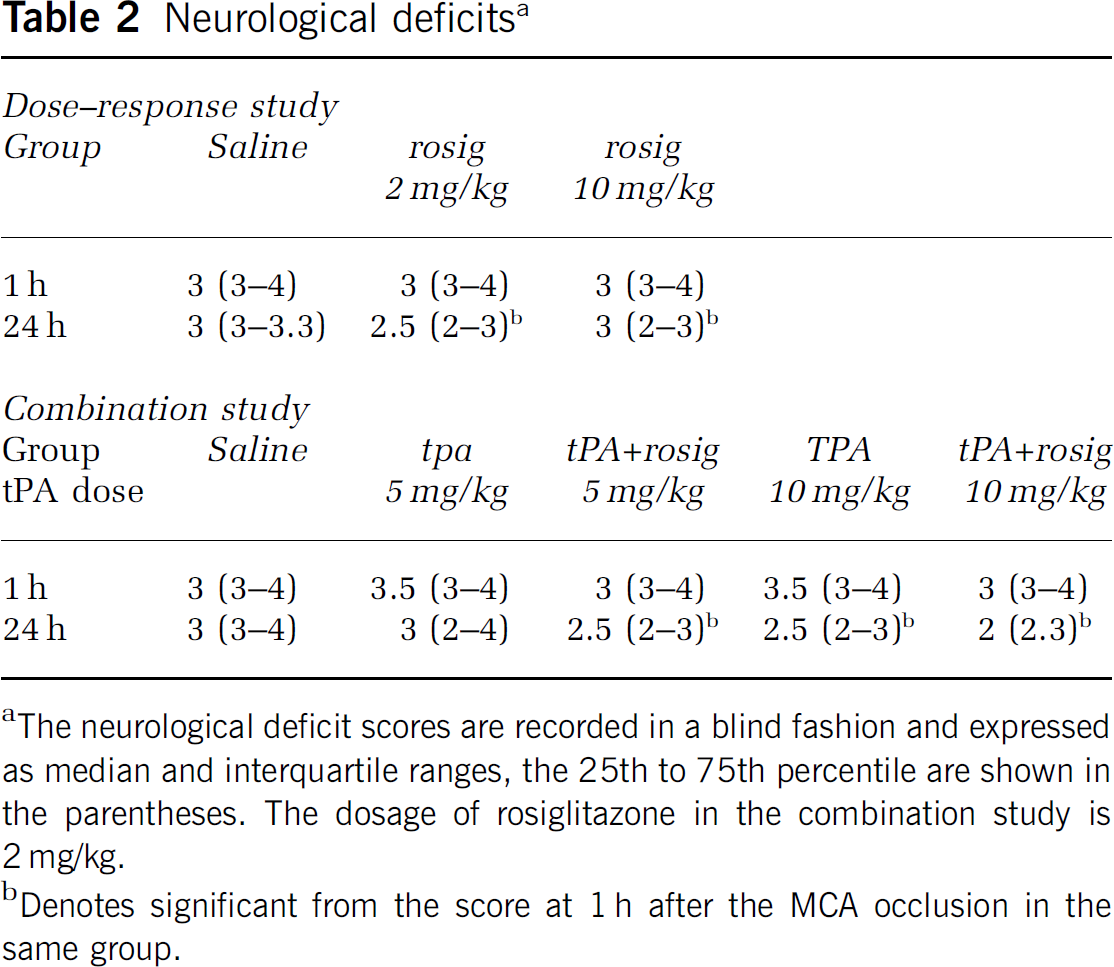
The neurological deficit scores are recorded in a blind fashion and expressed as median and interquartile ranges, the 25th to 75th percentile are shown in the parentheses. The dosage of rosiglitazone in the combination study is 2 mg/kg.
Denotes significant from the score at 1 h after the MCA occlusion in the same group.
Time course study
In the second series, we examined whether treatment with rosiglitazone only after the MCA occlusion still protects the ischemic injured brain (Figures 1C and 1D). Compared with the saline group, the infarct volume, brain edema, neurologic deficits, and brain hemorrhage (data not shown) did not change significantly in all groups examined when rosiglitazone treatment was started after the MCA occlusion (P>0.05).
Combination study
In the third series, we examined whether treatment with rosiglitazone could enhance the therapeutic effects of tPA (Figures 1E and 1F). Compared with the saline group treatment with tPA 5 mg/kg alone did not significantly reduce infarct volume (P>0.05). However, combination treatment with tPA 5 mg/kg plus rosiglitazone reduced infarct volume significantly (P<0.05). Treatment with tPA (10 mg/kg) either alone or in combination with rosiglitazone also significantly reduced infarct volume (P<0.05). The infarct volume was not significantly different between tPA alone (10 mg/kg) and tPA plus rosiglitazone (P>0.05). Compared with the saline group, treatment with either tPA alone (5 mg/kg) or in combination with rosiglitazone did not significantly reduce brain swelling (P>0.05). However, compared with the tPA (10 mg/kg) alone group, treatment with tPA (10 mg/kg) plus rosiglitazone decreased brain swelling significantly (P<0.05). Brain hemorrhage was observed in three rats in the saline group, five rats in tPA (5 mg/kg) alone group, four rats in the tPA (5 mg/kg) plus rosiglitazone group, seven rats in tPA (10 mg/kg) alone group, and four rats in the tPA (10 mg/kg) plus rosiglitazone group. Statistical analyses showed that the hemorrhage rate in the drug-treated groups was not significantly different from the saline group (P>0.05). Compared to the scores at 1 h, treatment with tPA (5 mg/kg) plus rosiglitazone, tPA (10 mg/kg) alone, or tPA (10 mg/kg) plus rosiglitazone significantly reduced the neurologic deficit scores at 24 h after the MCA occlusion (P<0.05), Table 2.
Rosiglitazone Did not Reduce Perfusion Deficit
To examine whether treatment with rosiglitazone can reduce microvessel occlusion, perfusion deficits were measured in the ischemic injured brain. In the rats killed at 5 min after the MCA occlusion the area of perfusion deficits was 45.6%±11.1% of ipsilateral hemisphere of the brain. Compared with the group killed at 5 min after the MCA occlusion, the perfusion deficit decreased significantly in the rats killed at 8 h after the MCA occlusion in all groups examined (P<0.05). Although perfusion deficit in the tPA and rosiglitazone plus tPA groups were smaller than the saline group but did not reach significant difference (P>0.05), Figure 2.
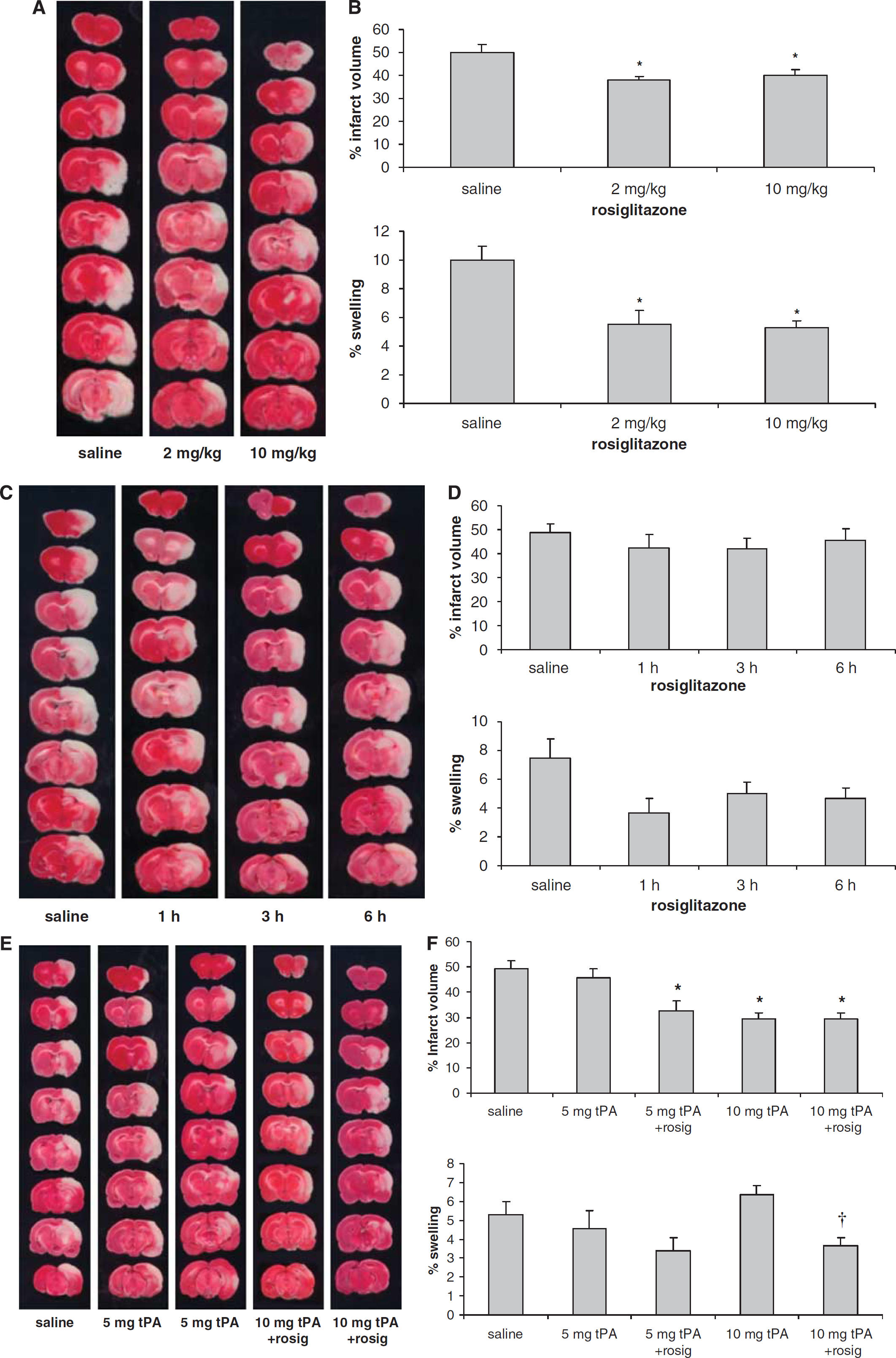
Treatment with rosiglitazone starting before the MCA occlusion protects the ischemic injured brain. Representative TTC-stained brain sections (
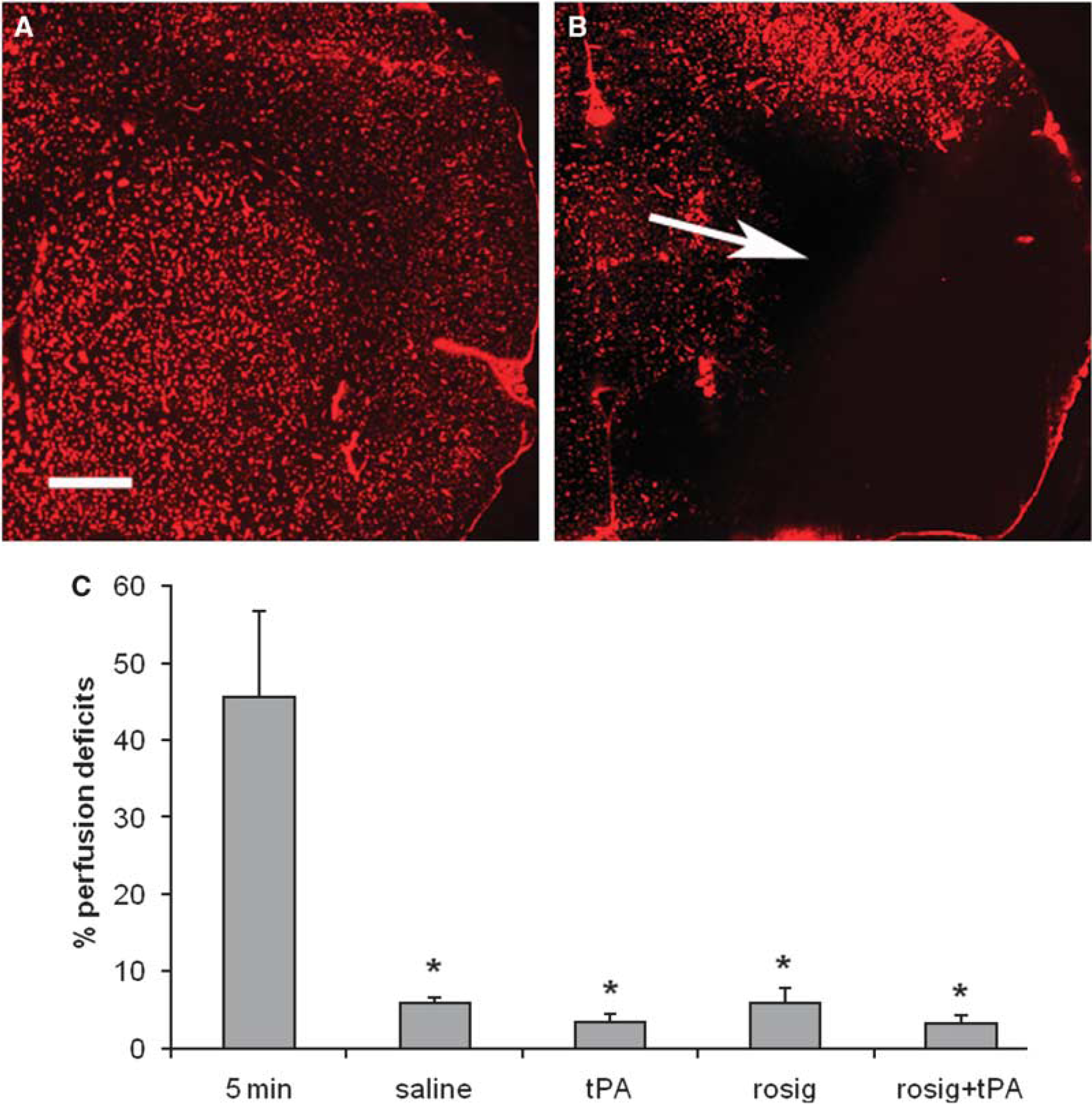
Treatment with rosiglitazone did not reduce perfusion deficits in the ischemic injured brain. Perfusion deficits were detected with Evans blue staining procedures. Under fluorescent microscopy perfusion deficits appear as black areas. (
Rosiglitazone Reduced the Damage of Microvascular Basal Membrane
Next, we examined whether treatment with rosiglitazone can reduce the damage to microvasculature in the ischemic injured brain (Figure 3). We first studied effects of rosiglitazone treatment on the integrity of the microvessels by measuring the changes in collagen type IV, a major structural protein of the microvascular basal membrane, with immunohistochemistry. When compared with saline group, treatment with tPA significantly decreased, and treatment with rosiglitazone significantly increased, the microvessel areas (P<0.05). Compared with the tPA alone group, the microvessel area was significantly higher in the animals that received either rosiglitazone alone or in combination with tPA (P<0.05). The changes in microvessel numbers paralleled those in the microvessel areas. In addition, the changes of collagen type IV detected with Western blot analyses were also in agreement with the results from immunohistochemistry study.
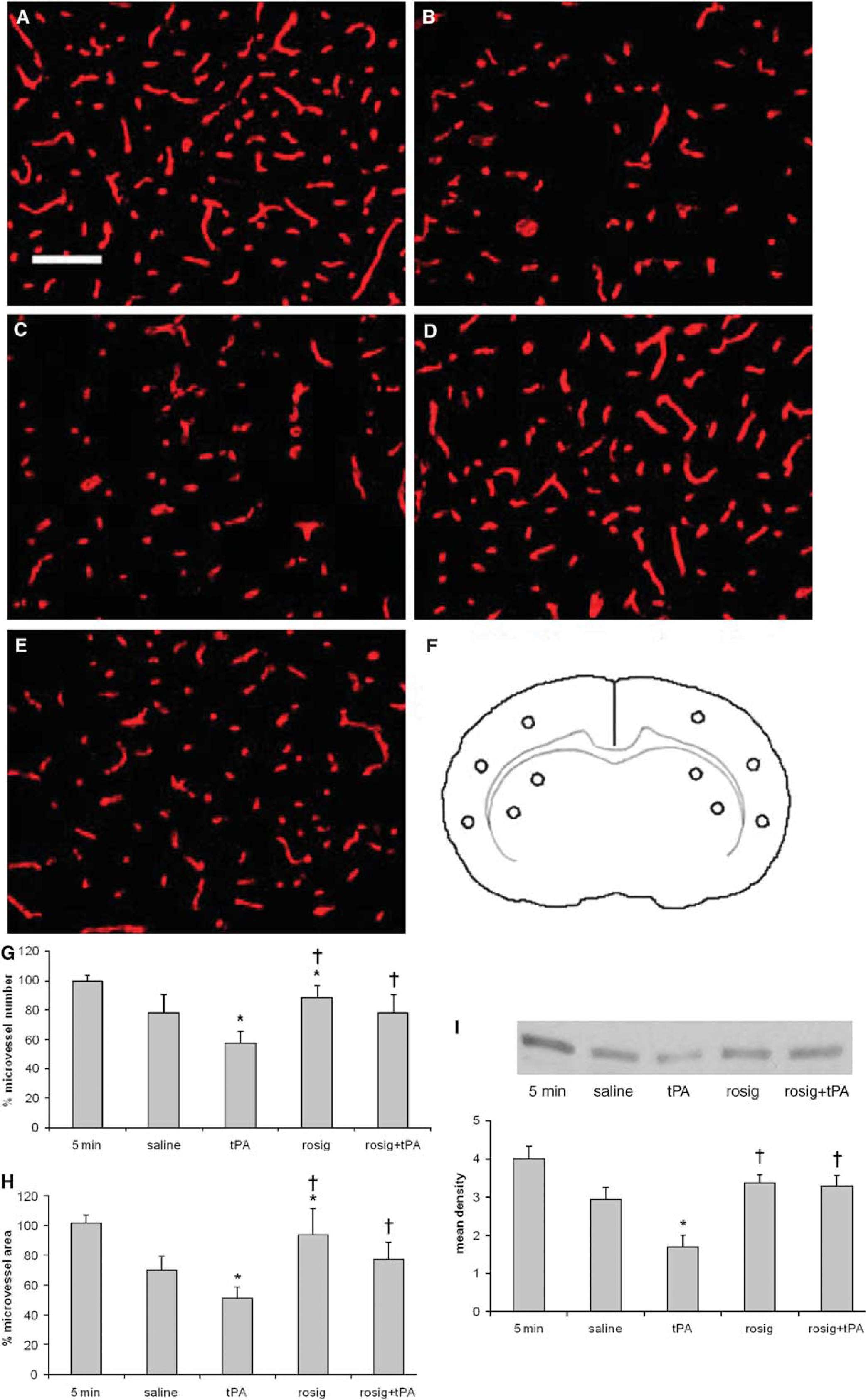
Collagen type IV changes in the ischemic injured brain. (
As collagen type IV is a substrate of MMP-9, a preotelytic enzyme involved in the dismantlement of the microvascular basal membrane, we also examined whether rosiglitazone treatment affects the activity of MMP-9. We found that ischemic injury resulted in an increase in MMP-9 activity in the ipsilateral hemisphere to the MCA occlusion (Figure 4A), but not in the contralateral hemisphere (data not shown). The MMP-9 activity increase started at 1 h and maximal activity was observed at 24 h after the MCA occlusion (Figure 4A). Compared with the saline group, treatment with tPA resulted in an increase in MMP-9 activity (P<0.05), Figure 4B. When compared with group that received tPA alone, the MMP-9 activity was decreased in the groups that received rosiglitazone alone or in combination with tPA (P<0.05). However, the MMP-9 activity in the rosiglitazone-treated group did not differ from the saline-treated group (P>0.05).
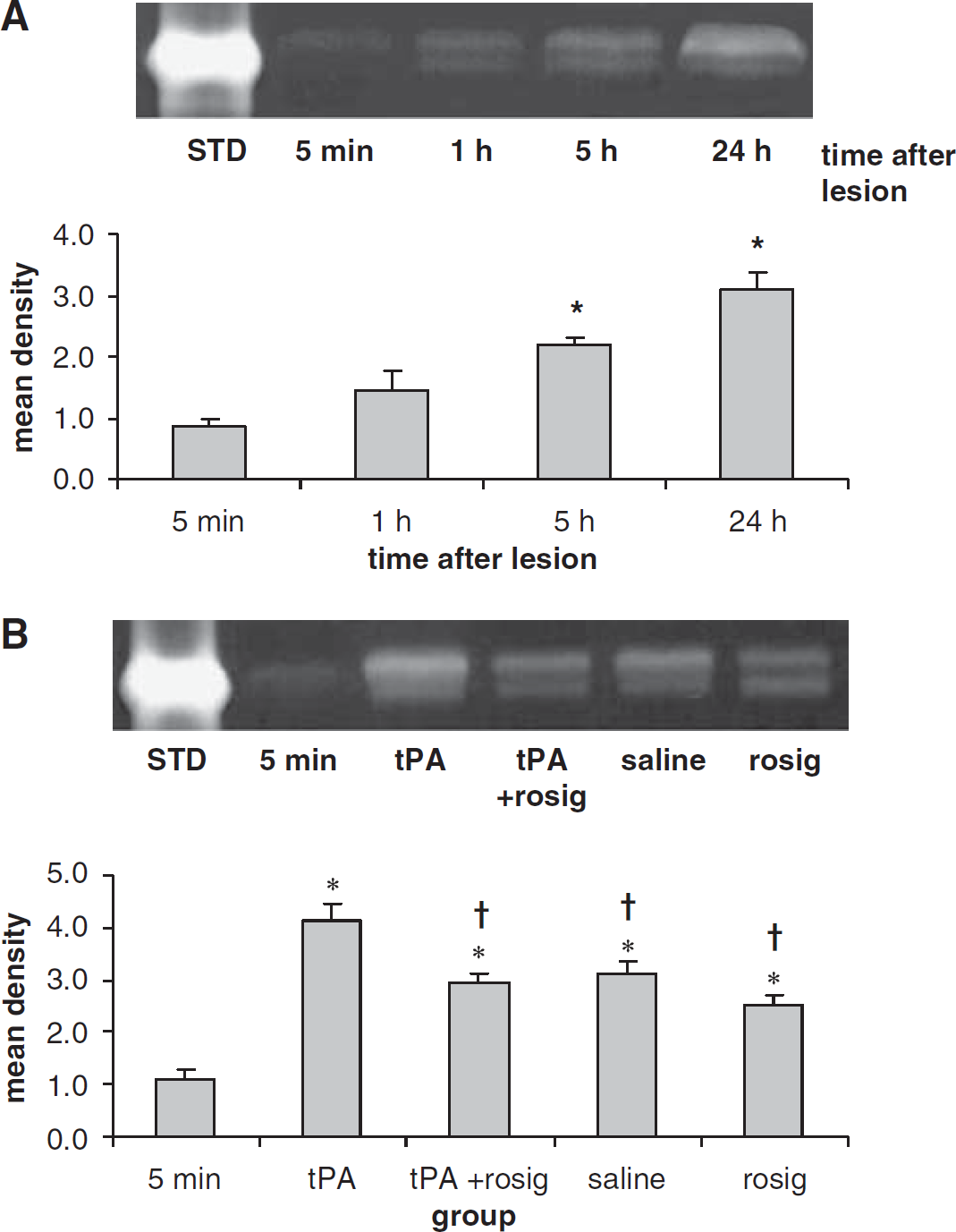
Zymograms show the gelatinolytic activities of MMP-9 in the ischemic injured brain. (
Rosiglitazone Reduced Inflammatory Reaction
To further study the mechanism of how rosiglitazone reduces ischemic brain injury, we next examined the actions of this drug on the levels of pro-inflammatory cytokines, IL-1β and TNF-α, in the ischemic injured brain (Figures 5A and 1B). Compared with the saline groups, treatment with rosiglitazone significantly reduced IL-1β in the cortex (P<0.05) but not in the striatum (P>0.05) at 8 and 24 h after the MCA occlusion. The rosiglitazone treatment also significantly reduced the TNF-α levels in both the cortex and striatum at 8 h, as well as in the cortex at 24 h after the MCA occlusion (P<0.05). As the productions of these cytokines are regulated by nuclear factors, we therefore examined the changes in NF-kB 65 after the rosiglitazone treatment. Western blot analyses showed that rosiglitazone treatment significantly reduced ischemia-induced increase in NF-kB 65 in the nuclear fraction (Figure 5C). Taken together, these data thus support that reduction in inflammatory reactions contributes to the protective actions of rosiglitazone in ischemic brain injury.
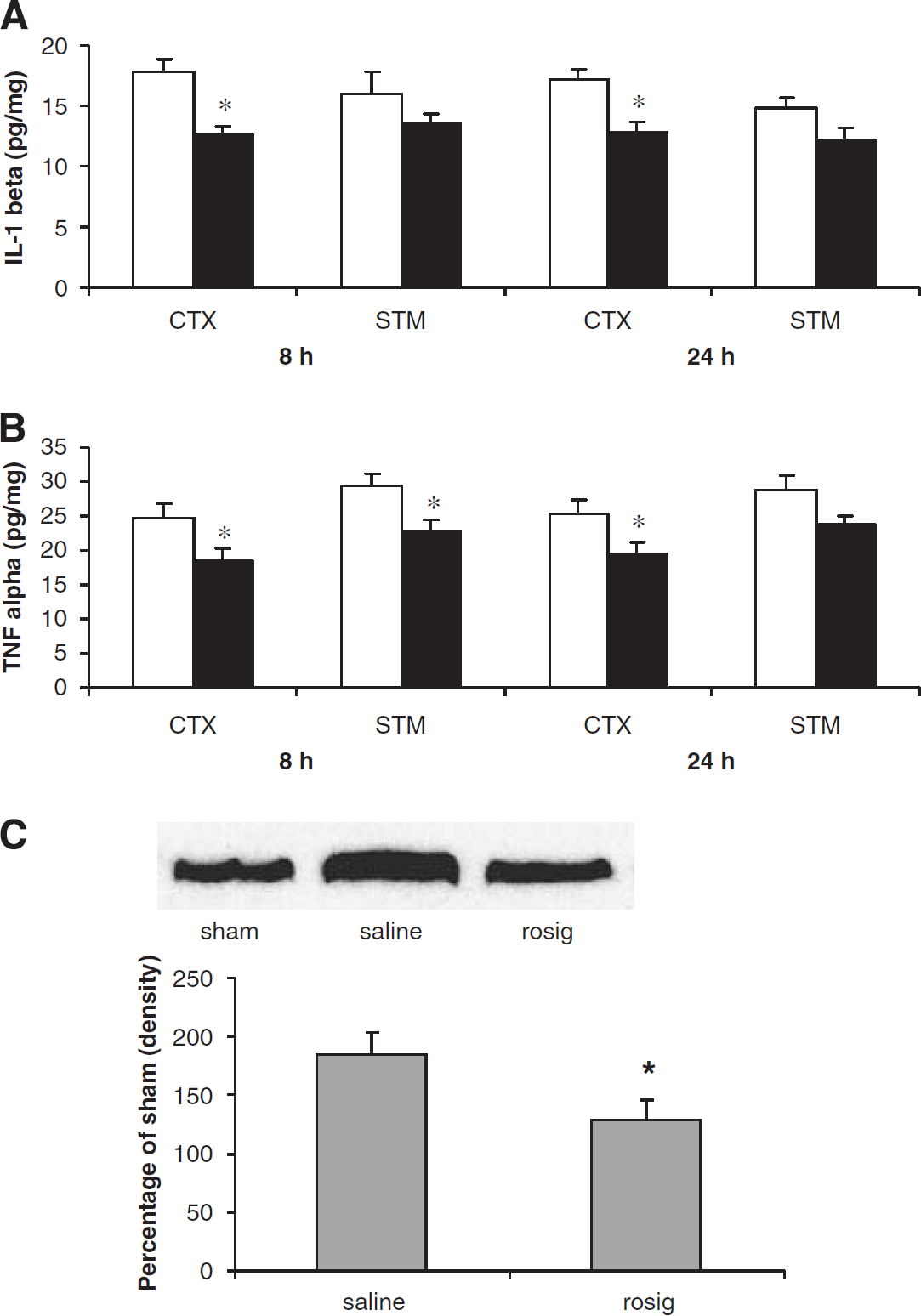
Changes of TNF-α (
Discussion
The data presented here show the neuroprotective effects of rosiglitazone in an embolic model of focal cerebral ischemia. Rosiglitazone, administered 2 weeks before the MCA occlusion and continued until the end of the experiment, significantly reduced infarct size and brain edema. Furthermore, this treatment also improved functional recovery after the ischemic brain injury, as measured by neurologic scores. Although it did not significantly improve stroke outcome when the medication was started after the MCA occlusion, this treatment did, however, enhance the therapeutic efficacy and lengthen the window of tPA administration without causing increase in cerebral hemorrhage. To our knowledge this study is the first to examine combination treatment with rosiglitazone and thrombolytic agent, tPA, in ischemic brain injury. The dosages of rosiglitazone used in this study were chosen based on information from published studies and they are comparable to the majority of studies in rodent models (Breidert et al, 2002; Tureyen et al, 2007; Chu et al, 2006; Allahtavakoli et al, 2007).
The early reestablishment of reperfusion is a logical first step in the treatment of acute ischemic stroke. Indeed, thrombolytic therapy with tPA improves neurologic outcome in animal models of ischemic brain injury and this therapy is the only proven effective pharmacological agent for acute stroke patients (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995; Hacke et al, 1995; Kwiatkowski et al, 1999). However, the increased risk of hemorrhagic transformation in the ischemic injured brain limits the clinical use of thrombolytic therapy with tPA. Experimental data suggest that inflammation associated with postischemic reperfusion might contribute to the hemorrhagic transformation and also the development of secondary brain damage (Jean et al, 1998). Leukocytes recruited to the ischemic zone after reperfusion produce proteolytic enzymes that, in addition to direct neuronal damage, injure the microvasculature. Thus, inflammatory reactions in the ischemic brain antagonize the efficacy of thrombolytic therapy in acute stroke. To maximize the beneficial actions of tPA and reduce reperfusion-induced inflammatory reactions, we examined combination therapy with rosiglitazone and tPA in the treatment of ischemic brain injury. These treatments produce synergistic effects on the reduction of ischemic brain injury. Interestingly, the synergistic effects of rosiglitazone with atorvastatin on the improvements of cardiovascular diseases have also been reported in diabetic patients (Tureyen et al, 2007). Together, these data support that adjuvant treatment with rosiglitazone can be used to enhance the effectiveness of other medications with different working mechanisms, such as tPA.
Inflammatory reaction has been shown to cause microcirculation obstruction (Hossmann, 1997). For example, blockage of the microvessels by inflammatory leukocytes has been observed in both focal and global ischemic brain injury models (del Zoppo et al 1991; Hallenbeck et al, 1986). The inhibition of leukocyte interference also reduces perfusion deficits (Mori et al, 1992). Therefore, we examined whether treatment with rosiglitazone can reduce the perfusion deficits in the ischemic injured brain. Although perfusion deficits in the animals received the treatment with tPA or tPA plus rosiglitazone are reduced, these reductions did not reach statistically significant difference. These results also support that the medications working through different mechanisms can produce synergistic neuroprotective actions for treatment of ischemic brain injury.
The damage to the microvasculature during ischemic brain injury has a significant role in hemorrhagic transformation and breach of blood-brain barrier (BBB) (Aoki et al, 2002; del Zoppo et al 1998; Pfefferkorn and Rosenberg 2003; Wang and Shuaib, 2007). MMP-9 is a proteolytic enzyme digesting components of the microvasculature (Lo et al, 2002; Cunningham et al, 2005), which contributes to the injury processes. Treatment with MMP inhibitors or MMP-neutralizing antibodies has been reported to reduce infarction in animal models of stroke (Romanic et al, 1998; Rosenberg et al, 1998; Asahi et al, 2000). Treatment with a broad-spectrum MMP inhibitor, BB-94, significantly decreased the rate of hemorrhage induced by tPA in a model of embolic focal ischemia, suggesting that inhibition of MMP activity can protect the integrity of microvasculature (Lapchak et al, 2000). Here, we show that treatment with rosiglitazone decreased the activity of MMP-9 and also reduced the degradation of collagen type IV. It is likely that rosiglitazone regulate the expression of MMP-9 by antagonizing the activities of the transcription factors such as AP-1, STAT, and NF-kB (Herzlich et al, 2008; Ricote et al, 1998). Whether the treatment with rosiglitazone directly regulates the MMP-9 expression is currently not clear and more studies are needed. Taken together, data presented here support that rosiglitazone contributes to the reduction in ischemic damage and the incidence of hemorrhagic transformation through the restriction of ischemia-induced destruction of the microvasculature.
Involvement of inflammatory reactions in the ischemic injury processes has been shown (Wang and Shuaib, 2002; Sandoval and Witt, 2008). Furthermore, sudden tissue reperfusion through recanalization by natural dissolving of the thrombus or by the treatment of thrombolytic agents can be deleterious, worsening BBB disruption, and inflammatory reactions because of the so called ‘reperfusion injury’ (Jean et al, 1998; Kelly et al, 2006; Molina and Alvarez-Sabín 2009). We, therefore, further studied whether rosiglitazone protects the ischemic injured brain through the inhibition of inflammation. We have chosen to detect two most studied pro-inflammatory cytokines, TNF-α, and IL-1β, because these cytokines mediated infarct enlargement, the breach of BBB, and increases in ischemic edema (Sandoval and Witt, 2008; Jean et al, 1998). Our data showed that treatment with rosiglitazone significantly reduced pro-inflammatory cytokines and also reactive oxygen species (data not shown) in the ischemic injured brain. Furthermore, our data also indicate rosiglitazone inhibited the ischemia-induced inflammatory reactions by modifying the upstream signal transduction through the regulation of nuclear factors. Although results from this study did not show significant change of PPARγ in whole cell extracts (data not shown), an earlier study has shown that PPARγ-specific DNA-binding activity in the nuclear extracts is enhanced markedly by rosiglitazone, which may regulate the injury processes after ischemia (Luo et al, 2006).
The microvascular basal lamina is a specialized part of the extracellular matrix that connects the endothelial cell compartment to the subjacent cell layers, end feet of astrocytes, and smooth muscle. The basal lamina is mainly composed of collagen type IV, laminin, fibronectin, and entactin (Wang and Shuaib, 2007). The basal lamina is essential for keeping the microvascular wall intact and the normal function of microvessels. After ischemic brain injury, the basal lamina is damaged, and the endothelial cells and the end feet detached from it. Detachment of these cell structures and dismantlement of the basal lamina result in a defect in the vascular wall. Subsequently, the microvessels collapse, which in turn results in increase in the microvascular permeability, hemorrhage transformation, and ischemic brain edema (Wang and Shuaib, 2007). Additionally, the basal lamina is the only barrier that prevents large amounts of protein-rich fluids and cellular blood elements from entering into the brain during ischemia (Hamann et al, 1999). Results from this study support that treatment with rosiglitazone reduced ischemic edema. Further study is needed to clarify whether this treatment also prevents the leakage of other blood components, such as proteins.
In summary, our extensive studies show that chronic treatment with rosiglitazone is neuroprotective. We also show that combination with tPA has additive effects. There is a 50% reduction in the dose of tPA required and this dose combination is effective when used up to 3 h after the insult. The protective effects of rosiglitazone may in part be secondary to its inhibitory effects on MMP-9 activities and activation of inhibitory mechanisms against inflammatory reactions after cerebral ischemia.
Footnotes
Acknowledgements
This work is supported by CIHR, HSFC, CINF career initiative funding and Kattner funding. Authors thank Jennifer Troyanovich for revising the manuscript.