
Abstract
The endothelial-specific transmembrane glycoprotein, vascular endothelial (VE)-cadherin, is required for the organization of a stable vascular endothelium. A number of cerebrovascular disorders are associated with mutations in genes that otherwise regulate vascular integrity through VE-cadherin dynamics. Hence, identification and characterization of regulatory pathways contributing to endothelial cell-cell adhesion is of clinical relevance, particularly in the treatment of aneurysms and cerebral cavernous malformations. The zebrafish (Danio rerio) have recently emerged as a powerful paradigm for studies geared toward elucidating the etiology of cerebrovascular disorders, principally in uncovering the genetic and mechanistic basis controlling endothelial adhesive barrier function.
VASCULAR ENDOTHELIAL CADHERIN MEDIATES CEREBROVASCULAR STABILITY
Nascent blood vessels are particularly fragile and prone to hemorrhages. 1 Timely and rapid stabilization of blood vessels, through establishment of endothelial cell (EC)–cell contacts, is critical to prevent bleeding into the brain parenchyma. 2 Endothelial cell–cell contacts are established and maintained by transmembrane adhesion molecules that are anchored to cytoskeletal elements near the plasma membrane, which confer structural support and, thus, contribute to vascular stability.3–5 Of these transmembrane proteins, the vascular endothelial (VE) Cadherin is enriched in the EC membranes, maintaining cell–cell interaction and barrier integrity. Vascular endothelial Cadherin interacts, through a number of intracellular binding partners, with the actin cytoskeleton, which is thought to coordinate the mechanical properties of EC–cell contacts. This is evidenced by the fact that exposure to thrombin, a clotting factor that induces endothelial barrier dysfunction, depletes both the VE-cadherin–catenin complex and actin filament organization from the EC periphery. 6 This linkage is under the tight control of tyrosine kinases and Rho/Ras guanosine triphosphatase (GTPase).7,8 Membrane localization and activation of Rho/Ras GTPases require prenylation to transport the inactive proteins to the cell membrane, a step mediated by a pathway involving 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) activity. Increased levels of the active (GTP-bound) Rho-GTPases, Rac1 and CDC42, stabilize cadherin-mediated cell–cell adhesion. 9 Similarly, activated CDC42 regulates permeability by enhancing the interaction of VE-cadherin with the actin cytoskeleton in mice-derived ECs. 10
Inactivation of VE-cadherin and truncation of the β-catenin-binding cytosolic domain of VE-cadherin induces EC-specific apoptosis, defective remodeling and maturation of the vasculature and early lethality in mice. 11 Endothelial-specific conditional deletion of β-catenin, a protein interacting with the cytoplasmic tail of VE-cadherin, induces altered vascular patterning, defective lumenization and frequent hemorrhaging in vivo, as well as decreased intricacy of EC-cell junctions in vitro.12,13 Congruent with findings derived from mammalian studies, work in zebrafish has shown that partial and transient loss of cdh5 expression, the zebrafish homolog of the VE-cadherin encoding gene, induces vascular instability, defective lumenization of vasculature, and cranial hemorrhages in embryos and larvae. 14 Complete depletion of VE-cadherin levels results in more profound defects, including total inhibition of EC sprouting activity, cardiac defects, and embryonic lethality. 14
WHY ZEBRAFISH?
The precise mechanistic basis as to how adhesive interactions between ECs confer a functional vasculature is not fully elucidated.15,16 As such, identification of the entire suite of genetic networks regulating VE-cadherin dynamics is particularly relevant for stroke research. However, some of the common encumbrances of traditional murine models of cerebrovascular research include the high cost, variable reproducibility of the desired phenotype, high mortality, and most notably, the difficulty of in vivo imaging of vascular dynamics. Accordingly, the use of zebrafish to model the etiology of cerebrovascular disorders would not only complement current in vitro and mammalian studies, but also accelerate drug discovery by facilitating the identification of gene regulatory networks and potential drug targets. Over the last decade, zebrafish have emerged as viable models for cerebrovascular research. Zebrafish are vertebrates and thus share conserved molecular mechanisms regulating vascular morphogenesis with mammals.17,18 A distinguishing feature of the zebrafish is their optical transparency through embryonic development, which facilitates noninvasive and in vivo visualization of any defects in vascular permeability through the use of bright-field microscopy. The presence of stable transgenic lines expressing fluorescent markers in vascular and erythroid cell lineages further enhances the sensitivity of phenotype-based screening, allowing for assessment of vascular structure and patterning in vivo (Figure 1). 19
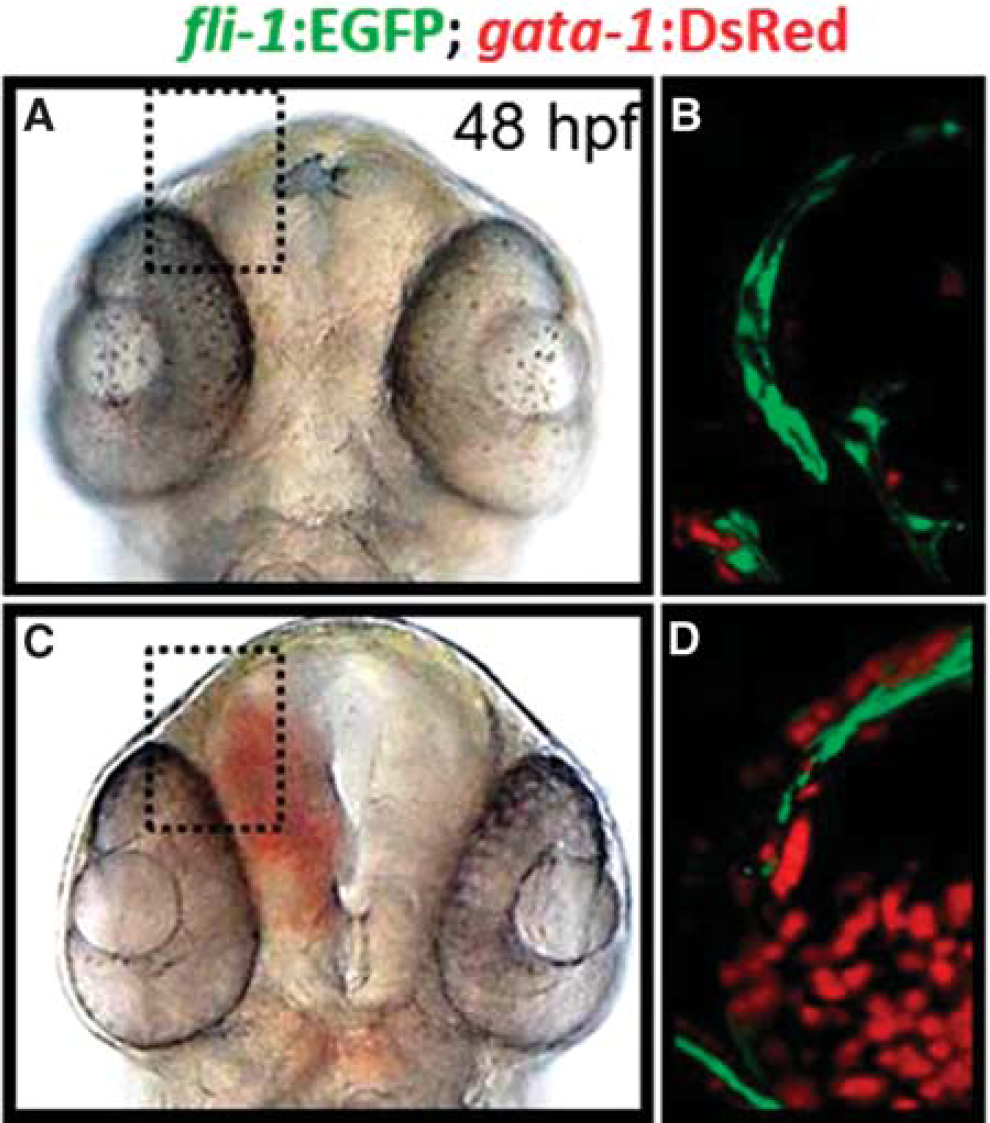
Phenotypes of zebrafish embryos with or without intracranial hemorrhage. (
GENES REGULATING CEREBROVASCULAR STABILIZATION IN ZEBRAFISH THROUGH VASCULAR ENDOTHELIAL CADHERIN DYNAMICS
Studies in zebrafish have uncovered a number of regulatory pathways that contribute to vascular stabilization through direct or indirect effects on the dynamics of VE-cadherin (Figure 2).
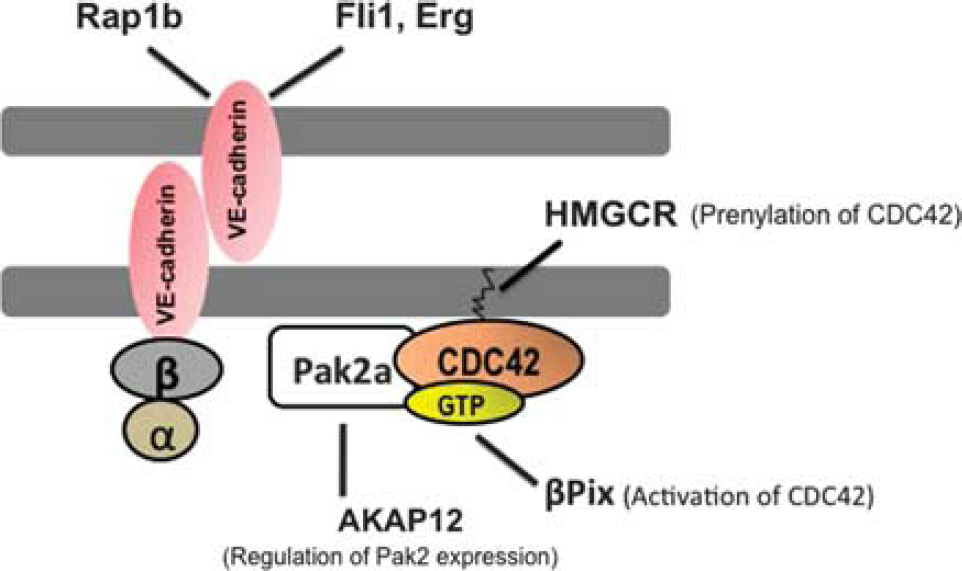
Stable endothelial cell (EC) junctions are maintained by a CDC42-dependent and vascular endothelial (VE) cadherin-mediated cell-cell adhesion. Vascular endothelial Cadherins are found on the surfaces of EC-cell junctions. Vascular endothelial Cadherins are associated with β and α catenins at their cytoplasmic domains, which connect them to the actin-based cytoskeleton (blue circles). CDC42 belong to the Rho-family of small guanosine triphosphatases (GTPases), which are the main regulators of VE-cadherin-based cell-cell adhesion. Additional genes and regulatory pathways regulating EC-junctional stability in zebrafish are indicated.
P21-activated kinase 2a
A recessive zebrafish mutant, termed redhead (rhd), which was identified in a chemical (N-ethyl-N-nitrosourea-derived) mutagenesis screen, 20 is characterized by cerebral hemorrhages during early development. 21 The mutation in this strain is shown to be a splice site error in the p21-activated kinase 2a (pak2a) gene, which otherwise encodes a member of the pak2a gene family. 21 The PAK gene family are kinases that act downstream of the Rho-family GTPases, CDC42 and RAC, and regulate actin organization at the membrane-cytosol interface (Figure 2). 22 The PAK gene family has been shown to regulate a multitude of relevant biologic processes in vitro, namely, cell motility, cytoskeletal rearrangements, and angiogenesis. 23 In light of the fact that the rhd mutants do not display discernable defects of morphogenesis,21,24 it is reasonable to speculate that the primary defects observed in these mutants are specific to blood vessels in the central nervous system, which is congruent with the preponderance of pak2a mRNA levels detected in the brain at 36hpf. 21
Pak-interacting exchange factor, βPix
Another vascular mutant identified in the same chemical mutagenesis screen is named bubblehead (bbh), presumably in reference to the hydrocephalus observed in these fish. 20 Additionally, the embryos are typified by cerebral hemorrhages. Molecular characterization, through positional cloning, highlighted the hypomorphic nature of this genetic defect owing to a point mutation in the splice donor site of exon 14 of βPix (Pak-interacting exchange factor), which is predicted to result in a nonfunctional product. 25 The functional role for βPix was confirmed through injection of a number of nonoverlapping oligonucleotides targeting distinct regions of βPix. At the ultrastructural level, the bbh mutants have been shown to exhibit ECs lacking contact with underlying mesenchyme. The βPix gene encodes the β isoform of the Pak-interacting exchange factor and a guanine nucleotide exchange factor, required for the activation of the Rho GTPases, Rac, and CDC42 (Figure 2). 25
Ras family small GTP-binding protein, Rap1b
Functional analyses of rap1b (Ras family small GTP-binding protein), one of the two isoforms of rap1 gene, which encodes a protein required for CCM1/KRIT1 localization to interendothelial junctions as well as an effector (kinase) for Ras GTPase, shows vascular-enriched expression in zebrafish. 26 Moreover, knockdown of rap1b in zebrafish induces cerebral hemorrhages, along with reduction in the expression and membrane localization of VE-cadherin and β-catenin. Interestingly, the incidence of central nervous system hemorrhages have been shown to increase when embryos were injected with combinations of morpholinos designed against βPix and pak2a, two previously identified Rac1/CDC42-interacting genes. 26 This is highly suggestive of synergy through genetic cross-regulation of components of Ras and Rho GTPases in vascular stabilization. 26
A-kinase anchoring protein 12, AKAP12
More recently, a role for A-kinase anchoring protein 12 (AKAP12) in the regulation of endothelial integrity has been reported in vivo. 27 AKAP12 is a central mediator of cAMP-dependent protein kinase A signaling pathway, 28 which, in turn, is a modulator of actin dynamics. 29 Genetic disruption of either of the splice variants of akap 12 in zebrafish is associated with reduced EC-cell contacts, giving rise to a discontinuous endothelial layer and abnormal sprouting phenotype, followed by frequent cerebral hemorrhages. 27 Analysis of AKAP12-deficient human umbilical vein ECs is associated with disrupted EC-cell contacts and reduced cortical actin filaments at the cell-cell contacts, all of which are shown to be because of reduced pak2 levels. 27 This study provides an additional regulator of VE-cadherin dependent interendothelial adhesion, mediated by pak2-dependent assembly and disassembly of actin cytoskeleton in ECs (Figure 2). 27
Ets-related gene, Erg and Friend leukemia integration 1, Fli 1
Molecular characterization of E26 transformation-specific family genes in zebrafish have led to the identification of two transcription factors involved in hematopoesis and angiogenesis, namely, erg and fli 1 (Ets-related gene and Friend leukemia integration 1), both of which have been associated with vascular stabilization. 30 Both erg and fli 1 exhibit expression profiles restricted to angioblasts and developing blood vessels in zebrafish embryos. 30 Injection of nonoverlapping morpholinos designed against erg or fli 1 induce identical hemorrhage phenotypes in the central nervous system. Moreover, whole-mount in situ hybridization analyses of both erg and fli 1 morphants show reduction of VE-cadherin mRNA levels in these embryos, 30 which is suggestive of erg and fli 1-dependent regulation of VE-cadherin transcription (Figure 2).
FUTURE DIRECTIONS
Studies over the last decade have been fruitful in identifying some of the genes required for developmental vascular stabilization through regulating VE-cadherin expression and membrane localization (Table 1). It is hoped that along with testing key scientific hypotheses relating to the genetic basis of cerebrovascular disorders, stroke research in zebrafish, which is still in a state of infancy, will help establish the grounds to design and test therapeutic strategies geared toward reversing defects in vascular stability. This is especially encouraging, given the recent advances in the successful application of high-throughput zebrafish drug screening platforms.31,32
Zebrafish genes regulating cerebrovascular stabilization via VE-cadherin dynamics

akap 12, A-kinase anchoring protein 12; βPix, Pak-interacting exchange factor; erg, Ets-related gene; fli 1, Friend leukemia integration 1; pak2a, P21-activated kinase 2a; rap 1b, Ras family small guanosine triphosphate-binding protein; VE, vascular endothelial.
Footnotes
RLM receives grant support from the Physicians Services Incorporated Foundation, Brain Aneurysm Foundation, Canadian Institutes for Health Research, and the Heart and Stroke Foundation of Canada; and is the Chief Scientific Officer of Edge Therapeutics.