
Abstract
Using in vivo two-photon imaging, we show that mice deficient in aquaporin-4 (AQP4) display increased fluorescence of nicotinamide adenine dinucleotide (NADH) when subjected to cortical spreading depression. The increased NADH signal, a proxy of tissue hypoxia, was restricted to microwatershed areas remote from the vasculature. Aqp4 deletion had no effects on the hyperemia response, but slowed [K+]o recovery. These observations suggest that K+ uptake is suppressed in Aqp4−/- mice as a consequence of decreased oxygen delivery to tissue located furthest away from the vascular source of oxygen, although increased oxygen consumption may also contribute to our observations.
Keywords
INTRODUCTION
Aquaporin-4 (AQP4) is the principal brain water channel and is concentrated in astrocytic endfoot membranes at the brain-blood and brain-liquor interfaces. 1 It is surprising that our understanding of the physiologic roles of AQP4 has evolved so slowly given its abundance in brain. Although AQP4 is engaged in interstitial fluid dynamics under physiologic conditions,2,3 several lines of work show that animals must be subjected to severe stress for a clear Aqp4−/- phenotype to become apparent. The role of AQP4 in mediating water exchange across the blood-brain interface was, for instance, initially demonstrated in models of brain edema that entail pronounced osmotic stress. 4
Here we used cortical spreading depression (CSD) as a model of severe metabolic stress to resolve whether oxygenation of brain neuropil depends on the presence of AQP4. Drawing on experience from studies of brain edema, we hypothesized that severe stress would be needed to disclose a link between AQP4 expression and tissue oxygenation. 4 Cortical spreading depression is a slowly spreading wave of depressed neuronal activity associated with a massive buildup of extracellular concentration of K+ ions ([K+]o), thought to occur during migraine headaches. The recovery of [K+]o involves several mechanisms, including inward rectifying K+-channels, (Na +)-K+-Cl_ cotransporters, and the Na +, K+-ATPase.5-7
Two-photon imaging of nicotinamide adenine dinucleotide (NADH) fluorescence can be used to provide high-resolution maps of tissue redox state in vivo. Increased NADH signal is a sensitive nonlinear proxy of tissue hypoxia. 8 Our analysis shows that Aqp4 deletion leads to a more pronounced oxygen deficit in microwatershed areas and a protracted [K+]o recovery. The most parsimonious explanation of these observations is that removal of AQP4 reduces oxygen supply and hence slows K+ re-uptake.
MATERIALS AND METHODS
Mouse Preparation
Aqp4 −/- and Aqp4+/+ mice of either sex were generated as previously described and anesthetized using intraperitoneal urethane (1 g/kg) and α-chloralose (50 mg/kg). 9 The mice were prepared for in vivo two-photon imaging as previously described. 7 Cortical spreading depression was evoked by either pressure injecting 1 M KCl through a micropipette or surface application of 1 M KCl (5 μl) through a small secondary craniotomy. Fixation of mice, preparation of tissue slices, and immunohistochemistry were performed as described previously. 9 All animal experiments were conducted in accordance with the ARRIVE guidelines and approved by the Animal Care and Use Committee of the University of Rochester.
In Vivo Recordings
A Mai Tai HP (Spectra Physics, Irvine, CA, USA) attached to a confocal scanning system (FV300, Olympus, Melville, NY, USA) and an upright microscope (IX51W) with an x 20 objective (0.95 NA, Olympus) was used. We performed combined imaging of NADH and intravascular fluorescein isothiocyanate-dextran (2000 kDa, 5%, intravenous). 7 Nicotinamide adenine dinucleotide was excited at 740nm and emission was detected using a 460 nm filter (50 nm bandwidth), while fluorescein isothiocyanate-dextran emission was detected using a 515 nm filter (50 nm bandwidth). The images were taken every 3 s at 50-150 μm depth. Images were filtered and converted to percent changes from baseline using ImageJ software (NIH, Bethesda, MD, USA) as described previously. 7 The NADH fluorescence changes during CSD reveal a characteristic biphasic response. 7 This response has a complex geometry, and perivascular ‘cylinders’ of low NADH signal can be seen whose size depends both on their vascular oxygen content and on their diameter. 8 We therefore calculated the change in signal intensity of individual pixels normalized to values in a baseline image (ΔF/F0), before the onset of CSD. By convention, the regions where pixel values became more negative (ΔF/F0<0) and positive (ΔF/F0 > 0) during the CSD wave were defined as ‘dip’ and ‘overshoot', respectively. 7 Direct current potentials and [K+]o were recorded using glass microelectrodes and tissue pO2 (tpO2) was measured using a Clark-type polarographic oxygen microelectrode (OX-4, Unisense, Aarhus, Denmark) as outlined before.7,10 Cerebral blood flow was assessed using a fiberoptic laser Doppler probe (PF5010, Perimed, Stockholm, Sweden) and connected to an infrared laser Doppler flowmeter. All signals were digitized (Digidata 1332A, Molecular Devices, Sunnyvale, CA, USA) and analyzed (pClamp 10.2, Molecular Devices, Sunnyvale, CA, USA).
RESULTS
Deletion of Aqp4 Increases Microwatershed NADH Fluorescence During Cortical Spreading Depression
We measured tissue NADH fluorescence, cerebral blood flow, tissue oxygen tension (tpO2), local field potentials, and [K+]o in living wild-type and Aqp4−/- mice after induction of CSD (Figure 1A). Immunofluorescence confirmed high perivascular AQP4 expression in wild-type mice and confirmed the efficacy of the gene knockout strategy (Figure 1B). The total and perivascular dip NADH responses in Aqp4−/_ animals did not differ from that in wild type during CSD (Figures 1C and 1D). However, the overshoot region of the NADH response was greater in the Aqp4−/- animals than in wild types almost from the onset of CSD, and became significantly higher ~ 2 minutes after the onset (total neuropil: wild type 14.36 ±4.02% vs. Aqp4−/- 19.61 ±6.21%; dip: wild type 17.33±3.43% vs. Aqp4−/- 23.23 ±4.78%;overshoot: wild type 17.09±3.71% vs. Aqp4−/- 39.48 ± 6.43%, all peak values, P <0.05 for 145 to 250 seconds post CSD onset) (Figures 1E and 1F). The rising phase of NADH fluorescence overshoot lasted 187.50 ± 36.54 seconds in wild-type and 133.33 ±49.09seconds in Aqp4−/- animals, and was not significantly different (data not shown). Thus, our data suggest that Aqp4 deletion selectively impairs tissue oxygenation in areas furthest away from the vasculature.
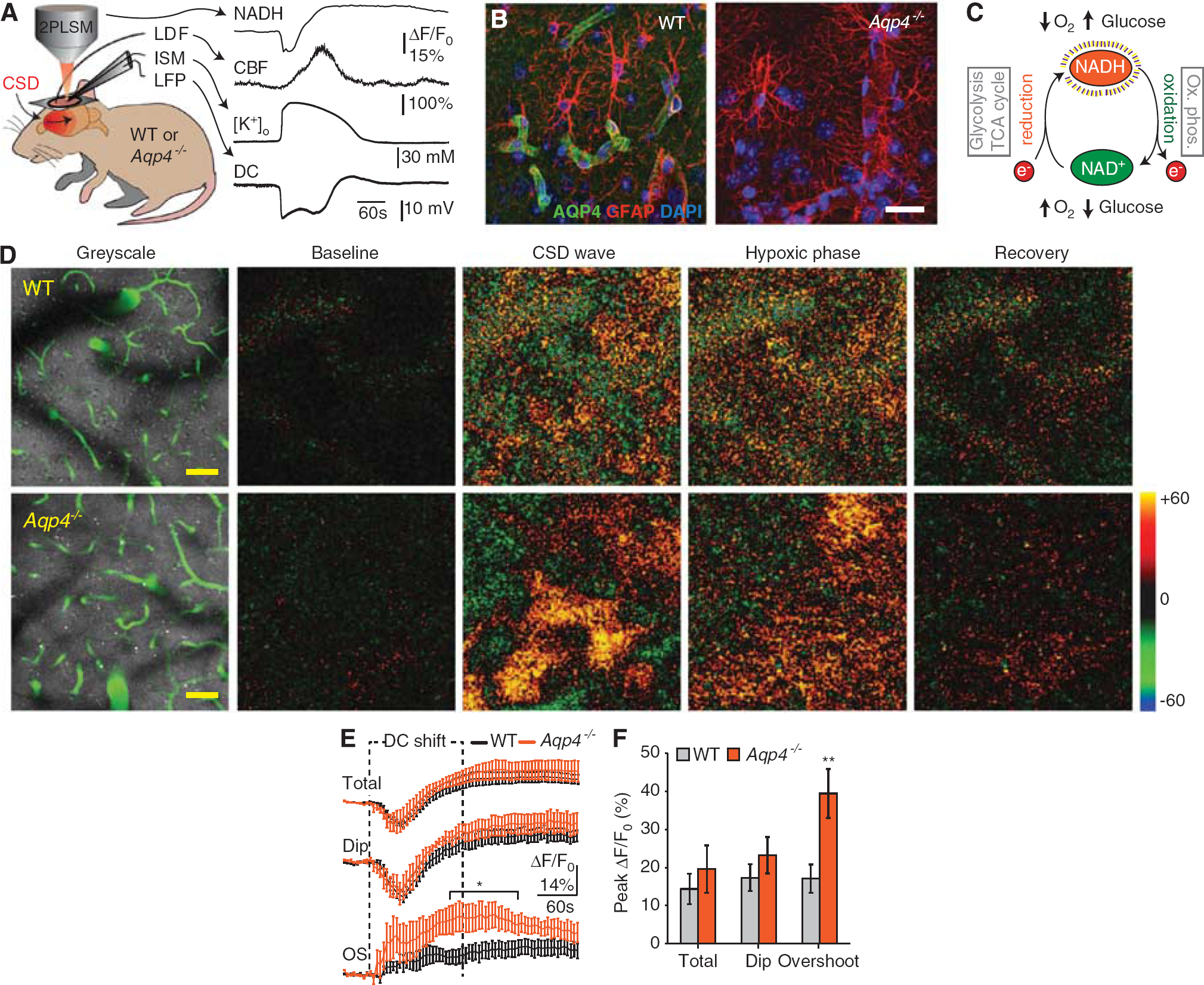
Deletion of aquaporin-4 (Aqp4) increased nicotinamide adenine dinucleotide (NADH) fluorescence in microwatershed areas during cortical spreading depression (CSD). (
Deletion of Aqp4 Does not Influence Hyperemia and Vascular Oxygen Supply in Cortical Spreading Depression
Cerebral blood flow is critical for maintaining adequate tissue oxygenation. Owing to the strong AQP4 expression around blood vessels, we tested whether Aqp4 deletion influenced the vascular response to CSD. Neither the amplitude nor the duration of the hyperemia phase differed between wild-type and Aqp4−/- animals (peak: wild type 164.50 ±4.57% vs. Aqp4 −/- 162.57 ± 7.54%;duration: wild type 174.50 ±11.56 seconds vs. Aqp4−/- 178.06 ±6.09 seconds) (Figures 2A–2C). Additionally, Aqp4 deletion did not affect overall tissue oxygen supply, which we assessed using oxygen-sensitive microelectrodes that integrate tpO2 from an -268-524 μm 3 volume encompassing both NADH dip and overshoot regions (baseline: wild type 38.60 ± 3.14 mm Hg vs. Aqp4−/- 45.98 ± 4.88 mm Hg;CSD: wild type 9.14 ± 2.05 mm Hg vs. Aqp4−/- 11.53 ± 3.03 mm Hg;Figure 2D). 7 The kinetics of these tpO2 recordings was not significantly different (Figures 2E and 2F) (AtpO2: wild type - 29.46 ±4.98 vs. Aqp4−/- - 34.45 ± 3.13;declining slope: wild type - 2.79 ± 1.22 mm Hg/second vs. Aqp4 −/- - 3.44 ± 0.74 mm Hg/second;recovery slope: wild type + 0.67 ± 0.38 mm Hg/second vs. Aqp4−/- + 0.58 ±0.12 mm Hg/second). Finally, when we used tpO2 and blood flow recordings to approximate the cerebral metabolic rate of oxygen (ACMRO2), this was not significantly different between the two genotypes during CSD (wild type 281.50 ±21.62 μmol/100 g per minute vs. Aqp4−/- 292.50±23.40 μmol/100g per minute, data not shown). However, this calculation uses equivalent coefficients for oxygen diffusion through brain in both genotypes, which may be an incorrect assumption if AQP4 alters gas diffusion. Taken together, we found no evidence that deleting Aqp4 altered hyperemia or overall oxygen supply during CSD.
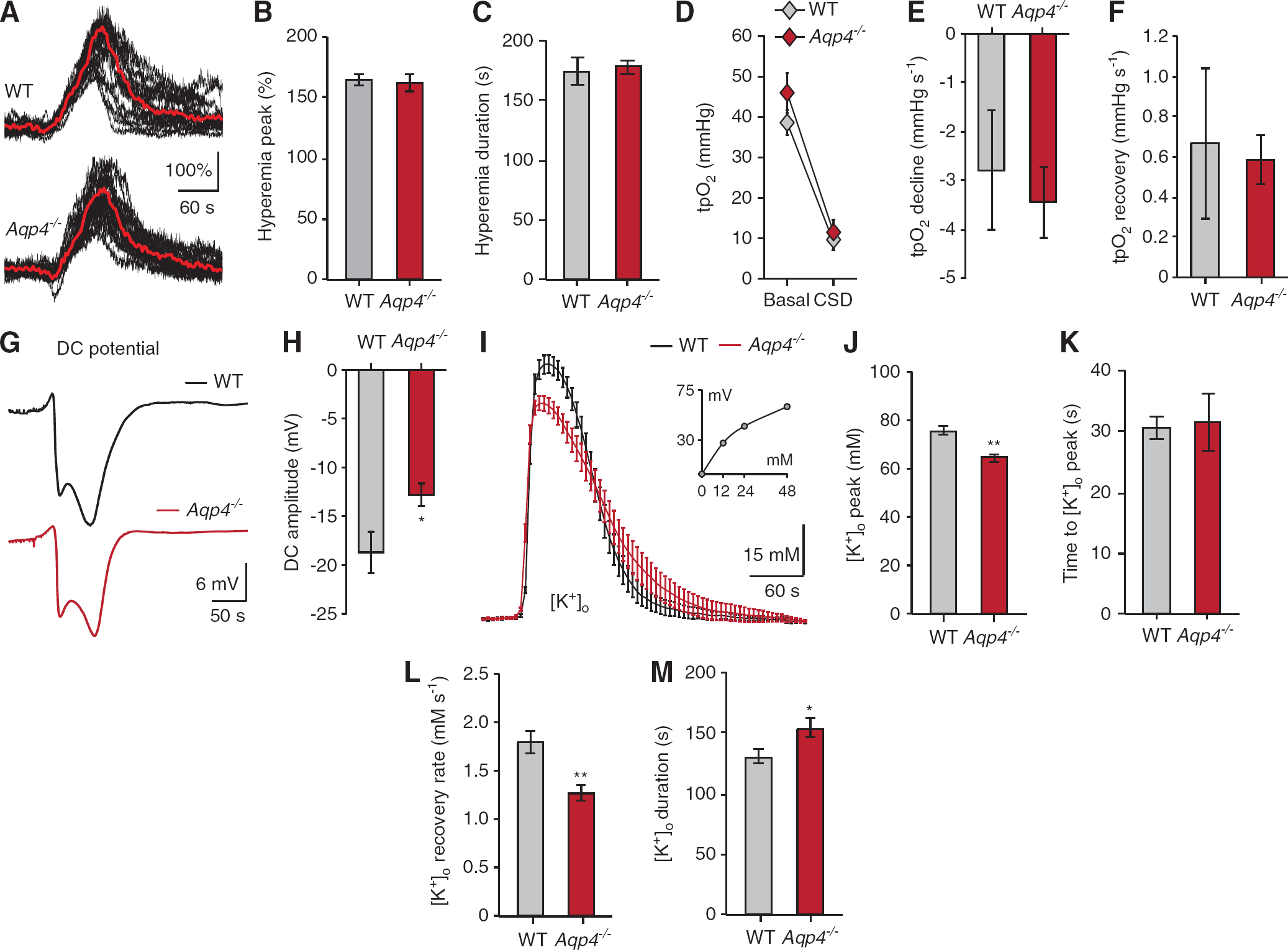
Deletion of aquaporin-4 (Aqp4) did not affect vascular oxygen supply but delayed extracellular K+ clearance in cortical spreading depression (CSD). (
Deletion of Aqp4 Delays [K+]o Recovery in Cortical Spreading Depression
The amplitude of the direct current potential shift, which is dependent on [K+]o, was significantly lower in Aqp4−/- than in wild-type mice (amplitude: wild type - 18.71 ± 2.11 mV vs. Aqp4−/- − 12.80± 1.16mV) (Figures 2G and 2H). In line with these data and the finding that Aqp4 deletion increases basal extracelluar space volume, 11 the amplitude of the [K+]o increase was significantly lower in Aqp4−/- animals (amplitude: wild type 75.54 ± 1.86 mM vs. Aqp4−/- 64.49 ± 1.73 mM) (Figures 2I and 2J). The rising phase of [K+]o in CSD lasted 30.53 ± 1.76 seconds in wild-type and 31.61 ± 4.75 s in Aqp4−/- animals, not significantly different (Figure 2K). However, Aqp4−/- mice showed a slower [K+]o recovery compared with wild types (recovery rate: wild type 1.79 ± 0.12 mM/s vs. Aqp4−/- 1.27 ± 0.08 mM/second;duration of [K+]o elevation: wild type 130.32 ± 5.42 seconds vs. Aqp4−/- 153.86 ± 7.54 seconds) (Figures 2L and M), suggesting that the capacity for [K+]o clearance is reduced by Aqp4 deletion.
DISCUSSION
Our study provides the first line of evidence that AQP4 impacts oxygenation of brain tissue. Using CSD as a model of severe metabolic stress, we show that Aqp4 deletion increases NADH fluorescence in areas furthest away from cerebral microvessels. NADH fluorescence is a sensitive indicator of tissue hypoxia with a p50 of 3.4 mm Hg, 8 and our data thus demonstrate that Aqp4 deletion enhances the already critical microwatershed hypoxia seen in CSD. 7 Measurements with laser Doppler flowmetry show that this impaired oxygenation was not due to inadequate blood supply, as Aqp4−/- and wild-type mice had a comparable hyperemia response. Moreover, oxygen-sensitive microelectrodes showed that the observed NADH changes did not reflect overall differences in tissue oxygenation, but were spatially restricted to microwatershed regions.
The observed increase of microwatershed hypoxia in Aqp4−/- mice can be explained by either enhanced oxygen consumption or reduced oxygen diffusion. Aqp4 deletion has previously been shown to impair [K+]o clearance during CSD, seizures, and neuronal stimulation.2,3,5,6,12 AQP4 can impact [K+]o clearance in several ways, including regulating extracellular space volume, dissipating osmotic gradients arising from ion transport, and enhancing interstitial bulk flow.2,3,6,12 Aqp4 deletion might therefore increase microwatershed hypoxia by making perivascular [K+]o homeostasis less efficient and consume excess oxygen. However, this hypothesis would imply that Aqp4−/- mice have a larger perivascular NADH signal dip and a higher overall oxygen uptake, neither of which we found in our study. Additionally, the hypothesis would predict that [K+]o increases in the Aqp4−/- mice prior to the observed NADH changes, whereas our data indicate the opposite.
The alternative hypothesis entails Aqp4 deletion impairing oxygen diffusion across perivascular endfoot membranes. Experimental analyses in oocytes and modeling studies have suggested that AQP4 might serve as a gas channel.13,14 Wang and Tajkhorshid 13 suggested that the central pore of the AQP4 tetramer could conduct NO and O2. However, the membrane permeability for gasses likely varies significantly depending on the type of gas studied (O2, CO2, NO), lipid composition of the membrane (e.g. endfoot vs. oocyte), and macromolecular organization of transmembrane proteins (e.g. AQP4 tetramers).
Aquaporin-facilitated gas transport is therefore an unresolved topic, where several studies have presented evidence for and against the hypothesis.13-15 If oxygen diffusion were rate limiting in our study, we would hypothesize that microwatershed hypoxia would be increased and involve a larger area, which the NADH imaging supports. We might also expect that inadequate oxygenation - through compromised Na + -K+-ATPase activity -precedes and potentially causes the slowed [K+]o clearance seen in Aqp4−/- mice, and our data show a tendency towards this. Finally, NADH is an indirect and nonlinear indicator of tissue hypoxia that can be influenced by factors unrelated to oxygenation (e.g. cell swelling, hypoglycemia). As we were unable to detect an overall difference in tissue oxygenation using microelectrodes, our data therefore do not directly show that mitochondrial oxygen uptake is inadequate or rate limiting in the Aqp4−/- mice.
In conclusion, our data suggest that Aqp4 deletion in CSD impairs the oxygenation of areas remote from brain microvessels. These observations are consistent with the hypothesis that AQP4 facilitates oxygen diffusion, but due to methodological limitations, we cannot conclusively differentiate this from the alternative explanation that Aqp4 deletion increases metabolic demand. Our study thus provides the first evidence to suggest that AQP4 is involved in facilitating oxygen diffusion in vivo.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.