
Abstract
Multimodality molecular brain imaging encompasses in vivo visualization, evaluation, and measurement of cellular/molecular processes. Instrumentation and software developments over the past 30 years have fueled advancements in multimodality imaging platforms that enable acquisition of multiple complementary imaging outcomes by either combined sequential or simultaneous acquisition. This article provides a general overview of multimodality neuroimaging in the context of positron emission tomography as a molecular imaging tool and magnetic resonance imaging as a structural and functional imaging tool. Several image examples are provided and general challenges are discussed to exemplify complementary features of the modalities, as well as important strengths and weaknesses of combined assessments. Alzheimer's disease is highlighted, as this clinical area has been strongly impacted by multimodality neuroimaging findings that have improved understanding of the natural history of disease progression, early disease detection, and informed therapy evaluation.
Introduction
The term multimodality imaging historically referred to imaging performed using different instrumentation platforms, although a given modality may provide multiple types of imaging outcomes. For brain imaging applications, example platforms include magnetic resonance imaging (MRI), computed tomography (CT), positron emission tomography (PET), single photon computed tomography (SPECT), optical, electroencephalography, and magnetoencephalography.
Instrumentation and software developments over the past 30 years have fueled advancements in multimodality imaging and combined assessments. Efforts by Hasegawa and colleagues in the late 1980s paved the way for combined SPECT/CT (in 1996) and PET/CT (in 1998) platforms for human imaging that generally facilitated sequential data acquisitions with high spatial correspondence of the multimodality data (Townsend, 2008). Simultaneous PET/MRI is now possible in humans (Catana et al, 2008; Herzog et al, 2011; Schlemmer et al, 2008) after an extensive technical development period that includes early study of magnetic field effects on positron range (Christensen et al, 1995; Iida et al, 1986) and subsequent development of a preclinical simultaneous PET/MR scanner by Cherry and colleagues (Townsend, 2008, for review). In the preclinical animal imaging arena, multimodality molecular imaging has been widely applied with dual and triple modality options (i.e., PET/MR, PET/SPECT/CT, or SPECT/CT/optical) possible or under development (Goorden and Beekman, 2010; Stout and Zaidi, 2009; Wehrl et al, 2009). Most preclinical systems in use require sequential imaging (Stout and Zaidi, 2009).
The term molecular imaging arose from early work in the ‘reporter’ gene technology field that utilized noninvasive in vivo imaging to visualize cellular processes at the molecular or genetic level (Blasberg, 2002, for review). The term has evolved to describe a multifaceted scientific approach that emphasizes the use of genetic and biomarker information in the development of new imaging probes and paradigms for the in vivo study of cellular and molecular events in normal and pathologic processes in living subjects (Massoud and Gambhir, 2003) with translation to clinical application (Jacobs et al, 2001; Paneda et al, 2011; Yaghoubi et al, 2009).
In 2007, the term further evolved to include the visualization, characterization, and measurement of biological processes at the molecular and cellular levels in humans and other living systems that typically consists of two- or three-dimensional imaging as well as quantification over time, and multiple imaging modalities that include radiotracer imaging in nuclear medicine, MRI, MR spectroscopy, optical imaging, ultrasound, and others (Mankoff, 2007).
This review emphasizes PET as a molecular imaging tool and MRI as a structural and functional imaging tool for human neuroscience research. Preclinical animal and optical imaging are briefly discussed in the context of molecular probe development. Example images are provided to exemplify complementary features and strengths of the imaging modalities and challenges that are relevant for both combined (sequential) and simultaneous multimodality imaging are discussed. Alzheimer's disease (AD) is emphasized because multimodality neuroimaging findings have begun to significantly impact the understanding and management of this disease.
Modalities
Optical Imaging
Optical imaging exploits the optical properties of tissue (i.e., absorption, scattering, luminescence, and fluorescence) to generate image contrast. Near infrared spectroscopy and diffuse optical imaging tomography allow functional brain imaging of hemodynamic parameters and neural activation based on the capability of these methods to study the dynamics of oxyhemoglobin and deoxyhemoglobin concentrations in the brain (Boas et al, 2004). Multiphoton microscopy applies a pulsed laser point source for the fluorescent excitation of molecules to achieve deeper penetration into scattering tissue than visible light microscopy and optical tomography but with a restricted excitation volume (Bacskai et al, 2002). This in vivo imaging method can be used to characterize functional parameters of potential molecular agents in animal models of disease during molecular probe development (Bacskai et al, 2003). Molecular optical imaging is possible through the use of optical contrast agents or ligands that bind to specific targets but this technology is not yet well developed for human brain (Balas, 2009).
Positron Emission Tomography Imaging
Positron emission tomography is an established molecular imaging tool that measures the radioactivity distribution of imaging agents labeled with positron-emitting radionuclides. After a positron is emitted from the nucleus of an atom, it travels a few mm in tissue (positron range) and interacts with an electron to generate two 511 keV photons (annihilation radiation) that are emitted ∼180° apart. Positron emission tomography scanners typically have multiple rings of scintillation detectors (e.g., bismuth germinate, BGO or lutetium oxyorthosilicate, LSO). The scanner registers an event when two photons are detected within a narrow few nanosecond time window (coincident event). The most widely used positron-emitting radionuclides for brain imaging are 15O (t1/2 ∼2 minutes), 11C (t1/2 ∼20 minutes), and 18F (t1/2 ∼110 minutes). Positron emission tomography spatial resolution ranges from about 3 to 8 mm (best for HRRT scanners; de Jong et al, 2007; Leroy et al, 2007).
Positron emission tomography offers a wide range of molecular imaging options (Table 1) that include numerous neuroreceptor system targets (e.g., presynaptic and postsynaptic neuroreceptors, transporter/reuptake sites, vesicular transporters), translocator proteins, and amyloid plaques. Positron emission tomography imaging of cellular processes, particularly relevant for neuro-oncology, include amino-acid uptake, hypoxia, protein synthesis, and cell proliferation. A unique strength of PET is its capability to perform quantitative in vivo imaging of ligand-binding interactions through application of tracer kinetic principles (Lassen and Perl, 1979) at low nM concentration levels. Figure 1A is a survey of example images of quantitative (Figure 1-A1) and semiquantitative (Figure 1-A2) blood flow (with MRI Figure 1-A3), serotonin (5-HT) neuroreceptor targets: 5-HT transporter ([11C]DASB; Figure 1-A4), 5-HT2A receptor ([18F]Altanserin; Figure 1-A5), 5-HT1A receptor ([11C]WAY100635; Figure 1-A6), and the dopamine (DA) transporter ([11C]PE21 PET; Figure 1-A7; Leroy et al, 2007). Zimmer and Luxen (2011) provides a current review of PET radiotracers for brain imaging.
Example of PET molecular imaging options for neurodegeneration (ND) and other diseases/disorders of braina

AD, Alzheimer's disease; DLB, dementia with Lewy bodies; PD, Parkinson's disease; PET, positron emission tomography.
See Zimmer and Luxen (2011), for review of PET radiotracers for molecular brain imaging.
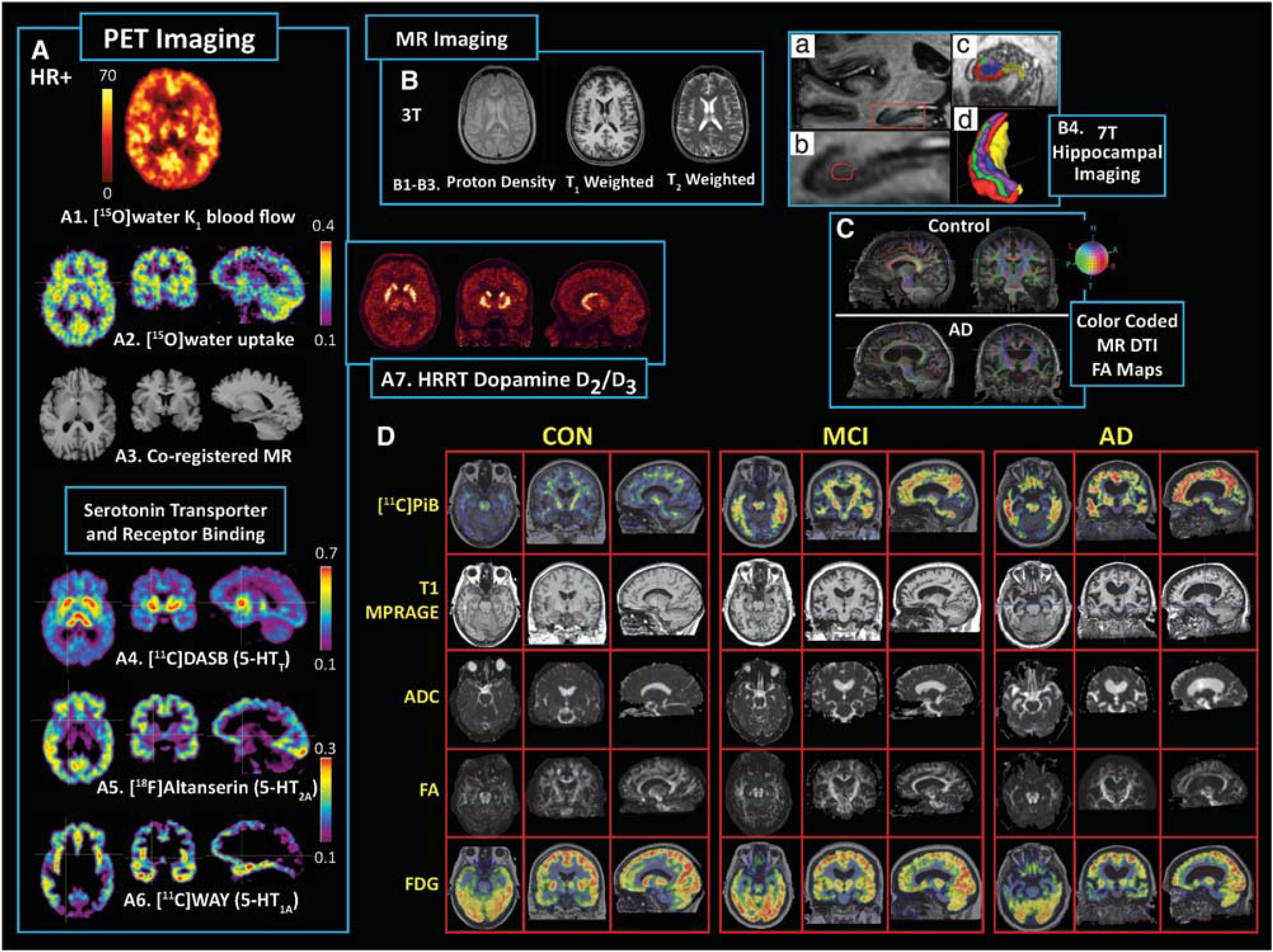
Composite survey of molecular positron emission tomography (PET) brain imaging targets and structural magnetic resonance imaging (MRI) correlates. (
Positron emission tomography has been widely applied to study in vivo pharmacology (e.g., drug specificity, selectivity, and/or occupancy) and endogenous neurotransmitter release (e.g., after drug administration or stimulus/task). The PET measure of endogenous neurotransmitter release has been most widely studied and validated for the DA system based on the in vivo sensitivity of D2/3 receptor binding radiotracers (e.g., [18F]-N-methylspiroperidol, [11C]raclopride, [18F]fallypride) to amphetamine-induced increases in extracellular DA (Dewey et al, 1991; Endres et al, 1997; Laruelle, 2000; Slifstein et al, 2010). This sensitivity is reflected by postintervention reductions in radiotracer binding relative to baseline binding that exceed the test–retest variation of the binding measure. Validation of the amphetamine/[11C]raclopride paradigm included a pharmacokinetic modeling study of DA microdialysis and [11C]raclopride PET data collected in nonhuman primates (Endres et al, 1997). Investigations of endogenous opioid neurotransmission have been conducted in studies of pain, reward, drug development, and include combined PET and functional MRI (fMRI) investigations (Apkarian et al, 2005; Rabiner et al, 2011) and use of [11C]carfentanil (mu-opiate receptor agonist) and [11C]diprenophine (nonselective opiate antagonist at tracer doses). Recent work shows preclinical evidence for the sensitivity of 5-HT1B PET ligands to endogenous serotonin (Finnema et al, 2011; Ridler et al, 2011).
Magnetic Resonance Imaging
Magnetic resonance imaging offers exquisite structural imaging with strong functional and molecular options. Magnetic resonance imaging exploits the nuclear magnetic resonance properties of paramagnetic nuclei (e.g., 1H, 13C, 19F, 23Na, 31P). Magnetic resonance imaging is operated at various magnetic field strengths (B0) that range from about 1.5 to 7 T for most human studies. This modality uses external radiofrequency pulses to excite and manipulate the magnetic properties of tissue to generate tissue contrast. Magnetic resonance imaging parameters of interest include the longitudinal magnetization time constant, T1 (seconds, ‘spin-lattice’ relaxation/tissue recovery), transverse magnetization time constant, T2 (milliseconds, ‘spin–spin’ relaxation/spin dephasing), and time constant T2∗ that reflects variations in both B0 and local internal magnetic field inhomogeneities in the tissue (with T2∗ less than T2). Blood oxygenation level dependent (BOLD) contrast reflects variations in the concentration of oxygenated blood through T2∗ because deoxyhemoglobin is paramagnetic, while fully oxygenated hemoglobin is diamagnetic. Image contrast can also be achieved by the use of exogenous contrast agents.
Structural MRI for neuroimaging applications is often T1- and/or T2-weighted (Figures 1B and 1D, row 2) with related sequences optimized for imaging of vasculature and brain pathology (Frisoni et al, 2010; Tartaglia et al, 2011). Diffusion-weighted imaging reflects water diffusivity and diffusion tensor imaging (DTI) (e.g., apparent diffusion coefficient and fractional anisotropy) assessments of white matter integrity and axonal architecture have become more routine (Figures 1C and 1D, rows 3 and 4). Sophisticated 3D algorithms enable white matter tractography based on standard DTI or advanced hybrid methods (i.e., diffusion spectrum imaging, DSI) (Thomason and Thompson, 2011).
As summarized by Duyn and Koretsky (2011), there are many advantages to high field MRI (e.g., 7 T), such as a 10- to 100-fold improvement in sensitivity over early 0.15 to 1.5 T MR scanners, the increased use of T2∗-weighted contrast and study of gray and white matter substructure (e.g., cortical laminar structure; Figure 1-B4), such that high field methods allow sensitivity to microscopic variations in magnetic properties of tissue and exploitation of endogenous iron and myelin for imaging, and the use of exogenous contrast agents (e.g., manganese and iron oxide) to study neural connectivity, neuronal tract tracing, and tracking of cell migration in vivo.
Blood oxygenation level dependent fMRI extends well beyond studies of neural activation and functional coupling and/or connectivity in humans, to include preclinical and human clinical applications, such as neurosurgical planning (Matthews et al, 2006). Magnetic resonance imaging of vascular hemodynamics (Zaharchuk et al, 1998) has undergone steady development with perfusion imaging most commonly performed by either dynamic susceptibility contrast or arterial spin labeling methods (Wolf and Detre, 2007).
Pharmacological MRI (phMRI) provides a functional assessment of pharmacological effects on brain function (Wise et al, 2002). An elegant validation of phMRI was performed early on for the DA system (e.g., amphetamine and DA transporter ligand administration) in rodents that included lesioning studies and assessment of vascular function, BOLD time course, physiological correlates, microdialysis, and behavioral outcomes (Chen et al, 1997). A wide range of phMRI studies has been performed (e.g., at rest and/or during stimulus/task), with strong interest in drug development (e.g., pain therapeutics). Most fMRI and phMRI studies utilize BOLD, although some also utilize arterial spin labeling (Newberg et al, 2005; Wang et al, 2011), such as the recent combined arterial spin labeling and BOLD study of the neural correlates induced by psilocybin (Carhart-Harris et al, 2012).
Molecular MRS (MRI spectroscopy) provides measures of important brain metabolites, such as N-acetyl aspartate (NAA: marker of neuronal health), choline (Cho: membrane synthesis and degradation), creatine (Cr: energy metabolism, can be elevated in disease), and lactate (not generally detected in the healthy brain) (Zhu and Barker, 2011). MRI spectroscopy provides important information for neuro-oncology applications (Heiss et al, 2011). Sodium MR imaging is a molecular method that can directly assess sodium ion homeostasis and cellular viability to detect metabolic changes that result from the loss of cell ion homeostasis due to cell death in the brain (Boada et al, 2005).
Multimodality Imaging
It is becoming more common to acquire multimodality neuroimaging data that include PET and fMRI, as well as other types of MR (and optical) outcomes (Apkarian et al, 2005; Catana et al, 2008; Dougherty et al, 2008; Herzog et al, 2011; Huppert et al, 2008; Newberg et al, 2005; Rabiner et al, 2011; Sperling et al, 2009; Tong et al, 2011). Combined fMRI and PET brain imaging will become even more commonplace with the increased availability of combined PET/MR imaging systems.
Challenges
Multimodality molecular imaging can be challenging because of the different nature of the imaging signals and analysis outcomes, pitfalls, and limitations. Many of these challenges are relevant for both combined sequential scanning and simultaneous scanning. Figure 1 exemplifies the complementary nature of PET and MRI in the context of information gained from combining molecular and structural imaging data to obtain a more comprehensive assessment of subject status. In addition to the serotonin imaging, Figure 1-D shows PET imaging of fibrillar amyloid plaques (row 1) in an elderly control, a subject with mild cognitive impairment (MCI) and an AD patient, along with anatomical and diffusion MRI outcomes (rows 2 to 4) and [18F]2-fluoro-2-deoxy-D-glucose (or FDG) metabolism (row 5). The limited spatial resolution of PET (relative to structural MR data) can be a serious issue for imaging of aging populations or in patients with severe disease, when substantial cerebral atrophy and cerebrospinal fluid (CSF) dilution of gray matter arises. Figure 1 shows a challenging example for an AD patient with extreme cerebral atrophy and advanced disease (Figures 1C and 1D).
Positron emission tomography resolution is generally limited by the scanner design and performance characteristics. The resolution of many PET scanners used for brain research is ∼5 to 6 mm (e.g., HR+ scanner). Partial volume averaging of the PET data (and lower resolution MRI outcomes) can complicate comparative image analyses. Issues to consider for PET drug challenges and/or measurement of endogenous neurotransmitter involve (1) efforts to minimize blood flow or radiotracer clearance confounds (i.e., bolus versus bolus+constant infusion radiotracer administration), (2) radiotracer half-life in the context of necessary study duration, (3) timing of drug/stimulus administration with respect to stability in drug response, and stability of receptor or target binding outcome, and (4) sufficient radiotracer-specific activity to ensure good signal-to-noise and to minimize contamination effects from carry-over of unlabeled ligand mass from baseline study to postintervention study when performed the same day.
A well-known challenge for fMRI is the incomplete understanding of the complex physiological changes caused by neural activation, such that the interpretation of BOLD signals, and other functional neuroimaging signals related to blood or tissue oxygenation, may be limited until it is possible to better understand brain oxygen metabolism and how it is related to blood flow (Buxton, 2010). Contamination of the BOLD signal by physiological noise can be a complicating factor (Tong et al, 2011). Technical challenges also can arise in the experimental fMRI paradigm (e.g., task, stimulus, pharmacologic challenge) and/or ‘resting state’ conditions. The choice of study design (i.e., event-related, block design, or mixed design) and the extent to which a block design may yield greater effect sizes (Friston et al, 1999) are important considerations. The spatial ‘functional’ resolution of fMRI (mm) depends on the voxel size. A larger image acquisition matrix (smaller voxels) improves spatial resolution but reduces the voxel signal, and short TR may improve temporal resolution but this depends on the temporal nature of the hemodynamic response function (HRF) (Huettel et al, 2009). Methods have been proposed to recover signal loss in fMRI, including the use of tailored radiofrequency pulses (Yip et al, 2006). Table 2 summarizes parameters for consideration for combined PET and fMRI.
Considerations for multimodality PET and MR brain imaging
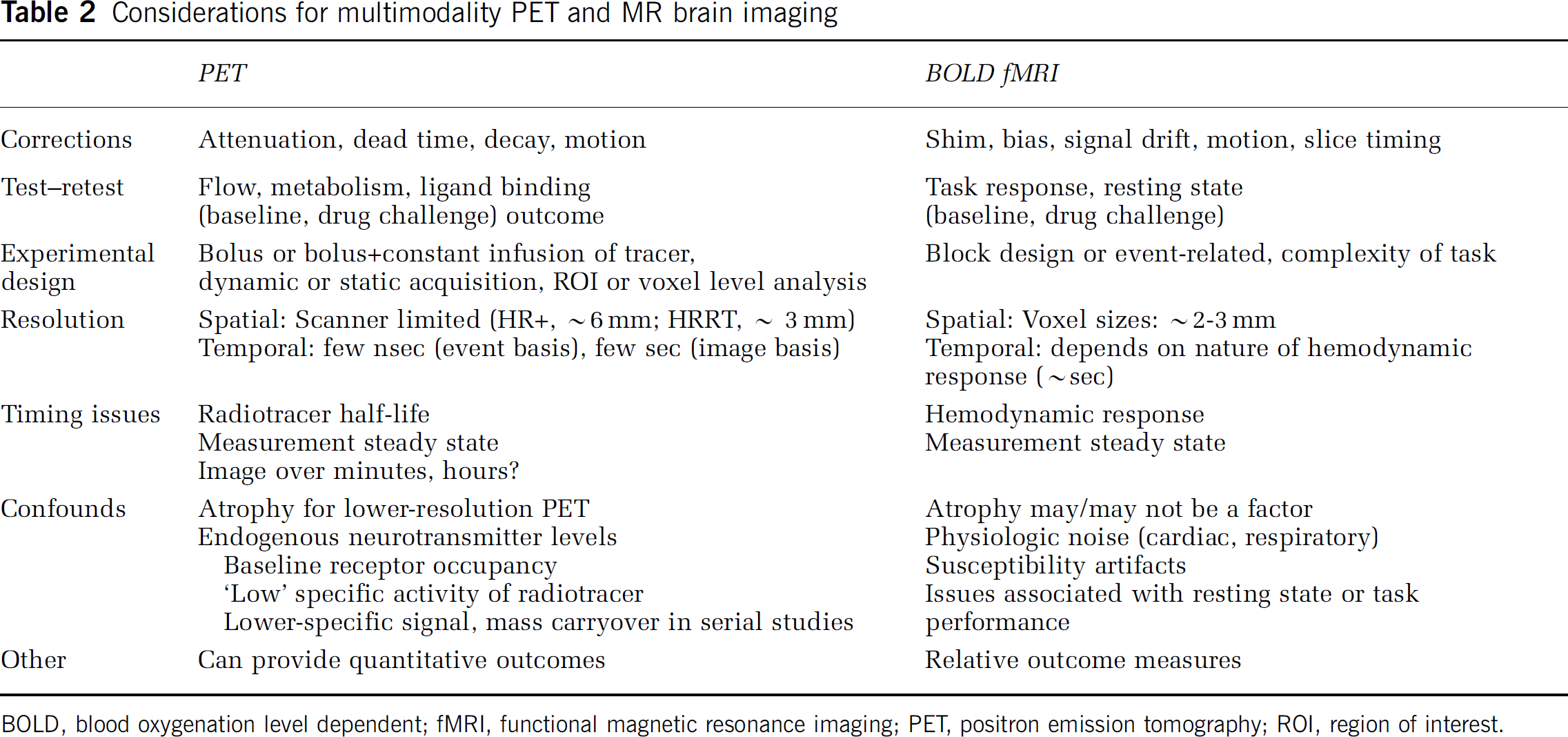
BOLD, blood oxygenation level dependent; fMRI, functional magnetic resonance imaging; PET, positron emission tomography; ROI, region of interest.
Analysis of multimodality studies can also be challenging, and knowledge of the technical and statistical strengths/weaknesses of the data can inform choice of analysis method. Multimodality outcomes, for example, can be related on an ROI (region of interest) basis, based on maps of statistical group differences, by categorizing subjects into molecular or fMRI outcome groups, or by incorporation of data from one modality into the analysis (or reconstruction) of another (Bayesian). It may be advantageous and more powerful to pursue an ROI-based approach when testing regionally specific hypotheses (Mitsis et al, 2008), although voxel-based and data-driven approaches are sometimes required (e.g., functional connectivity). Numerous methods have been used to enable the statistical analysis of multimodal and intramodal data sets that include statistical parametric mapping methods and specialized toolboxes with correlative, concordant, or integrative techniques and modeling-based approaches (Buckner et al, 2005; Casanova et al, 2007; Hayasaka et al, 2006; Krishnan et al, 2011; Ye et al, 2009). As an example, concordance–discordance analysis (Hayasaka et al, 2006) can be powerful for the comparison of group difference t-maps within and across imaging modalities.
Combined
A novel study design was recently implemented that combined molecular and functional imaging with in vivo pharmacology and food stimuli. Rabiner et al (2011) described the use of [11C]carfentanil PET to measure mu-opiate receptor occupancy and fMRI to measure activation of brain reward centers by palatable food stimuli—before and after a single oral dose of either naltrexone (opioid receptor antagonist) or GSK1521498 (inverse agonist at mu-opioid receptor) across a dose range of 0.4 to 100 mg and 2 to 50 mg, respectively. The investigators observed that GSK1521498 (but not naltrexone) attenuated the fMRI activation of amygdala by palatable food stimuli, providing an example of how the pharmacological properties of opiate receptor antagonists can be characterized directly in humans by novel integration of molecular and functional neuroimaging techniques (Rabiner et al, 2011).
Simultaneous
The ability to perform true simultaneous molecular and structural/functional imaging in humans became possible with the prototype hybrid BrainPET insert within a 3T MR scanner. The BrainPET was operational at a limited number of sites, but the design was space limited and did not allow for a full range of neuroimaging options. The utility of the instrument was demonstrated (Schlemmer et al, 2008), particularly for neuro-oncology (Heiss et al, 2011). Figure 2 shows examples of simultaneous imaging in tumor patients and the suitability of hybrid imaging for monitoring of disease progression, treatment response, and fMRI assessment of motor function (Herzog et al, 2011).
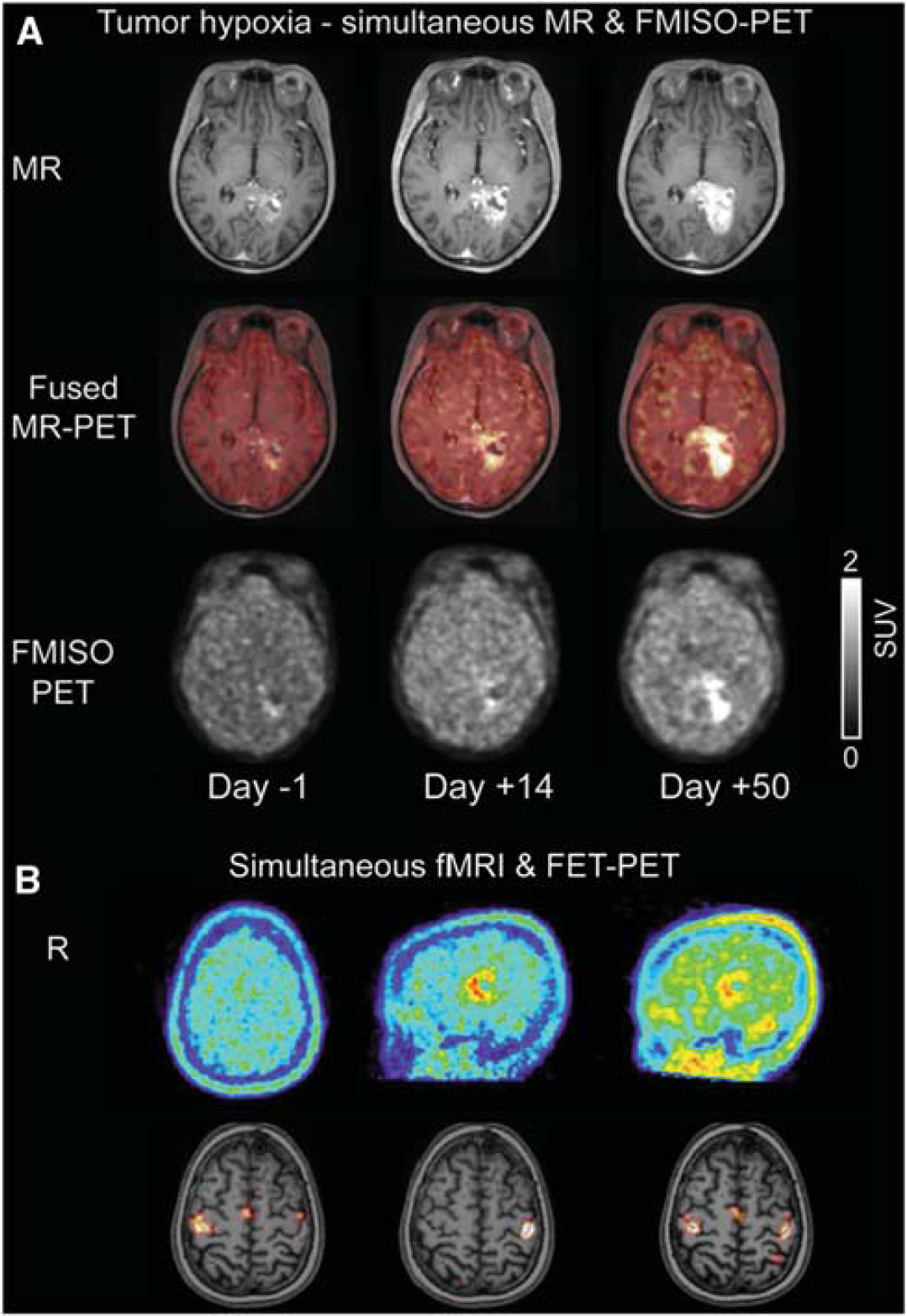
Examples of simultaneous magnetic resonance/positron emission tomography (MR/PET) imaging achieved using BrainPET insert. (
There is now the capability to perform simultaneous whole body PET/MR imaging (Delso et al, 2011). This platform will allow for the strengths of simultaneous imaging to be fully realized with capability that is well beyond that of sequential multimodality imaging. This platform enables multimodality imaging for the same drug challenge, in vivo pharmacology condition(s), endogenous neurotransmitter release, resting state, or clinical state and this is not possible for sequential imaging platforms.
Multimodality Imaging in Alzheimer's Disease
Background
Alzheimer's disease is the most common type of dementia and it is estimated that AD accounts for 60% to 80% of dementia cases (Alzheimer's Association, 2010). Alzheimer's disease is characterized by the neuropathological accumulation of amyloid-β (Aβ) plaques and neurofibrillary tangles (NFTs). The diagnosis of AD requires autopsy confirmation of sufficient amounts of Aβ plaques and NFTs. Mild cognitive impairment can be closely related to AD and characterized by either isolated memory impairment or impairment in several cognitive domains, but not severe enough to meet diagnostic criteria for AD. The MCI diagnostic group is heterogeneous in its symptomatology. The MCI amnestic subtype is the most well-defined and well-studied group and is most commonly defined as those with a singular, substantial deficit in episodic memory, accompanied by otherwise normal levels of function in other cognitive domains and no measurable functional impairment in daily life (Petersen et al, 1997). Many studies of the amnestic subtype have found that 10% to 15% convert to a diagnosis of AD annually (Wolk and Klunk, 2009, for review).
Amyloid plaques are composed primarily of aggregated Aβ peptide, while NFTs are intraneuronal lesions of paired helical filaments of hyperphosphorylated tau protein (Spires and Hyman, 2005). The postmortem distribution of Aβ plaques and NFTs in the brains of AD patients has shown that, compared to NFTs, plaques occur mainly in neocortex, while NFTs appear first in entorhinal cortex before progressing to the neocortex. Thus, while limbic areas have early and severe tangle pathology, the medial temporal lobe has relatively little neuritic plaque pathology early in the disease (Mathis et al, 2007, for review).
Although the greatest risk factor for AD is age, there is a major susceptibility gene for late-onset sporadic AD that is the apolipoprotein E (ApoE) e4 allele. The ApoE gene (on chromosome 19) has a role in cholesterol transport, uptake, and redistribution and both ApoE and its receptor (low-density LRP (lipoprotein receptor-related protein)) are found in plaques in AD (Spires and Hyman, 2005, for review). There are also individuals with rare chromosomal mutations in regions that code for the amyloid precursor protein (APP) or the secretase complex (Presenilin 1 or 2) that yield a dominant inheritance pattern for AD that is 100% penetrant with early onset before the age of 65 (Pastor and Goate, 2004). Down's syndrome subjects who have an extra copy of chromosome 21 (codes for APP) exhibit Aβ deposition at autopsy; these observations support a long-standing hypothesis, the ‘amyloid cascade hypothesis,’ that it is the accumulation of Aβ in the brain that drives AD pathogenesis (including NFT formation) as a result of an imbalance between Aβ deposition and clearance (Hardy and Selkoe, 2002). Research in the molecular genetics of AD has fueled development of transgenic animal models for in vivo investigations.
Animal Imaging
Transgenic (Tg) mouse models serve as a major experimental tool for preclinical neuroimaging research in AD. Spires and Hyman (2005) provided a piece-wise summary of the molecular pathology of Tg animal models of AD, such that APP cleavage by β- and γ-secretases leads to production of Aβ, APP overexpression leads to plaque formation in mice, coexpressing either β- and γ-secretase components with APP accelerates plaque formation, Aβ increases NFT formation in tau overexpressing mice, studies of ApoE knockout and Tg mice indicate that ApoE may be involved in both Aβ deposition and clearance, and reducing LPR in mouse models increases Aβ deposition, indicating a role for LRP in clearance or degradation of Aβ. These animal models may express Aβ plaques but not NFTs (PDAPP, Tg2576, PSAPP), NFTs only, and mixed pathologies (Gotz et al, 2004; Spires and Hyman, 2005, for reviews).
Positron emission tomography imaging in Tg mice has focused recently on imaging of Aβ plaques. The majority of studies used Pittsburgh compound B (or PiB), an agent that labels fibrillar Aβ plaques in the brain and has intrinsic fluorescent properties (Mathis et al, 2007). Bacskai et al (2003) used multiphoton microscopy to image PiB in PDAPP and Tg2576 mice (both 18 to 20 months old) and PSAPP mice (12 months old). PiB was imaged in real time as it appeared in vessels, cleared across the blood–brain barrier and first labeled amyloid angiopathy, followed by parenchymal dense-core plaques (first labeled at periphery then gradually filled in to the core), and eventually cleared from the brain (3D volume: 615 × 615 μm2, depth: ∼150 μm) (Bacskai et al, 2003). This provided early evidence that PiB was suited for the in vivo detection of amyloid deposits in transgenic mice (Bacskai et al, 2003).
Despite the multiphoton microscopy success, microPET imaging of transgenic mice yielded poor [11C]PiB retention in the brain (Klunk et al, 2005; Toyama et al, 2005). Klunk et al (2005) did not observe significant [11C]PiB retention (specific activity > 37 GBq/μmol) in older PS1/APP mice expected to exhibit Aβ deposition that exceeded levels in the AD brain. This group performed in vitro evaluations of PS1/APP mice brain tissue that revealed less than one high affinity (Kd ∼1 to 2 nm) binding site per 1,000 molecules of Aβ, as compared with AD brain with >500 binding sites for 1,000 molecules of Aβ in the AD brain. In contrast to this, Maeda et al (2007) observed high retention of [11C]PiB in Aβ plaque-rich areas of APP mice that was consistent with the significantly higher specific activity (291±10.3 Gbq/μmol) achieved by this group. These investigators additionally reported that autoradiographic signals indicated that the detectability of Aβ by [11C]PiB PET may be dependent on the accumulation of specific Aβ subtypes (Maeda et al, 2007).
Magnetic resonance imaging studies of Tg mice also include vascular imaging, hippocampal and ventricular volumes, DTI, fMRI, and MRS—with additional PET imaging of metabolism, acetylcholinesterase activity, and neuronal integrity via benzodiazepine receptor binding (Waerzeggers et al, 2010). It can be challenging to detect age- and neuropathology-related abnormalities in the small mouse brain. Magnetic resonance imaging requires high magnetic field strength and advanced imaging methods, while PET is limited by spatial resolution despite high sensitivity capabilities. Efforts are ongoing to achieve better correspondence between neuroimaging results in AD-Tg mouse models, relative to what is observed in human studies (Waerzeggers et al, 2010). Recent efforts report alternative MRI methods that enable detection of Aβ plaques in Tg mice (Chamberlain et al, 2011). Detailed summaries of this body of work are available in recent reviews (Delatour et al, 2010; Waerzeggers et al, 2010).
Human Imaging: Structural Magnetic Resonance Imaging, Functional Magnetic Resonance Imaging, and Molecular Positron Emission Tomography Imaging in Alzheimer's Disease
Structural Magnetic Resonance
Atrophy in the hippocampus and entorhinal cortex is a well-established imaging finding of AD-related neurodegeneration (De Leon et al, 1997; Jack et al, 1999, 2000). This atrophy can be highly correlated with severity of cognitive dysfunction. Significant reductions in the volume of the hippocampus and related structures have also been noted in MCI. In longitudinal studies, MRI-based measurements of hippocampal volume loss have been highly correlated with the conversion of MCI to AD. High interindividual variability in cerebral volume loss of normal aging has led to the development of brain-mapping techniques to further enable detection and tracking of atrophy in crosssectional and longitudinal studies (Thompson et al, 2004). Further comprehensive detail is available in recent reviews (Ewers et al, 2011a; Frisoni et al, 2010; Tartaglia et al, 2011).
Functional Magnetic Resonance Imaging
A large body of research has established functional alterations in brain areas linked with memory processing in AD, MCI and in some at risk for AD (APOE e4 carriers), with involvement of hippocampus and mesial temporal lobe (MTL) (Mevel et al, 2011; Tartaglia et al, 2011). In an example study, Dickerson et al (2005) applied a face-name encoding task and observed greater hippocampal activation in MCI subjects than controls, while hippocampal or entorhinal volumes were not different. The AD subjects, in contrast, showed reductions in activation and also atrophy in both brain areas, relative to controls. The authors concluded that there was a phase of increased MTL activation early in the course of prodromal AD that was followed by a subsequent decrease with disease progression. This work was one of several timely fMRI studies that probed memory function in AD at that time (see Sperling et al, 2009).
Studies of cortical function revealed a distributed network of regions that characterize a default mode network (Raichle and Snyder, 2007) that is more active during rest (nontask condition) than during active task condition. The default network is active when engaged in internally focused tasks including autobiographical memory retrieval, envisioning the future, and conceiving the perspectives of others, with an MTL subsystem and a medial prefrontal subsystem that converge on important nodes of integration (Buckner et al, 2008). This network includes posterior cingulate and parietal lobe (Buckner, 2004, 2005; Greicius et al, 2003, 2004; Wang et al, 2006). Greicius et al (2004) reported decreased default mode network activity in posterior cingulate and hippocampus of AD subjects that suggested disrupted connectivity between these areas. Wang et al (2006) reported disrupted connectivity between specific areas of hippocampus and prefrontal cortex, anterior cingulate, infero-temporal cortex, and cuneus that extended into precuneus and posterior cingulate areas.
Molecular Positron Emission Tomography Imaging
Early PET imaging in AD revealed alterations in SPECT blood flow and FDG metabolism that showed (1) parietotemporal hypoperfusion that sometimes coincided with frontal lobe hypoperfusion (Herholz et al, 2007; Jagust et al, 1997); (2) bilateral metabolic reductions in parietotemporal association cortices and relative sparing of primary neocortical and subcortical areas until later stages (Friedland et al, 1983); and (3) decreased posterior cingulate FDG metabolism that was more prominent than typical parietotemporal reductions (Minoshima et al, 1994). Metabolic reductions in posterior cingulate were found in cognitively normal subjects homozygous for the APOE e4 allele (Reiman et al, 1996, 2001). FDG PET can identify regional deficits in MCI subjects, relative to age-matched control subjects, and predict conversion of MCI subjects to AD (de Leon et al, 2007; Drzezga et al, 2005; Mosconi et al, 2008). Lastly, although a key neurodegenerative feature of AD is loss of cholinergic projections from the basal forebrain, PET imaging of neurotransmitter system dysfunction in AD has not been well established and is partly reflective of methodological limitations of available radiotracers for this system (Herholz et al, 2007).
With the advent of PET Aβ imaging, much has been learned about the presence and accumulation of fibrillar Aβ plaques in the brain of living humans (Wolk and Klunk, 2009, for review). Most PET Aβ imaging studies utilized [11C]PiB (or PiB). Initial studies showed that PiB retention in AD subjects was nearly twice that of controls in specific cortical areas (P<0.002) but similar in areas without Aβ deposition (i.e., subcortical white matter, cerebellum). These studies showed that specific PiB uptake correlated inversely with FDG metabolism (e.g., parietal) (Klunk et al, 2004). Fully quantitative arterial-based studies provided absolute PiB retention measures that supported results of simpler methods (e.g., uptake measures). PiB retention in MCI subjects ranged from control to AD levels with evidence of early amyloid deposition in frontal and precuneus areas (Lopresti et al, 2005; Price et al, 2005).
PiB retention in AD brain correlated with the rate of brain atrophy (Archer et al, 2006) and hypometabolism (Edison et al, 2007). In another study, specific PiB retention was observed in about 20% of normal elderly controls (Mintun et al, 2006) (e.g., Figures 3A and 3B) and correlated well with decreases in amyloid (Aβ42) levels in CSF in both cognitively normal and demented subjects (Fagan et al, 2006). Further study of control subjects revealed an APOE4 gene dose effect that was associated with increased cortical PiB retention in control subjects (Morris et al, 2010; Reiman et al, 2009) and lowered CSF Aβ that appeared to begin earlier than the elevations in cortical PiB retention (Morris et al, 2010).
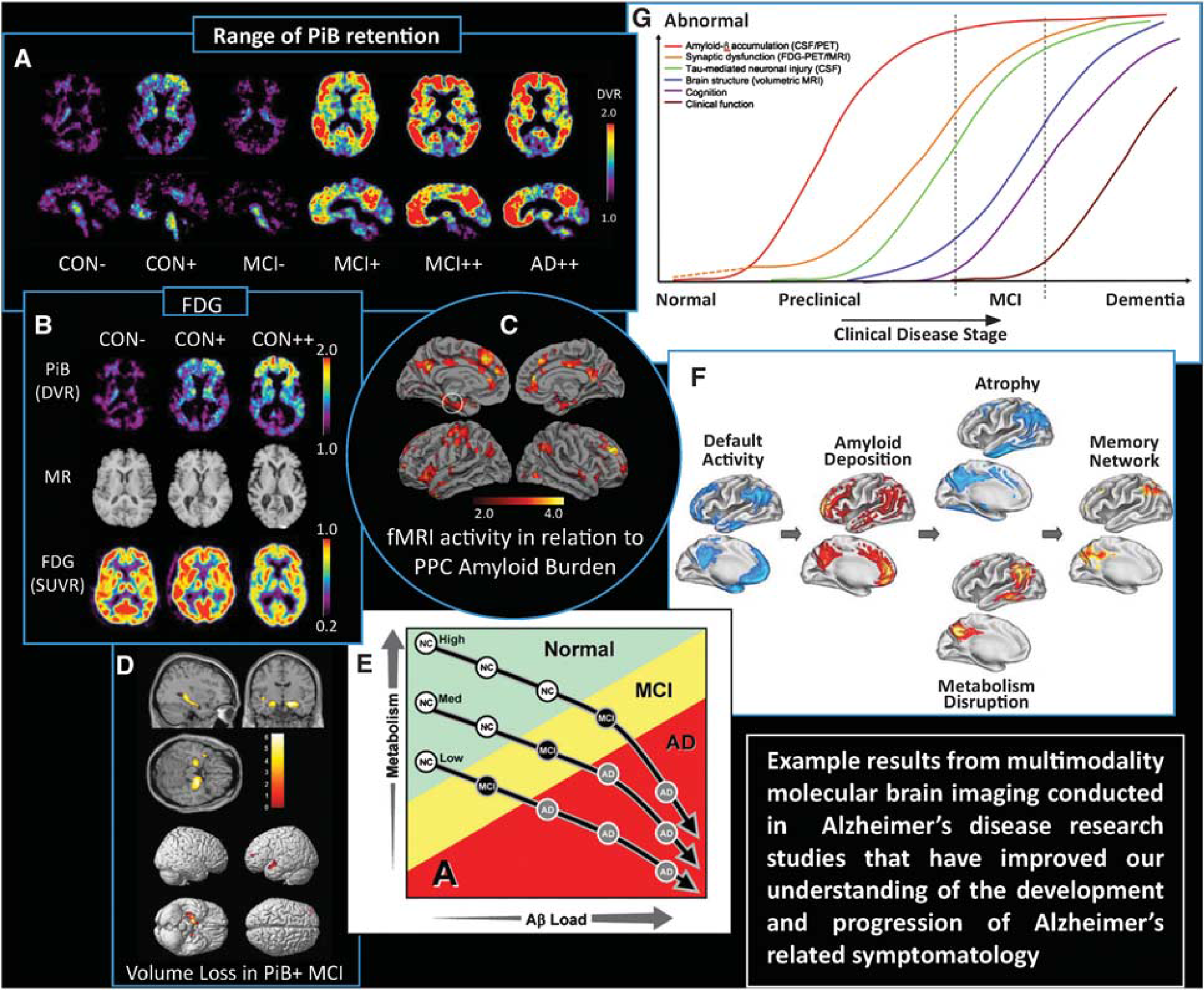
Composite figure showing examples of multimodality neuroimaging findings in Alzheimer's disease (AD)-related research that have advanced knowledge of normal aging, the natural history of amyloid deposition and the development of AD. (
A 2-year follow-up study found little progression of PiB uptake in symptomatic AD patients (Engler et al, 2006). Results emerging from this and other longitudinal studies (Villemagne et al, 2011; Weiner et al, 2010; Jack et al, 2009) are beginning to elucidate the sequence of pathological events in AD. At 20-month follow-up, Villemagne et al (2011), reported small significant increases in PiB uptake value ratios in AD and MCI groups, and in controls with high PiB retention at baseline. Lastly, PiB PET could detect posttreatment reductions in amyloid load in AD subjects who participated in a longitudinal antiamyloid therapeutic trial (Rinne et al, 2010).
Impact of Multimodality Neuroimaging on Alzheimer's Disease Hypothesis Development: Example Studies
Sperling et al (2009) noted that posterior components of the default network (i.e., precuneus and posterior cingulate (or PPC) were particularly vulnerable to early Aβ deposition. This group performed a combined PiB PET and fMRI study (mixed block and memory event-related design) of older asymptomatic and minimally impaired subjects. The authors reported that high levels of Aβ deposition were associated with an aberrant default network fMRI activity pattern that was similar to the pattern of dysfunction reported in AD. These findings indicated that Aβ pathology is linked to neural dysfunction in the brain regions that serve memory function, thereby supporting the hypothesis that cognitively intact older individuals with evidence of Aβ pathology may be in early stages of AD (Sperling et al, 2009) (see Figure 3C).
A recent comparison of PiB PET and structural MRI in 26 MCI patients who were amnestic (a-MCI) and nonamnestic MCI revealed a significant proportion of amyloid-positive (or Aβ(+)) patients. There were no obvious differences in the PiB distribution in the nonamnestic MCI group. Predictors of conversion to clinical AD in a-MCI, that included poorer episodic memory and medial temporal atrophy (see Figure 3D), were observed in the Aβ(+) a-MCI patients, relative to Aβ(–) a-MCI patients. Longitudinal follow-up suggested that (1) a significant percentage of all MCI subtypes develop AD and (2) Aβ(+) patients are likely to have early AD. The authors concluded that PET Aβ imaging may be important for determining which patients may benefit from disease-specific therapies (Wolk et al, 2009).
Cohen et al (2009) explored associations of Aβ deposition with FDG metabolism in Aβ(+) controls and patients with MCI or AD. In Aβ(+) controls, no clear correlation patterns were observed. In MCI patients, metabolism in anterior cingulate showed positive correlations with PiB in most brain areas, while metabolism and PiB retention were positively correlated locally in precuneus/parietal cortex. However, there was no significant increase in metabolism in MCI compared with age-matched controls, negating the possibility that Aβ deposition directly caused reactive hypermetabolism. The authors proposed that in MCI, higher basal metabolism could either be exacerbating Aβ deposition or increasing the level of Aβ necessary for cognitive impairment sufficient for an AD diagnosis. It was only after extensive Aβ deposition had been present for longer periods of time that Aβ becomes the driving force for decreased metabolism in clinical AD and, only in more vulnerable brain regions such as parietal and precuneus cortices (Cohen et al, 2009) (see Figure 3E).
Buckner et al (2005) sought to better understand how cortical patterns of metabolic disruption relate to structural atrophy and MTL neuropathology in AD and to the observed correlation between Aβ load, atrophy and hypometabolism in posterior cingulate cortex. The authors integrated results across five neuroimaging data sets (n=764) that were maps of: (1) [15O]water PET default mode activity; (2) PiB PET Aβ load; (3) MRI structural atrophy; (4) FDG PET glucose metabolism; and (5) fMRI retrieval success effect. The authors reported convergence of effects in posterior cortex including areas active in default states in young adults and that show Aβ deposition in older adults with AD. The authors proposed that during the early progression of AD, atrophy and metabolic abnormalities emerged in these posterior regions, along with atrophy in MTL, while event-related fMRI revealed that these cortical regions are active during successful memory retrieval in young adults. The authors proposed that lifetime cerebral metabolism associated with regionally specific default activity predisposes cortical regions to AD-related changes, including Aβ deposition, metabolic disruption, and atrophy. These cortical regions may be part of a network with the medial temporal lobe whose disruption contributes to memory impairment (see Figure 3F).
Lastly, Jack et al (2009) submitted a hypothetical model of dynamic biomarkers of the AD pathological cascade that were based on a body of scientific research results from [11C]PiB PET, CSF Aβ42, structural MRI, FDG PET and measures of cognition. These authors proposed that the initiating event in AD is related to abnormal processing of β-amyloid peptide, ultimately leading to formation of Aβ plaques in the brain (i.e., reductions in CSF Aβ42 and increased amyloid PET tracer retention). This process was hypothesized to occur while individuals are still cognitively normal. After a lag period, that is variable across individuals, neuronal dysfunction and neurodegeneration become the dominant pathological processes, as evidenced by increased CSFtau and structural MRI measures of cerebral atrophy. Neurodegeneration is accompanied by synaptic dysfunction, which is indicated by decreased fluorodeoxyglucose uptake on PET. This model was later reviewed in the context of different stages of disease and the predictive value of multimodal neuroimaging for AD dementia (Ewers et al, 2011b) and also in consideration of the long preclinical phase of the disease. This led to a revision of the Jack model that included a synaptic dysfunction phase, evidenced by abnormal FDG PET and fMRI, before tau-mediated neuronal injury (Sperling et al, 2011) (see Figure 3G).
This body of work led to a joint National Institute on Aging—Alzheimer's Association task force that established a process for revising diagnostic and research criteria for AD involving three workgroups that formulated new diagnostic criteria that incorporated biomarkers of the underlying disease state for three phases of AD (i.e., preclinical stages of AD (Sperling et al, 2011), MCI due to AD (Albert et al, 2011); and dementia due to AD (McKhann et al, 2011)). The core clinical criteria regarding AD dementia and MCI due to AD were intended to guide diagnosis in the clinical setting, while the preclinical AD recommendations were intended purely for research purposes (Jack et al, 2011). As an example, the ‘MCI due to AD’ criteria (Albert et al, 2011) listed biomarkers under investigation for AD in the order of (1) Aβ deposition (CSF Aβ42, PET amyloid imaging); (2) neuronal injury (CSFtau/phosphorylated tau, hippocampal volume or medial temporal lobe atrophy, rate of brain atrophy, FDG PET, SPECT perfusion, and less well-validated biomarkers (fMRI activation, resting state BOLD functional connectivity, MRI perfusion, MRS, DTI, voxel-based, and multivariate measures); and (3) associate biochemical change (inflammatory biomarkers, oxidative stress, and markers of synaptic damage and neurodegeneration, i.e., cell death). The final set of criteria for MCI due to AD has four levels of certainty based on the presence and nature of the biomarker findings, that are (1) MCI—core clinical criteria; (2) MCI due to AD—intermediate likelihood; (3) MCI due to AD—high likelihood; and (4) MCI—unlikely due to AD. Figure 4 shows a possible neuroimaging ‘decision tree’ scheme for MCI due to AD that reflects the new criteria. These criteria would likely classify the MCI subject in Figure 1D as ‘MCI due to AD—high likelihood.’

Example neuroimaging ‘decision tree’ scheme for mild cognitive impairment (MCI). This scheme was derived to be consistent with the role of neuroimaging in the list of biomarkers under investigation for Alzheimer's disease (AD) described by Albert et al (2011) in the National Institute of Aging—Alzheimer's Association workgroup criteria for MCI to AD (Albert et al, 2011, Tables 1 and 2). The final set of criteria for MCI due to AD has four levels of certainty that are depicted in the scheme. The certainties are based on the outcomes of various biomarkers that include structural magnetic resonance imaging (sMRI), positron emission tomography (PET) amyloid imaging (Aβ PET), and [18F]2-Fluoro-2-deoxy-D-glucose (or FDG) PET imaging. (∗) Positive Aβ PET has been reported for about 50% to 70% of MCI subjects (Rowe et al, 2010; Wolk et al, 2009).
Future directions
The brain mapping field rapidly expanded with the availability of BOLD contrast fMRI methods in the 1990s and multimodality imaging became commonplace in clinical settings with the availability of the combined PET/CT scanner over the last decade. It is possible that true simultaneous PET/MRI will enable similar advances in multimodality molecular brain imaging methods, research, and clinical applications. Future directions for molecular brain imaging include the development of multimodality nanoparticle hybrids (De et al, 2011), application of hyperpolarization for MRI (i.e., to enhance the signal of nuclei, such as 13C (Viale et al, 2009)), continued development of optical contrast agents, and identification of new PET imaging targets and validation of novel radiotracers. This raises exciting new possibilities for molecular imaging in the multimodality era.
Footnotes
Acknowledgements
The author thanks all those who provided example figures or permission for figure reuse (Drs H Aizenstein, R Buckner, C Catana, A Cohen, H Herzog, C Leroy, C Meltzer, B Rosen, R Sperling, and D Wolk) and to Drs C Mathis and W Klunk for additional supportive resources. The author thanks all those who commented on this work including W Bi, Y Chen, and Dr J Mountz, and to C Matan for help with the figures. Lastly, the participation of research study volunteers and their families is gratefully acknowledged.
Disclosure/conflict of interest
The author declares no conflict of interest.