
Abstract
The role of tumor necrosis factor (TNF) and its receptors after traumatic brain injury (TBI) remains unclear. We evaluated the effects of genetic deletion of either p55 or p75 TNF receptor on neurobehavioral outcome, histopathology, DNA damage and apoptosis-related cell death/survival gene expression (bcl-2/bax), and microglia/macrophage (M/M) activation in wild-type (WT) and knockout mice after TBI. Injured p55 (– / –) mice showed a significant attenuation while p75 (– / –) mice showed a significant worsening of sensorimotor deficits compared with WT mice over 4 weeks postinjury. At the same time point, contusion volume in p55 (– / –) mice (11.1 ± 3.3 mm3) was significantly reduced compared with WT (19.7 ± 3.4 mm3) and p75 (– / –) mice (20.9 ± 3.2 mm3). At 4 hours postinjury, bcl-2/bax ratio mRNA expression was increased in p55 (– / –) compared with p75 (– / –) mice and was associated with reduced DNA damage terminal deoxynucleotidyl transferaseYmediated dUTP nick end labeling (TUNEL-positivity), reduced CD11b expression and increased Ym1 expression at 24 hours postinjury in p55 (– / –) compared with p75 (– / –) mice, indicative of a protective M/M response. These data suggest that TNF may exacerbate neurobehavioral deficits and tissue damage via p55 TNF receptor whose inhibition may represent a specific therapeutic target after TBI.
INTRODUCTION
The role of cerebral inflammatory responses in mediating behavioral and cellular outcome after traumatic brain injury (TBI) has received increasing attention. Post traumatic inflammatory cascades have been documented both in clinical and experimental TBI, including the entry of leukocytes into the injured brain,1,2 and upregulation and release of cytokines, such as tumor necrosis factor-α (TNF), interleukin (IL)-1β, IL-6, IL-10 and IL-12.3–5 Pro and antiinflammatory cytokines interact in a complex but not yet fully understood network involving both damage and recovery processes, suggesting that post traumatic inflammation may have a dual role after TBI.610
In the brain, TNF is produced by microglia, astrocytes, endothelial cells, and also by neurons, and its effects range from induction of apoptotic and necrotic cell death to stimulation of cell growth and differentiation. Tumor necrosis factor is synthesized as a monomeric type-2 transmembrane protein (tmTNF) that is inserted into the membrane as a homotrimer and is cleaved by the matrix metalloprotease TNF converting enzyme to a 51 kDa soluble circulating trimer (solTNF); both tmTNF and solTNF are biologically active. 11
Tumor necrosis factor binds two different surface receptors—TNFR1 (p55) and TNFR2 (p75).12,13 These membrane glycoprotein receptors specifically bind TNF, but the two receptors differ in their expression profiles, ligand affinity, cytoplasmic tail structure, and downstream signaling pathway activation. Several biologic responses classically attributed to TNF are mediated by p55, which is expressed in most cell types, and can be activated by binding of either solTNF or tmTNF (with a preference for solTNF). 11 Downstream events resulting from p55 receptor activation include apoptosis, and activation of two transcription factors: nuclear factor kB (NF-KB) and c-Jun N-terminal kinase (JNK). 14 In contrast, the p75 receptor is expressed primarily by cells of the immune system, endothelial cells, and specific neuronal populations, and is preferentially activated by tmTNF. To date, p75 receptor activation is believed to initiate proinflammatory and prosurvival signaling. 11
In various models of central nervous system (CNS) disease and injury, TNF has shown to possess both neurotoxic and neuroprotective activity. This is believed to depend on several factors including specific cell expression, the timing of TNF expression and receptor activation, ligand concentration and cross talk amongst the different signaling pathways. Experimental models of TBI have revealed that TNF is upregulated at early time points, reaching a peak within a few hours after the initial injury. 15 TNF also appears to be released faster than other proinflammatory cytokines and initiates the activation of several cytokines and growth factors as well as the recruitment of immune cells. Specific pharmacological manipulations, aimed at reducing post traumatic TNF production and antagonizing its activity acutely have been associated with improved post traumatic outcome.1,17 However, subsequent studies using genetically engineered mice lacking TNF or its receptors have provided apparently contradictory results, suggesting that this cytokine might be detrimental in the acute phase after TBI while participating in regenerative/pleiotropic processes in the chronic postinjury phase.18,19
The differential involvement of TNF receptors in TBI may be one of the keys to our understanding the complex role of TNF in the injured brain.20,21 In a murine model of retinal ischemia, activation of the p55 receptor was found to be associated with increased tissue destruction, 22 while activation of the p75 receptor was, conversely, found to be protective. In a model of kainic acid-induced seizures, p55 activation exacerbated, while p75 inhibited seizures. 23 Similarly, in a cryolesion model, deletion of p55 receptor leads to a decreased inflammatory reaction and reduced apoptosis. 24 Conversely, TNF-associated neuroprotection after cerebral ischemia has been shown to occur via the p55 receptor25–27 indicating that the functional role of TNF receptors in brain injury is still to be fully elucidated.
We have previously reported that deletion of p55 receptor was associated with decreased cognitive impairment after TBI. 28 In a similar model, Yang et al. 29 have also shown a protective role for p75/Fas. In order to more definitively clarify the role of the two TNF receptors in the response to TBI, we compared neurobehavioral and histologic outcomes after TBI in wild-type (WT) mice and mice genetically engineered to be deficient in either the p55 receptor (p55 (– / –)) or the p75 receptor (p75 (– /–)). In order to more completely characterize the molecular mechanisms underlying the effects of genetic manipulation of TNF receptors in the postinjured brain, we also evaluated changes in mRNA expression of proapoptotic and antiapoptotic genes and assessed the microglia/macrophage (M/M) response in the injured cortex in the three mouse strains after TBI.
MATERIALS AND METHODS
Experimental Brain Injury
All procedures conformed to institutional guidelines in compliance with National (D.L. n.116, G.U. suppl. 40, 18 February 1992) and International laws and policies (EEC Council Directive 86/609, OJL 358,1; Dec.12, 1987; NIH Guide for the Care and Use of Laboratory Animals, U.S. National Research Council 1996). The study was reviewed and approved by the local ethics committee of the IRCCS-Istituto di Ricerche Farmacologiche Mario Negri. The protocol used and details of this report are also in accordance with ARRIVE guidelines. Before beginning any procedure, all mice (n = 5/cage, 8 weeks old, 20-25 g) were housed for at least 1 week in their home cages at a constant temperature, with a 12-hour light-dark cycle, and ad libitum access to food and water in a specific pathogen-free vivarium.
For both Study 1 and Study 2, 8-week old male WT C57Bl/6 (n= 46), p55 (– / –) (n= 46), and p75 (– / –) (n= 44) (both generated on C57Bl/6 background, kindly provided by Immunex Seattle, WA, USA) mice were anesthetized with intraperitoneal (i.p.) administration of sodium pentobarbital, (65 mg/kg), and placed in a stereotactic frame. An eye lubricant ointment (Xantervit ophthalmic ointment, Sifi, CT, Italy) was applied to protect corneal membranes during surgery. Mice were subjected to a craniectomy followed by induction of controlled cortical impact (CCI) brain injury.30,31 Sham-injured WT mice (n= 25), p55 (– / –), (n= 27) and p75 (– / –) (n= 22) received identical anesthesia and surgery without brain injury to serve as uninjured controls. Our model of CCI brain injury uses a 3 mm rigid impactor driven by a pneumatic piston, rigidly mounted at an angle of 20° from the vertical plane and applied perpendicularly to the exposed dura mater over the left parietotemporal cortex, midway between bregma and lambda, at a velocity of 5 meter/second and depth of 1 mm. To obtain the zero point, the impactor tip was lowered until it touched the intact dura mater. A transducer positioned at the upper rod of the device was connected to a linear velocity displacement transducer (model ATC-101, Schaevitz, Pennsauken, NJ, USA), which produces an analog signal transmitted to a computer through an analog-digital (AD) converter (Power Lab with Chart Pro, AdInstruments Ltd, Oxfordshire, UK) for storage and analysis of the impact velocity. At the end of the procedure, the craniotomy was covered with a cranioplasty and the scalp sutured. During and after all surgical procedures, mice were kept on a heating pad and maintained at a body temperature of 37 °C degrees.
Experimental Design and Blinding
In study 1, we evaluated the effects of TNF receptor deletion on long-term (4 weeks) neurobehavioral dysfunction and histopathological outcome after either CCI brain injury or sham injury in WT, p55 (– / –) and p75 (– / –) mice. Experimental groups were as follows: (1) brain-injured WT mice (n= 12); (2) brain-injured p55 (– / –) mice (n= 12); (3) brain-injured p75 (– / –) mice (n= 12); (4) sham-injured WT mice (n= 12); (5) sham-injured p55 (– / –) mice (n= 10); (6) sham-injured p75 (– / –) (n= 9) mice. Animals were evaluated at 48 hours postinjury and then weekly over a 4-week period for sensorimotor function. At 4 weeks, all animals were killed, brains removed, and processed for evaluation of the extent of tissue loss in the injured cortex.
Study 2 was designed to begin to evaluate the cellular mechanisms underlying our observations made in study 1. Experimental groups were identical to those detailed above for study 1. At various time points (that will be detailed thereafter) postinjury or sham injury, separate groups of animals were killed for analysis of regional cell death/survival gene expression, DNA damage and M/M response. To minimize the variability, all surgeries and injuries were performed by the same investigator, who was masked to the genotype of each mouse. At the end of the procedure, a second investigator assigned a masked code to each mouse (including the sham-injured group). All subsequent behavioral and histologic evaluations were done by masked investigators unaware of genotype or injury status of the animals.
Physiology
One subgroup of brain-injured WT, p55 (– / –) and p75 (– / –) mice (n= 6/genotype) was used for blood pressure measurement and blood gas analysis to test whether genotype could influence physiologic response to TBI. Systolic, diastolic, and mean arterial pressure and heart rate were measured in mice during recovery from anesthesia (range 60-90 minute) in a noninvasive manner using a computerized system (BP-2000 Blood Pressure Analysis System, 2 Biological Instruments, Varese, Italy). Additional animals (n= 6) were subjected to carotid artery exposure during recovery from anesthesia (range 45-60 minute); 100 μL of blood were withdrawn and loaded in cartridges, and blood gas analysis was then performed by the i-STAT 1 analyzer (Oxford Instruments S.M, Burke e Burke, MenfisBiomedica, Milan, Italy) as previously described. 30
Study 1: Effects Of P55 and P75 Deletion on Post Traumatic Neurobehavioral Dysfunction and Lesion Volume
Assessment of Sensorimotor Function
Prior to killing for assessment of injured area and contusion volume, all brain-injured and sham-injured animals were evaluated for sensorimotor function using the well-established composite neuroscore. Neurobehavioral assessments were conducted beginning 48 hours postinjury and then weekly from 1-4 weeks postinjury. In our hands, the composite neuroscore has proven to be a sensitive paradigm for measuring post traumatic neurologic deficits in mice up to 1 month postinjury.30–32 The neuroscore generates a score for each individual animal from four (normal) to zero (severely impaired) for each of the following indices: (1) forelimb function; (2) hindlimb function; (3) resistance to lateral right and left pulsion, as previously described.30–32 The maximum score per animal is 12.
Tissue processing for histopathological analysis
At 4 weeks postinjury (after the last behavioral evaluation), mice were killed for histologic analysis of contusion volume. Under deep anesthesia (Equitensin 120 μL/mouse), all animals were transcardially perfused with 20mL of phosphate buffer saline (PBS) 0.1 mol/L, pH 7.4, followed by 50 mL of chilled paraformaldehyde (4%) in PBS, and then the brains were carefully removed from the skull. The brains were post fixed overnight at 4 °C, then transferred to 30% sucrose in 0.1 mol/L phosphate buffer for 24 hours until equilibration. The brains were frozen by immersion in isopentane at − 45 °C for 3 minutes before being sealed into vials and stored at − 80 °C until use. Forty-micron-thick serial sections were cut using a cryostat from bregma + 1 mm to bregma − 4 mm. Finally, eight sections (bregma + 0.6; 0; − 0.8; − 1.5; − 2.25; − 2.65; − 3.25, and − 4 mm) were stained with neutral red (Neutral Red Sigma-Aldrich, St. Louis, Missouri, USA).
Contusion volume
The extent of traumatic brain damage in the injured hemisphere was quantified at 4 weeks postinjury by acquiring color images on a computer and using the image analyzer Analytical Image System (Imaging Research Inc., BrockUniversity, St Catharines, Ontario, Canada), as previously described.30,31 The ipsilateral and contralateral hemispheres were manually outlined. Subsequently, the injured area was calculated by subtracting the contralateral hemisphere minus the ipsilateral hemisphere at the coordinates mentioned above. Finally, the contusion volume was calculated by integration of the injured area according to the following formula:

where CA is the area in the contralateral hemisphere, IA is the area in the ipsilateral hemisphere, and d is the distance from a given section (k) to the subsequent section (k + 1).
Study 2: Cellular Mechanisms Underlying Genotype-Related Cell Death and Neurobehavioral Changes
Post traumatic expression of cell death genes in wild type, p55 (– / –) and p75 (– / –) mice
In order to gain an insight into the mechanisms leading to the different outcomes of the three strains observed in study 1, we evaluated the expression of genes involved in post traumatic cell death and protection that occurs in the injured pericontusional cortex. At either baseline (T = 0), 4 or 18 hours postinjury, subgroups of brain-injured and sham-injured animals (n= 6/genotype/time point) were killed and ipsilateral cortical areas (including all the tissue above the rhinal fissure) were dissected out, rapidly frozen on dry ice, and stored at 80 °C until gene expression analysis. 32 Total RNA was obtained from tissue specimen using the Trizol reagent (Gibco BRL, MD). Samples of total RNA (1.5 μg) were reverse transcripted with random hexamer primers using MultiScribe Reverse Transcriptase (TaqMan Reverse transcription reagents, Applied Biosystems, Foster City, CA, USA). Real time RT-PCR was conducted according to manufacturer instructions. Beta-actin was used as reference gene and relative gene expression levels were determined according to manufacturer's AACt method (Applied Biosystems). Primers were designed using the Primer Express Software (Applied Biosystems) based on the following GenBank accession numbers: NM031144 (β-actin, the housekeeping gene), NM009741 (BCL2), and NM007527 (BAX). Gene expression profiles are expressed as fold induction compared with control group. In addition, Bcl-2 and Bax are presented also as a ratio since this has been considered to better reflect the apoptotic status. 33
DNA damage in brain-injured WT, p55 (– /–) and p75 (– /–) mice
To assess the presence of injured cells showing DNA damage, a second subgroup of brain-injured animals (n= 10) was killed at 24 hours postinjury. Terminal deoxynucleotidyl transferaseYmediated dUTP nick end labeling staining was performed on 20 μm sections by in situ cell death detection kit (Roche, Mannheim, Germany) according to the manufacturer instructions at 24 hours postinjury. Adequate negative and positive controls were performed.
Immunohistochemical analysis of macrophage/microglial activation
At 24 hours postinjury, brain-injured (n= 6/genotype) or sham-injured (n = 6/genotype) mice were killed. Immunohistochemistry was performed on 20 μm brain coronal sections using mouse anti-CD11b (1:1000, kindly provided by Dr Doni 34 ), mouse anti-Ym1 (1:400, Stem Cell Technologies, Vancouver, BC, Canada), in order to measure M/M activation and the expression of the protective M2 M/M phenotype. Positive CD11b and Ym1 cells were stained by reaction with 3, 3 diaminobenzidinetetra-hydrochloride (DAB, Vector laboratories, Burlingame, CA, USA). For each reaction adequate negative controls were performed.
Slice selection and quantitative analysis
Three brain coronal sections per mouse at − 1.2, − 1.8, and − 2.2 mm from bregma were used to quantify CD11b and Ym1 stained area. One coronal section per mouse at − 1.5 mm from bregma was selected to quantify TUNEL-positive cells. To ensure unbiased operator independent sampling, we used the contusion edge in the cortex as a reference point and acquired the same focal plan throughout the samples by positioning acquisition fields as shown previously.31,32,35,36 Thirty-three and 42 fields (for CD11b and Ym1, respectively) per mouse were acquired at 40 ×magnification over a cortical region proximal to the lesion. For CD11b, the first row of acquisition fields was positioned at the edge of the contused cortical tissue, spacing each field by 236 μm (distance between centres of the fields). Additional rows of acquisition fields were positioned with an interfield distance of 472 μm. There was no overlapping between the fields. Since the Ym1 marker was found to be exclusively located in close proximity to the lesion, a single row of fields was positioned at contusion edge with no spacing or overlapping between fields. For TUNEL, eight fields per mouse at 20 × were acquired over a region proximal to the cortical lesion with no spacing or overlapping between fields.
An Olympus BX61 microscope equipped with a motorized stage and managed with AnalySIS software (Olympus, Tokyo, Japan) was used for image acquisition. Immunostained areas for CD11b and Ym1 were expressed as positive pixels/total assessed pixels and indicated as staining percentage area for subsequent statistical analysis.31,32,36 TUNEL-positive cells were expressed as cell density (number of cells per mm2).31,37 All quantifications were performed using ImageJ software (http://rsbweb.nih.gov/ij/).
Statistical analysis
The data are presented as mean±standard deviation. Comparisons between groups for neuroscore, injured area, and apoptotic genes were performed using a two-way repeated measurements analysis of variance (ANOVA) followed by Bonferroni post hoc test. Comparisons between groups for contusion volumes, physiologic variables, CD11b and Ym1 immunostaining and TUNEL were performed using a one-way ANOVA followed by Bonferroni post hoc test. A P value < 0.05 was considered statistically significant.
RESULTS
Acute (24 hours) post traumatic mortality was minimal: a total of 5 mice died on day 1 postsurgery, 3 p75 (– /–) brain-injured, 1 p75 (– / –) sham-injured, and 1 p55 (– / –) brain-injured. Following CCI brain injury, blood pressure, heart rate, and room air blood gases were similar in the three genotypes (Table 1).
Physiologic data in WT, p55 (– / –) and p75 (– / –) brain-injured mice assessed as detailed in the methods section

Abbreviations: DAP, diastolic arterial pressure; HR, heart rate; MAP, mean arterial pressure; SAP, systolic arterial pressure; WT, wild type. n = 6.
Study 1
Sensorimotor function
No difference in motor performance was observed among the three groups of sham-injured mice. Composite neuroscore evaluation revealed that all brain-injured mice showed sensorimotor deficits that persisted for the entire duration of the study. The score of all groups of these mice recovered slightly from 48 hours to 4 weeks postinjury. During the entire observation period, p55 (– / –) mice showed significantly attenuated sensorimotor deficits when compared with either WT (P < 0.001) or p75 (– / –) mice (P < 0.001). Moreover, p75 (– / –) mice showed significantly exacerbated sensorimotor deficits when compared with WT mice (P < 0.001) at all time points (Figure 1).
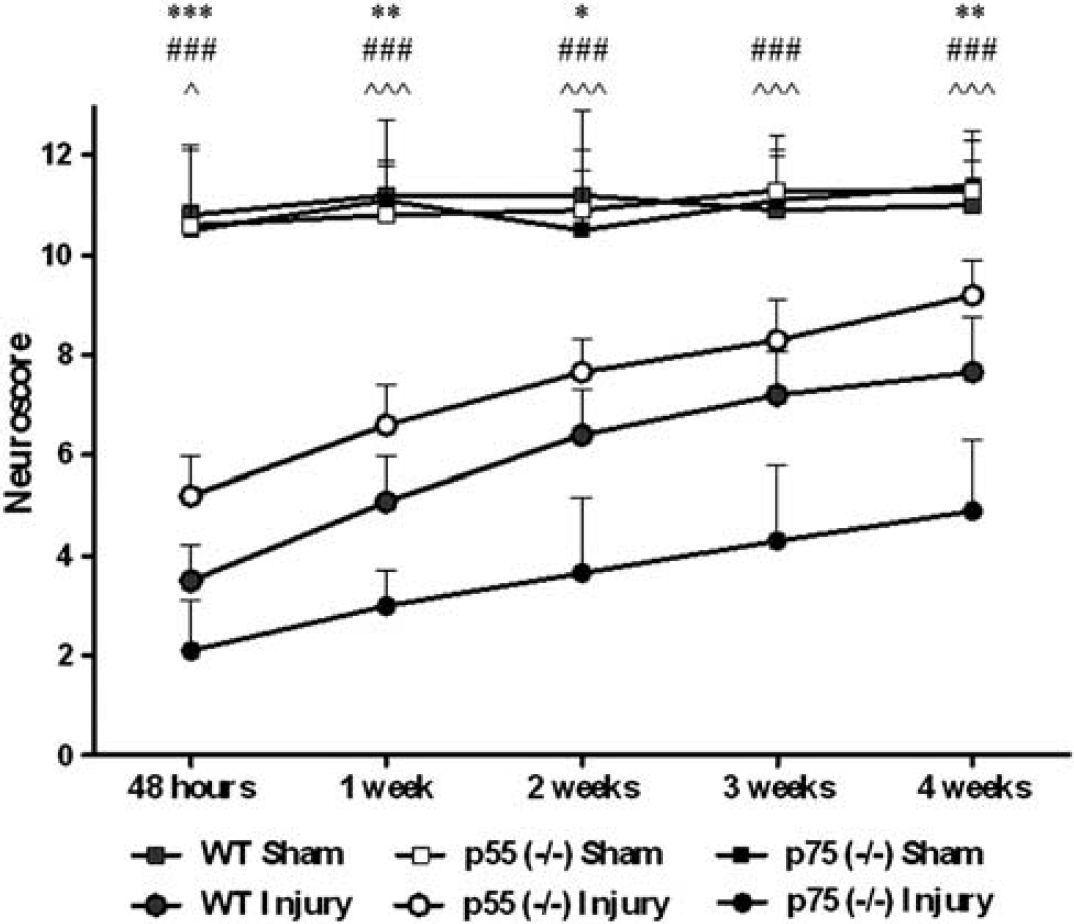
Sensorimotor function. Neuroscore during 4 weeks postinjury in wild type (WT), p55 (– / –) and p75 (– / –) mice. Data are reported as mean + s.d. Two-way analysis of variance for repeated measurements followed by Bonferroni post hoc test for individual comparison. Wild type sham and WT injury: n = 12; p55 (– / –) sham: n = 10; p55 (– / –) injury: n = 12; p75 (– / –) sham: n = 8; p75 (– / –) injury: n = 9. *P < 0.05, **P < 0.01 and ***P < 0.001: injured p55 (– / –) versus injured WT mice; ###P<0.001: injured p55 (– / –) versus injured p75 (– / –) mice; ^ P < 0.05, ^^^P < 0.001: injured p75 (– /–) versus injured WT mice.
Histopathology
Cortical tissue loss was not observed in the brains of sham-injured animals, regardless of genotype. At 4 weeks postinjury, we observed an extensive macroscopic area of cortical tissue loss extending rostrocaudally, from bregma + 0.6 to − 4 mm in all three genotypes. The core of the traumatic lesion was observed from bregma − 1.5 to − 2.25 mm in all injured groups. More importantly, p55 (– / –) mice showed a significantly reduced injured area compared to WT (48%) and p75 (– / –) mice (49%, Figures 2A and B, P < 0.01). At 4 weeks postinjury, p55 (– / –) mice showed a contusion volume of 11.1 ± 3.3 mm3 that was significantly reduced when compared with those of WT mice (19.7 ± 3.4 mm3, P < 0.01) and p75 (– / –) mice (20.9 ± 3.2 mm3, P < 0.01, Figure 2C).

Histopathology. (
Study 2
Expression of cell death genes and DNA damage
The cortical expression of bcl-2 and bax and their ratio was similar in sham-injured WT, p55 (– / –) and p75 (– / –) mice. At 4 hours postinjury bcl-2/bax mRNA ratio was significantly increased in brain-injured p55 (– / –) compared with p75 (– / –) mice (P < 0.05). This effect was no longer significant at 18 hours (Figure 3A).

Cell death genes and DNA damage. (
At 24 hours postinjury, we observed intense TUNEL-positive labeling in the injured cortex, indicative of the presence of apoptotic cells. Quantification of TUNEL-positive labeling showed that brain-injured p55 (– /–) mice had a significantly reduced number of damaged cells compared with brain-injured p75 (– / –) mice (P < 0.05, Figure 3B).
Macrophage/microglial activation
At 24 hours postinjury, all brain-injured mice, regardless of genotype, displayed extensive CD11b immunoreactivity in the ipsilateral cortex (Figure 4A). CD11b staining revealed three zones characterized by a different M/M morphology: (1) border of the contusion where ameboid morphology was clearly prevalent (Figure 4B); (2) transition zone where ramified (hypertrophic soma with thick branches) and ameboid morphology coexisted (Figure 4C); and (3) peripheral zone far from the contusion with the typical ramified morphology of resting microglia (Figure 4D).

Immunohistochemical analysis and quantitation of CD11b expression. (
Quantitation of CD11b immunoreactivity in the zone 1 and zone 2 revealed a significant reduction of M/M activation in p55 (– / –) compared with both WT and p75 (– / –) mice (P < 0.001, Figure 4E–H). In contrast, at 24 hours postinjury, mice displayed positive Ym1 immunoreactivity only in the border of the contusion (Figure 5A–C), indicating that the overall signal was reduced compared with that of CD11b. Quantitation of Ym1 immunoreactivity revealed significantly increased immunoreactivity in p55 (– /–) compared with p75 (– /–) mice (P < 0.05, Figure 5D).
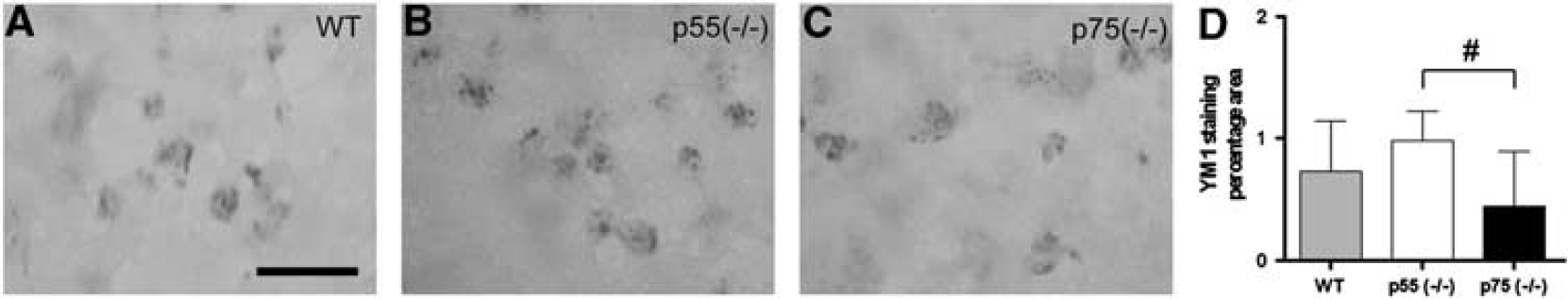
Immunohistochemical analysis and quantitation of Ym1 expression. (
DISCUSSION
The results of the present study demonstrate that, after TBI, mice genetically engineered to be deficient in the p55 TNF receptor show (1) sustained improvement in neurologic motor function over 4 weeks postinjury when compared with long-lasting neurobehavioral deficits observed in brain-injured WT and p75 (– /–) mice; (2) markedly reduced histologic damage at 4 weeks postinjury when compared with WT and p75 (– / –) mice that was associated with; (3) an increase in bcl-2/bax gene expression ratio, favoring cell survival, at 4 hours postinjury with evidence of reduced DNA damage (TUNEL-positive cells) in the injured cortex at 24 hours postinjury when compared with brain-injured p75 (– / –) mice; (4) reduced M/M activation (CD11b expression) when compared with WT and p75 (– / –), and increased Ym1 expression (M/M protective phenotype marker) when compared with p75 (– / –) mice at 24 hours postinjury. Conversely, p75 (– /–) brain-injured mice showed increased neurologic deficits over 4 weeks postinjury when compared with WT mice. These data, demonstrating that the lack of p55 receptor is associated with a lower degree of injury and a better outcome after TBI, suggest that the p75 TNF receptor may possess a neuroprotective/repair function after brain injury.
The overall role of TNF in mediating postinjury cell death or survival after TBI is not fully understood. Early pharmacological studies showed that TNF antagonism was associated with reduced neurobehavioral dysfunction.16,17 However, subsequent studies using genetically engineered mice are suggestive of a more complex picture. Scherbel et al. 18 showed that in the acute post traumatic period, TNF (– / –) mice exhibited significantly reduced deficits in both memory and neuromotor function when compared with WT mice. This advantage disappeared over time since functional recovery was found to be impaired and histopathological damage worsened in the same TNF (– / –) mice at 4 weeks when compared with WT, indicating that excessive TNF may be detrimental in the acute phase but involved with regenerative/healing processes in the more chronic postinjury period. In addition, when compared with WT mice, mice lacking both p75 and p55 receptors showed a greater lesion volume, alteration in blood– brain barrier (BBB) permeability, delayed NF-kB activation, and reduced manganese superoxide dismutase (MnSOD) expression in the acute post traumatic period after TBI, suggesting that TNFR-mediated NF-kB activation may initiate neuroprotective pathways early in the postinjury cascade through antiapoptotic/antioxidative mechanisms. 19
In our study, we tested the hypothesis that the two individual TNF receptors might have distinct roles in TBI by evaluating the effects of TBI in mice engineered to lack either the p55 or the p75 receptor. We observed that p55 (– / –) mice had markedly improved sensorimotor neurobehavioral performance up to 4 weeks after injury when compared with WT and p75 (– / –) mice and that p75 gene deletion significantly worsened post traumatic sensorimotor dysfunction. Additionally, we have previously reported also an attenuated cognitive dysfunction in the Morris Water Maze learning task in p55 (– / –) when compared with p75 (– / –) mice at 4 weeks after TBI. 28
Taken together, these results suggest that the dual role of TNF in post traumatic sequelae might be a function of the receptor type activated at any given time after injury, p55 activation being responsible for the early deleterious effect of TNF, while p75-related pathway being associated to long-term recovery. Further support of the specific roles of the two TNF receptors after TBI is provided by our histopathological data demonstrating that p55 (– / –) mice showed significantly reduced contusion volume in the injured cortex after TBI when compared with brain-injured WT and p75 (– / –) mice.
Recently Yang et al. 29 evaluated the effects of genetic deletion of individual p55, p75, p55/Fas, p75/Fas receptors using the same model of TBI employed in our study. Contrary to our observations, these authors failed to observe differences in motor function during the first week postinjury and lesion volume at 2 weeks postinjury between p55 (– / –) and p75 (– / –) mice. These discrepant results might be related to differences in the surgical procedure used in the two laboratories leading to different injury severities. In our studies, we induced a higher degree of injury (1 mm depth of deformation when compared with 0.6 mm) and sealed the craniectomy, a procedure that may be associated with a more severe post traumatic pathophysiological outcome (leading to higher intracranial pressure, more severe functional deficits, and tissue damage when compared with an open skull procedure38,39). Our results are in accordance with those previously reported from studies investigating the effects of genetic deletion of p55 and p75 receptors using experimental models of retinal ischemia, seizures, cold injury, and spinal cord injury22–24,40 and support the idea that selective inhibition of the p55 receptor after acute brain injury is associated with postinjury neuroprotection. The possible involvement of the p55 receptor after TBI has also been shown by Lotocki et al 41 who observed increased levels of p55 receptors in cortical lysates of the injured hemisphere within 5 minutes after fluid percussion brain injury in rats (in the presence of unchanged p75 receptor levels). These investigators concluded that acute TNF signalling after TBI is dependent upon increased p55 TNF receptor expression. Interestingly, these same authors subsequently reported that hypothermia, which is known to be neuroprotective after experimental TBI, could attenuate post traumatic increases in p55 receptor and suggested that this receptor may represent a novel therapeutic target after TBI. 42
Various mechanisms have been associated with p55 receptor-deletion-associated neuroprotection in different models of acute brain injury, including reduced inflammatory response, reduced astrocytosis, and reduced apoptosis.22,24,40 To more fully elucidate the possible cellular mechanisms underlying our observations of reduced cell death and improved neurobehavioral function in brain-injured p55 (– / –) mice, we evaluated the post traumatic changes in mRNA expression of genes involved in apoptotic cell death, which is known to occur after clinical and experimental TBI.43–46 We focused on the bcl-2 family genes, key regulators of the intrinsic apoptotic pathway known to be influenced by TNF signaling. The proapoptotic bax and the antiapoptotic bcl-2 regulate the permeability of the outer mitochondrial membrane and the release of cytochrome c from the mitochondria, a key step in the intrinsic apoptotic pathway.47,48 The association between levels of bcl-2, bax and outcome after clinical and experimental TBI has been previously demonstrated. Clinically, the outcome in TBI patients was shown to be inversely related to cerebrospinal fluid levels of bcl-2. 49 Studies using genetically engineered mice investigating individual cell death/survival genes after TBI have shown that when compared with brain-injured WT mice, reduced histologic damage occurs in bcl-2 overexpressing 50 and in bax (– / –) mice. 51 Taken together, the data regarding the role of bcl-2 and bax after TBI suggest that their ratio is a key regulator for post traumatic cell death/survival. 33 In the present study, baseline expression of proapoptotic and antiapoptotic genes were similar in uninjured WT, p55 (– / –) and p75 (– / –) mice as previously shown. 28 However, we observed a significant difference in bcl-2/ bax ratio between p55 and p75 (– / –) mice at 4 hours postinjury, suggestive of a shift towards a more neuroprotective/antiapoptotic balance in the brain-injured mice lacking the p55 receptor. We also compared the number of TUNEL-positive cells in the injured cortex, to assess the degree of DNA damage associated with apoptotic cells, and observed a significant reduction of TUNEL-positive cells in brain-injured p55 (– / –) mice when compared with p75 (– / –) at 24 hours postinjury further indicating a reduced apoptotic response after brain injury in p55 (– /–) mice.
To characterize the involvement of TNF receptors in the activation/recruitment of inflammatory cells in the traumatically injured brain, we chose to evaluate M/M phenotype after injury. Traumatic brain injury induces activation and proliferation of both macrophages and microglia, which appear to attempt the formation of a penumbral layer between damaged and healthy surrounding tissue, possibly representing an early defence against injury progression. Excessive M/M activation leads to the release of cytokines, chemokines, nitric oxide, free radicals, and reactive oxygen species detrimental for the injured brain.10,52,53 However, activated microglia may have also beneficial effects, and it is now widely recognized that at least two different polarization states exist, the classically activated (M1) state, associated with proinflammatory properties, and the alternatively activated (M2) microglia state, associated with antiinflammatory activity, extracellular matrix remodelling, and tissue repair. 52 We assessed CD11b expression, as marker of general M/M activation and expression of Ym1, a marker of M2 protective polarization. 54 Similar to previous observations made in experimental models of brain injury,36,55 M/M showed three different morphologies relative to their distance from the contusion/injury site, changing from ramified, to ramified but with hypertrophic soma and thick branches, to amoeboid morphology, mainly located in proximity of the contusion and suggestive of an increased phagocytic activity. The quantification of M/M markers was performed in the lesioned area that corresponds to the zones where CD11b positive cells showed amoeboid and ramified morphology with hypertrophic soma and thick branches. In brain-injured p55 (– / –) mice, the area of CD11b-immunopositivity was significantly reduced when compared with brain-injured WT or p75 (– / –) mice. Moreover, in the same mice, the protective marker Ym1 was increased when compared with p75 (– / –). These data suggest that in brain-injured p55 (– /–) mice, there was an overall reduction in M/M activation with a concomitant switch towards a more protective phenotype.
In conclusion, using gene-deletion technique, our study shows that the absence of the TNF p55 receptor in brain-injured mice dramatically improves post traumatic functional outcome and markedly reduces histologic damage. Mechanistically, these events appear to be associated, in part, with reduced apoptosis and DNA damage, a decreased M/M activation and induction of a protective M/M phenotype. These results underscore the possible role of the p55 receptor as a specific therapeutic target. Thus administration of a specific p55 antagonist, which has recently become available, 56 may represent a possible novel tool for the treatment of traumatic brain injury.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Tracy McIntosh, PhD for his valuable advice.