
Abstract
Excitotoxicity is a major contributor to cell death during the acute phase of ischemic stroke but aggressive pharmacological targeting of excitotoxicity has failed clinically. Here we investigated whether pretreatment with low doses of memantine, within the range currently used and well tolerated for the treatment of Alzheimer's disease, produce a protective effect in stroke. A coculture preparation exposed to modeled ischemia showed cell death associated with rapid glutamate rises and cytotoxic Ca2+ influx. Cell death was significantly enhanced in the presence of high memantine concentrations. However, low memantine concentrations significantly protected neurons and glia via excitotoxic cascade interruption. Mice were systemically administered a range of memantine doses (0.02, 0.2, 2, 10, and 20 mg/kg/day) starting 24 hours before 60 minutes reversible focal cerebral ischemia and continuing for a 48-hour recovery period. Low dose (0.2 mg/kg/day) memantine treatment significantly reduced lesion volume (by 30% to 50%) and improved behavioral outcomes in stroke lesions that had been separated into either small/striatal or large/striatocortical infarcts. However, higher doses of memantine (20 mg/kg/day) significantly increased injury. These results show that clinically established low doses of memantine should be considered for patients ‘at risk’ of stroke, while higher doses are contraindicated.
Introduction
Cerebral stroke is the third leading cause of death in developed countries and the leading cause of adult disability. The only licensed pharmacological treatment available for acute cerebral stroke is thrombolysis with recombinant tissue plasminogen activator but because of its narrow therapeutic window (<4.5 h) and safety concerns, approximately only 15% of stroke patients receive recombinant tissue plasminogen activator. 1 Focal cerebral ischemia initiates a cascade of complex pathophysiologic events including excitotoxicity, acidotoxicity, ionic imbalance, oxidative stress, inflammation, and apoptosis.2, 3, 4 In an attempt to limit the acute cell death after the onset of ischemia a variety of neuroprotective strategies have been developed, which aim to antagonize injurious biochemical and molecular events that culminate in neuronal death. However, such strategies have failed to translate effectively into clinically available treatments for stroke patients (see Davis et al 5 ) and the development of safe and effective treatments remains a major challenge to stroke research.
The most rapid, and possibly the most severe, pathophysiologic mechanism initiated after ischemic stroke is that of excitotoxicity, which triggers widespread necrosis and subsequent functional impairment.6, 7 Historically, the focus of neuroprotection research has been to use strategies that supress the excitotoxic response after induction of ischemia and primarily target the glutamate system.
8
Various NMDA (N-methyl-
To exert a beneficial effect after ischemic stroke the majority of NMDA receptor antagonists have to be administered at high doses, which increases the possibility of producing unwanted psychotomimetic and cardiovascular effects. 18 However, not all NMDA receptor antagonists produce unwanted side effects at clinically effective doses and some are in current clinical use. One such NMDA receptor antagonist, memantine (1-amino-3,5-dimethyladamantane), has been approved since 2002 in Europe (and since 2003 in the USA) for the treatment of moderate to severe Alzheimer's disease, 19 and has been shown in clinical trials to be a safe and effective treatment for vascular dementia. 20 Memantine is distinct from other NMDA receptor antagonists as it possesses fast on/off kinetics, low-moderate receptor affinity and is able to block the effects of excessive glutamate without interfering with the physiologic activation of NMDA receptors. 21 Although acute postischemic administration of other NMDA receptor antagonists has been shown to be protective in experimental models (for review see Lipton, 2004) there is a wealth of evidence showing that this therapeutic approach of trying to limit cell death during the acute phase of ischemia is difficult to achieve. Thus, a more realistic approach may be prophylactic administration of memantine to those patients identified at being at risk of ischemic stroke, at a dose already tolerated in clinical practice, e.g., for the treatment of Alzheimer's disease. The aim of this study was to determine if systemic administration of the antiexcitotoxic drug, memantine hydrochloride, at doses currently administered for moderate to severe Alzheimer's disease, could produce protection from ischemic stroke.
Material and Methods
Cell Culture
High-density cultures (HDCs): Cortices were obtained from E16 balb-c mice after humane cervical dislocation under UK Home Office regulations. UK home office regulations were followed for all experimental work which was conducted in accordance with the relevant guidelines and regulations. The animal welfare and ethics committee of University of Leicester approved all the experimental protocols. The tissue was placed in Hank's balanced salt solution, trypsinized (1% trypsin/DNase), triturated, centrifuged (250g), resuspended in growth medium (Neurobasal+
Cell Culture Characterization
Cover slips were washed (0.1 mol/l phosphate-buffered saline (PBS)), fixed (4% paraformaldehyde/PBS or methanol/acetone 1:1), permeabilized (PBS/10% goat serum (Dako, Cambridge, UK)/0.5% Triton-X (Sigma-Aldrich, Gillingham, UK), PBSGT), incubated (PBSGT overnight at 4°C with primary antibody), exposed to appropriate secondary antibody (60 minutes PBSGT), mounted (SuperFrost Plus slides, Menzel-Glaser, Braunschweig, Germany) in PermaFluor (Thermo Fisher Scientific, Loughborough, UK), and imaged using a Leica (Milton Keynes, UK) TCS SP2 confocal microscope. Primary antibodies: glial fibrillary acidic protein and neuron specific enolase (1:400, Sigma-Aldrich); NSE (prediluted, Sigma-Aldrich); CNPase (1:100, Chemicon, Nottingham, UK); IB-4 (1:100, Molecular probes, Fisher Thermo Scientific). Projections (eight slices) were viewed using Leica software, Fluoview (Olympus, Southend-on-Sea, UK), or Metamorph (Molecular Devices, Sunnyvale, CA). Multiple slide areas were imaged and total cell number established either using the Hoechst 33,342 nuclear stain or light images of the cells; both methods produced similar cell counts (Supplementary Figure S1). At least three separate cell cultures and 3 to 8 slides were analyzed from each culture.
Cell Imaging
Oxygen—glucose deprivation (OGD)-induced intracellular Ca2+ ([Ca2+]i) changes were assessed using FURA-2FF (Invitrogen, Fisher Thermo Scientific), a low affinity dye that does not affect cell viability during ischemia. 22 The more sensitive FURA-2 was used for agonist responses. In both cases, cells were acetoxymethyl-loaded (see Fern 23 for more details of imaging methods). FURA dyes tended to leak from astrocytes over longer recording periods so, cell viability during OGD was assessed using 5-chloromethylfluorescein diacetate (Invitrogen) acetoxymethyl-loaded at 2.5 μmol/l. Dye-loaded cultures were mounted into a perfusion chamber (atmosphere chamber, Warner Instruments, Hamden, CT, USA), perfused at 2 ml/min (artificial cerebrospinal fluid in mmol/l: NaCl, 126; KCl, 3; NaH2PO4, 2; MgSO4, 2; CaCl2, 2; NaHCO3, 26; and glucose, 10; pH, 7.45, bubbled with 5% CO2/95% O2) and maintained at 37°C on the stage of an epifluorescence microscope (Nikon, Kingston Upon Thames, UK). Oxygen—glucose deprivation involved switching to artificial cerebrospinal fluid containing no glucose prebubbled with 95% N2/5% CO2, while chamber atmosphere was switched from 95% O2/5% CO2 to 95% N2/5% CO2. Cell temperature was monitored and maintained via flow-through, objective, and room heaters. A rapid exchange perfusion system was used for short application agonist experiments (ValveBank8.2, AutoMate Scientific, Berkeley, CA, USA). Oil immersion x20 images were collected at 520 nm or 508 nm using appropriate filter sets (Chroma Technology Corporation, Bellows Falls, VT, USA). For FURA-loaded cultures, cells were illuminated at 340, 360, and 380 nm; 5-chloromethylfluorescein diacetate cells were illuminated at 489 nm (Optoscan, Cairn Research). Images were captured by a coolSNAP HQ camera (Roper Scientific, Sarasota, FL, USA) controlled via MetaFluor (Molecular Devices) with background signal subtracted. For FURA-2 imaging, 340:380 was converted to [Ca2+]i using a calcium calibration kit (Invitrogen). Cell death was characterized by sudden collapse of the fluorescent signal to the background level and this phenomenon was used to calculate cell death rates and precise time points of cell death for all cells within the field of view. Cells were also imaged before and after the experiment in quadrants surrounding the field of view, where initial cell counts and surviving cell counts were measured. Cell death data plotted as a time series represents the real-time recordings from the field of view, while total cell death data includes the cells from the surrounding quadrants.
Cultured astrocytes and neurons are morphologically distinct under phase contrast. The majority of neurons are high contrast which have either pyramidal, fusiform, or multipolar characteristics. 24 Astrocytes show low phase-contrast with a flat morphology forming a continuous layer once confluent. These criteria for identification were tested and confirmed using immunohistochemical staining of fixed cocultures for glial fibrillary acidic protein and neuron specific enolase, (Supplementary Figure S2). The criteria were used to distinguish cells using initial phase-contrast and fluorescence images. Any unidentifiable cells were excluded from subsequent analysis.
Biosensors
Glutamate microelectrode biosensors (Sarissa Biomedical, Coventry, UK), 25 amplified via a Duo-Stat ME-200+ potentiostat (Sycopel International, London, UK) were used to record real-time glutamate or adenosine triphosphate concentration changes in vitro from the unstirred fluid layer surrounding cells. This layer is in communication with the bath solution and is likely to vary between recordings depending on the placement of the electrodes. These factors may contribute to the degree of variance observed between data sets, but reliable mean recordings were achieved. Signals were differential to a null electrode and both active and null electrodes were carefully inserted into a modified atmosphere chamber until the sensor tips rested directly on the cell layer. An Ag/AgCl reference electrode was introduced at a distal site. Oxygen—glucose deprivation and control experiments were performed as described above for cell imaging. Sensors were recalibrated in the chamber at the end of the OGD period after retraction from the cell layer. Values from the null, sensor, and sensor-minus-null outputs were recorded at 0.5 Hz and subsequently converted into Δ adenosine triphosphate or Δ glutamate, rather than absolute concentrations. Experiments were repeated a minimum of three times, all values collected for a time point during a specific condition/experiment were averaged using Prism (Graphpad, La Jolla, CA, USA).
Focal Cerebral Ischemia
This study was conducted in accordance with the UK Animals (Scientific Procedures) Act, 1986 (Project License 60/4315). Male adult C57 BL6 mice (Charles River, Oxford, UK) weighing between 22 and 32 g at the time of surgery were randomly assigned to a treatment group (the surgeon was masked to treatment and subsequent analyses). A total of 69 mice were used, 7 mice were excluded because of severe blood loss during surgery or poor recovery after middle cerebral artery occlusion (MCAO). Treatment groups were vehicle (n=17) or NMDA GluR antagonist, i.e., memantine, at the daily doses: 20 (n=9), 10 (n=7), 2 (n=7), 0.2 (n=16), or 0.02 mg/kg (n=6). All drugs were dissolved in 10% dimethyl sulfoxide and 90% saline, loaded into mini pumps (Alzet, Charles River. Model: 1003D; total volume 100 μl, flow rate 1 μl/h) and implanted subcutaneously 24 hours before MCAO. Antagonists or vehicle (10% dimethyl sulfoxide; 90% saline) were administered for 3 days in total.
Mice were anesthetized with isoflurane (induction 4%; maintenance 1.5% in N2O/O2 70/30%). A small subcutaneous incision was made on the midflank and the osmotic mini-pump inserted. The wound was sutured and animals recovered for 24 hours before focal ischemia. Focal cerebral ischemia was induced for 60 minutes by occlusion of the right middle cerebral artery as previously described. 26 Body temperature was monitored throughout surgery (rectal probe) and maintained at 37.0°C±0.6°C using a heating mat (Harvard Apparatus, Holliston, MA, USA). Laser Doppler flowmetry (Moor Instruments, Axminster, UK) was used to monitor relative cerebral blood flow for 5 minutes before and 5 minutes after MCAO. After 60-minute MCAO, mice were reanesthetized and the occluding filament withdrawn. Mice were weighed at 24 and 48 hours after surgery and neurologic status assessed using a 28-point neurologic score. 27 At 48 hours after surgery, mice were killed via cervical dislocation and brains were removed, sectioned (10 × 1 mm coronal slices) and stained (2% 2,3,5-triphenyltetrazolium chloride in saline) for 30 minutes. 2,3,5-Triphenyltetrazolium chloride is a marker of mitochondrial function and has been shown to be a reliable indicator of ischemic areas for up to 3 days after ischemia. 28 Sections were stored in 10% formalin solution at 4°C and photographed for analysis. Infarct areas were calculated as previously described 29 using an indirect method whereby overestimation of the infarct area because of edema is avoided.
Statistical Analysis
In focal ischemia experiments, lesion volume, behavior, and body weight assessment were performed (by MT who was masked) and are presented as mean±s.e.m. The population distribution for lesion volume in the vehicle infusion group was not uniformly distributed, falling into two groups: small lesions located in the striatum and large lesion encompassing both the striatum and cortical areas. When separated into these two patterns of injury, lesion volume in the vehicle group had a normal distribution and a parametric one-way analysis of variance (Fisher's) test was therefore used to determine significance between vehicle and test groups in these data sets. For in vitro data, all experimental protocols were repeated a minimum of three times and results are presented as means±s.e.m. Experiments were compared using either a t-test (when comparing two groups) or analysis of variance (Tukey's), with differences being significant when P<0.05.
Results
Establishing a Coculture Model of In Vitro Ischemia
To facilitate biosensor recording of neurotransmitter release in vitro, HDCs were developed (Figure 1) for astrocyte (93%±2.7% pure), neuronal (96%±2.7% pure) or mixed cells (51.9%±2.7%/43.8%±2.8%, respectively; 196.4±7.9 cells per field of view). High-density cultures were subjected to 90-minute OGD evoking biphasic [Ca2+]i rises and acute cell lysis (Figures 2A–2C). Neurons were less sensitive than astrocytes to injury in both mixed and mono-HDCs (Figure 2D) and neuronal death was potentiated by coculture with astrocytes suggesting the influence of a cytotoxic glial factor (Figure 2E). The astrocyte death rate was not significantly different in mono- and coculture conditions. High-density cocultures were found to release recordable quantities of glutamate during continuous perfusion using biosensor recordings from the unstirred fluid layer surrounding cells (Figure 2F, top). Glutamate levels increased synchronously with the first phase of the [Ca2+] rises occurring before the second phase or [Ca2+] rises/cell death events in both mixed (Figure 2F, top) and astrocyte (Figure 2F, bottom) HDCs. A similar rise in glutamate was not found in neuronal HDCs (Figure 2F, middle). Neuron injury in HDC was significantly reduced by the NMDA GluR blocker MK-801 (10 μmol/l) and the non-NMDA GluR blocker 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (30 μmol/l; Figure 2D top). In contrast, GluR antagonists failed to protect astrocytes (Figure 2D bottom), although the absence of any significant protection in the presence of 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione may be a product of the small sample size in this data group. Exogenous glutamate-evoked [Ca2+]i rises in all HDC neurons, mediated by both Mg2+-sensitive NMDA and Mg2+-insensitive non-NMDA GluRs (Figure 3A). In contrast, only 13.9%±6.8% of astrocytes showed glutamate-evoked [Ca2+]i rises, which were smaller and almost exclusively non-NMDA GluR-mediated (Figure 3B).
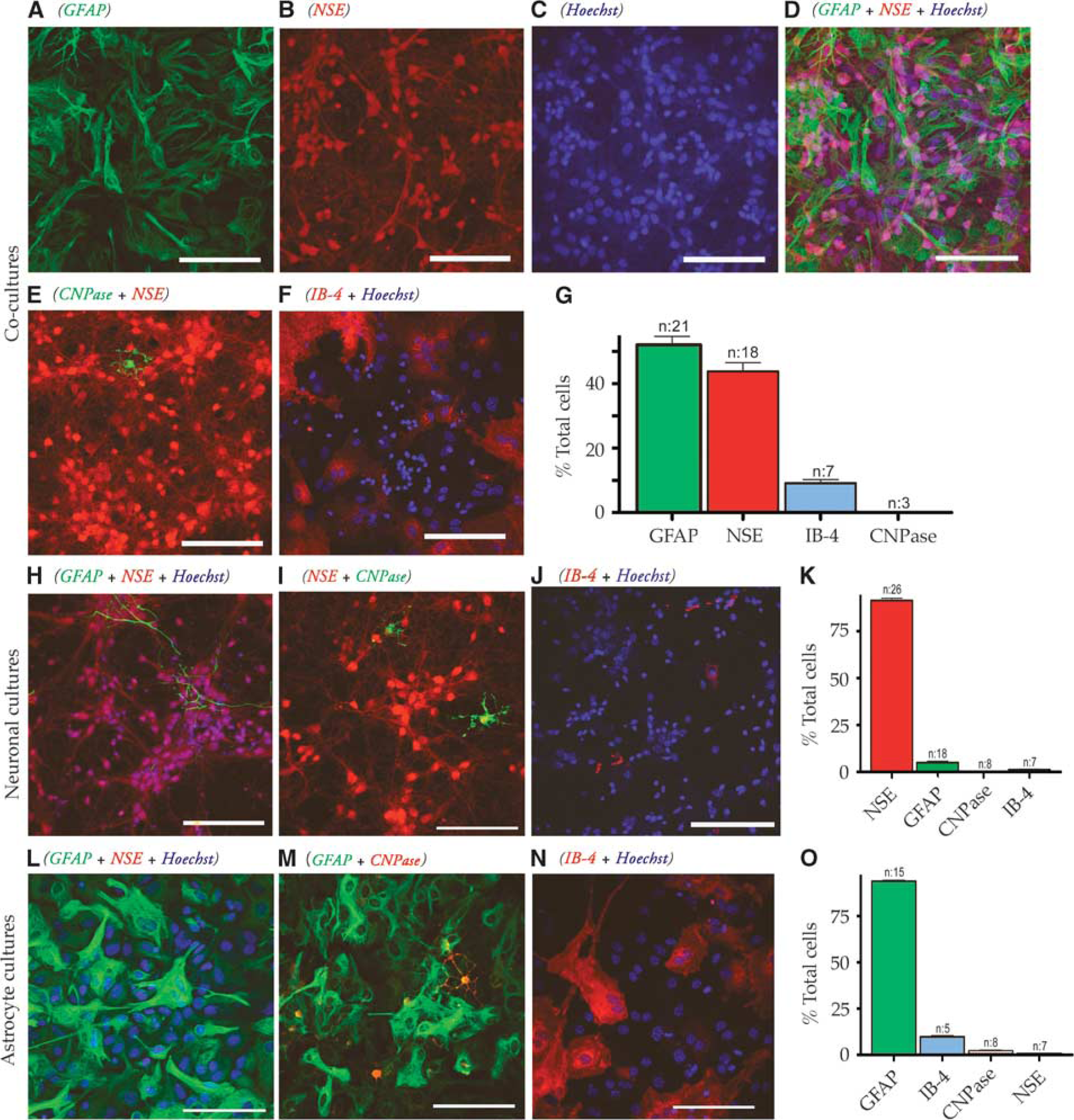
Characterization of high-density cultures (HDCs). (
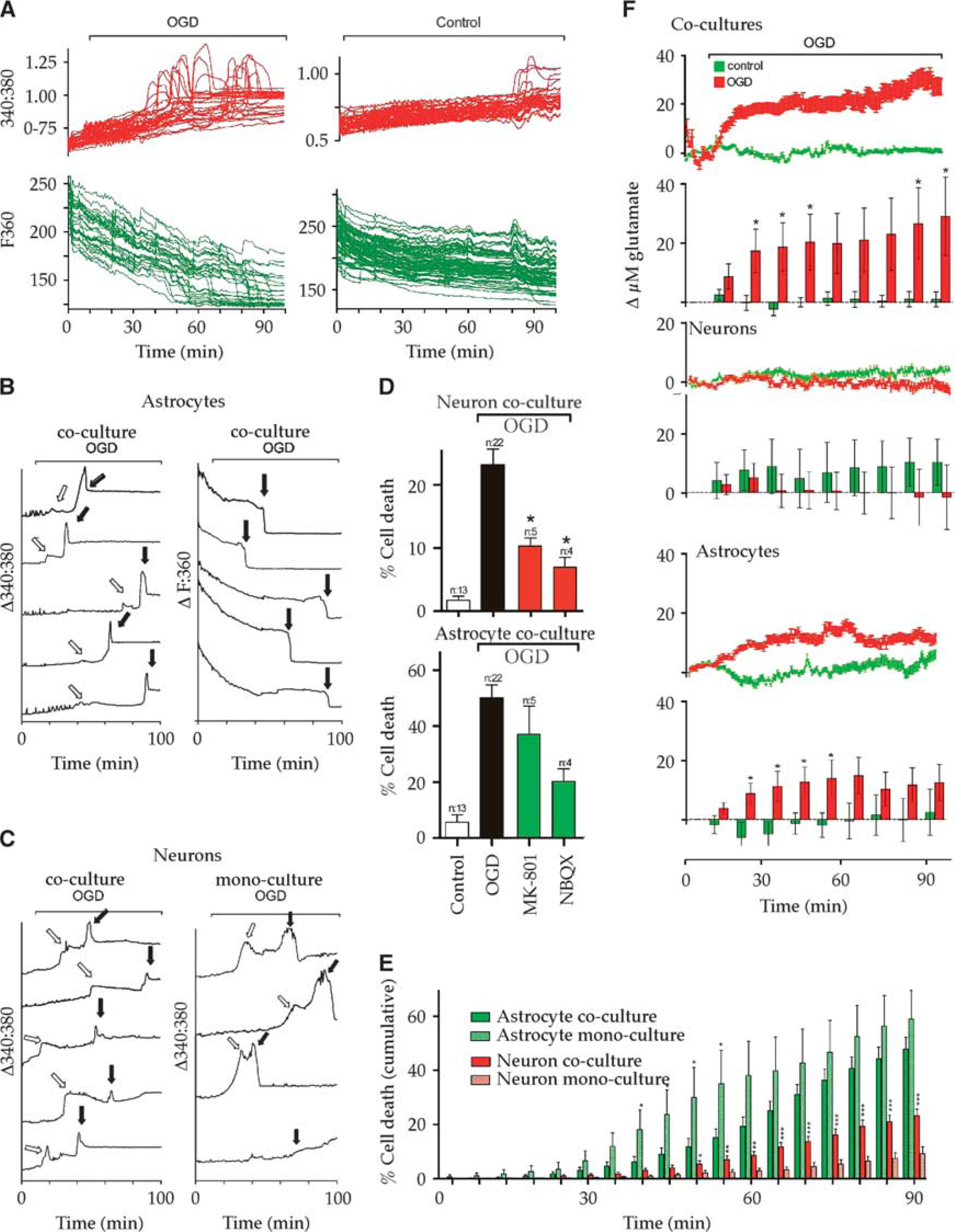
Cellular interactions during ischemia in high-density cultures (HDCs). (
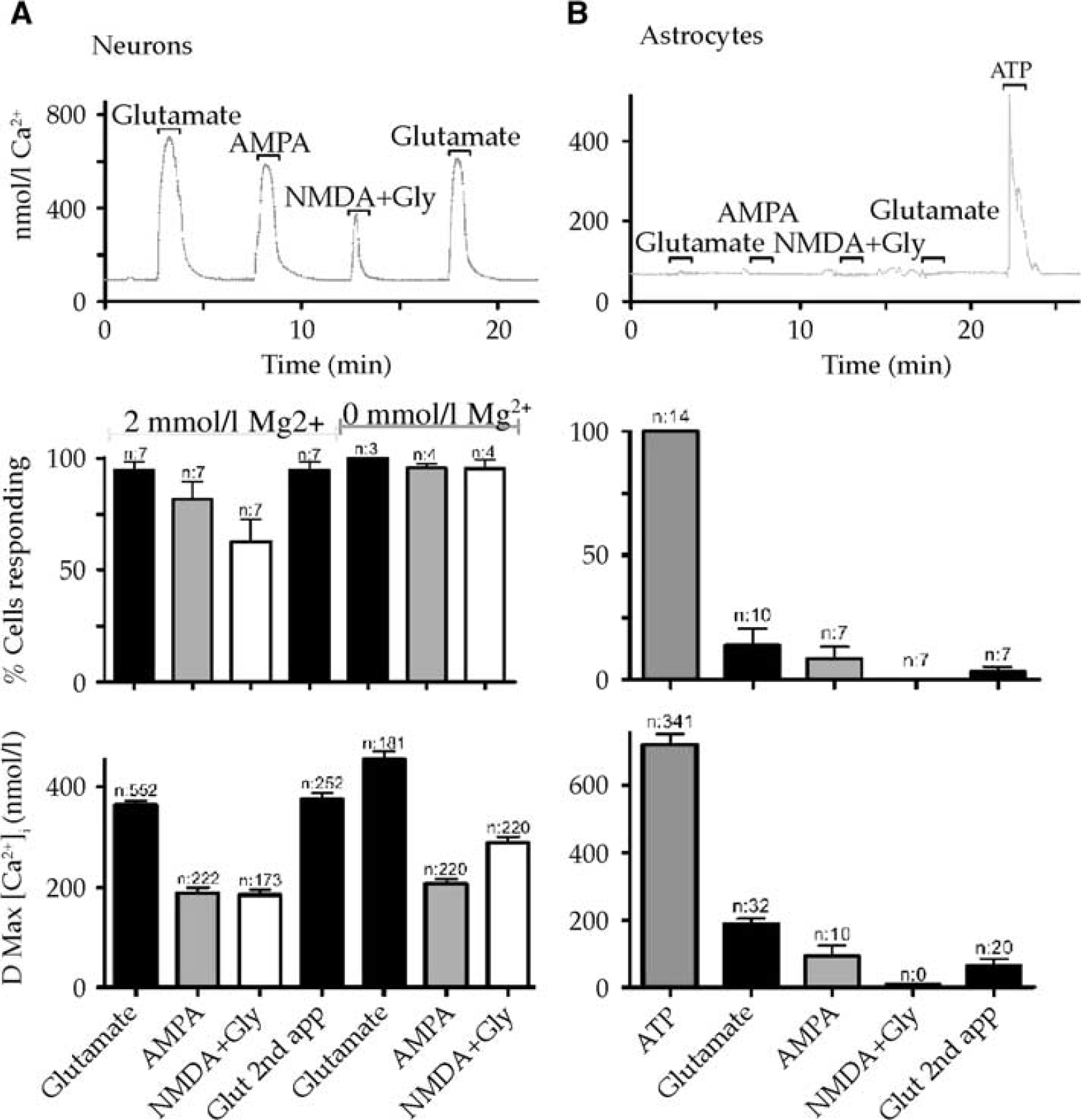
Intracellular Ca2+ ([Ca2+]i) changes evoked in neurons (
Dose-Dependent Effects of Memantine after In Vitro Ischemia
High concentrations of memantine (100 to 300 μmol/l) produced toxicity in normal-density cocultured cells when perfused for 40 to 90 minutes, affecting both astrocytes and neurons (Figures 4A and 4B). Although toxic effects of NMDA receptor antagonists have been noted previously (see Longuemare et al 30 ) and deleterious effects of memantine have been observed in vivo,31, 32, 33 there are no previous reports of toxicity associated with memantine in vitro where vascular factors are eliminated. Memantine toxicity in neural cell cultures was not potentiated by high extracellular [K+] and was not mimicked by exposure to high MK-801 concentrations (100 μmol/l; Supplementary Figure S3) and the underlying mechanisms are not known. However, low concentrations of memantine (0.5 to 2 μmol/l) applied to normal-density cultures replicated the protection seen in HDCs with MK-801 (Figure 4C), although high concentrations (300 μmol/l) retained toxicity under ischemic conditions resulting in a ‘U’-shaped curve. Although findings based on reduced preparations are highly dependent on technical factors, these results show that while low doses of memantine resulted in protection, as a consequence of successfully blocking the excitotoxic injury cascade, higher doses have a neurotoxic effect and significantly increased the amount of cell death.

Protection and toxicity of memantine in normal-density cultures. (
Establishing the Dose-Dependent Effects of Memantine after In Vivo Cerebral Ischemia
Preinfusion for 24 hours with low-dose memantine reduced whole lesion volumes but significance levels were not achieved using the appropriate nonparametric comparison (Figure 5A). Significant reduction in both striatal and striatocortical lesions (examples shown in Figures 5B and 5C) were found in the 0.2-mg/kg memantine infusion group when the two lesion types were separated (Figure 5C). Significance was tested using a parametric analysis of variance post hoc test (Fisher's). Reduced lesion volume was associated with improved behavioral scores at 24 hours after MCAO (Figures 5D–5F) and significantly reduced body weight loss in the striatal lesion group at 24 and 48 hours after MCAO (Figure 5F). Higher memantine concentrations (2 to 10 mg/kg) failed to provide any protection while 20 mg/kg potentiated injury (Figure 6), correlating with the inverse concentration dependence of the protective effect of memantine over the 0.2 to 10 mg/kg range.
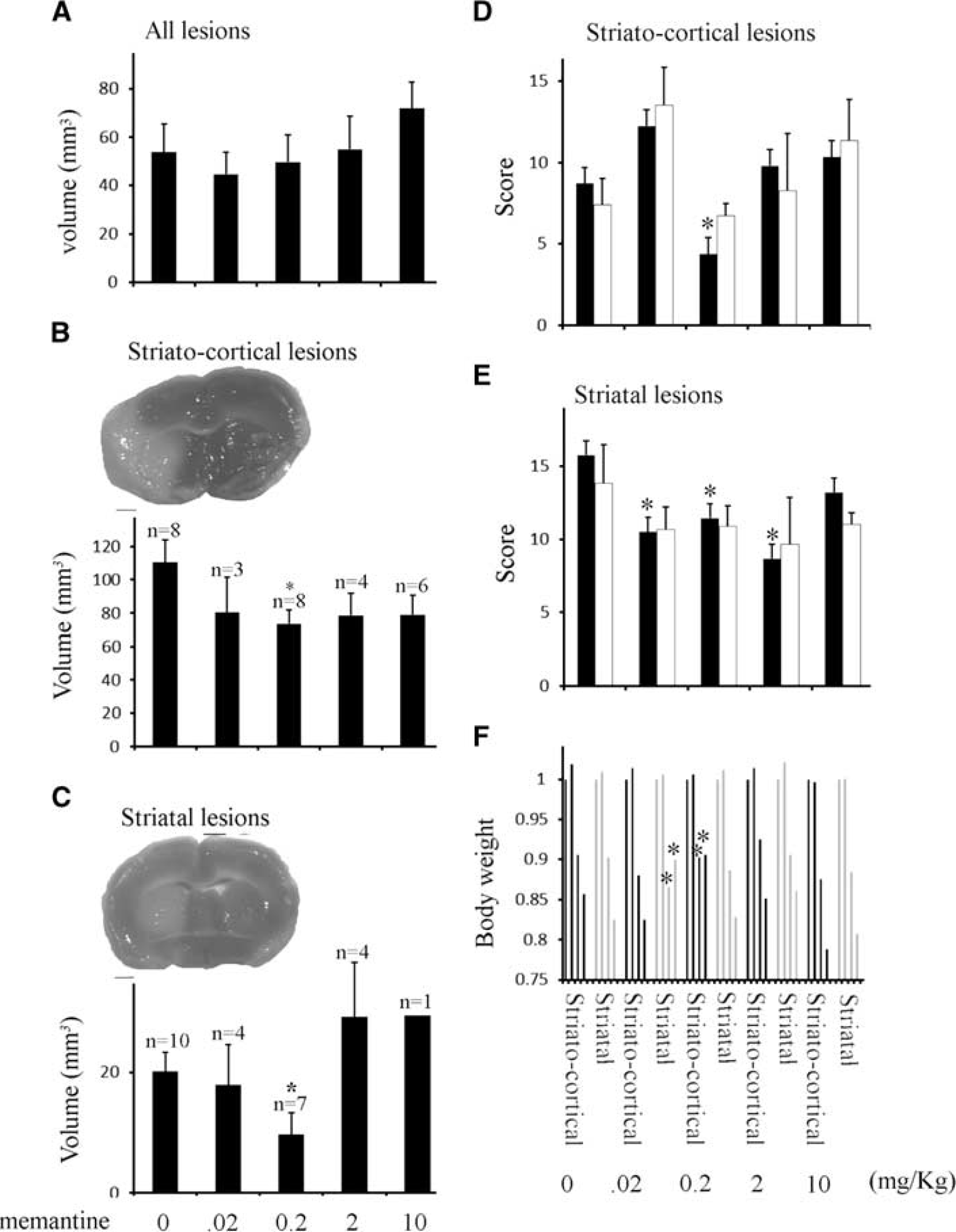
Pretreatment with low doses of memantine are highly protective in both striatal and striatocortical lesions. (

Potentiation of injury at high memantine doses. (
Discussion
In the current study, memantine exerted a dose-dependent effect in both the degree of protection provided against ischemic injury and the degree of toxicity directed against neural cells. A broad range of concentrations/doses were tested, revealing significant protection at levels several orders of magnitude lower than previously tested in in vivo studies of stroke, bringing prophylactic pretreatment for patients at risk of stroke within the therapeutic window. Importantly, this study replicated these dose-dependent effects both in vitro and in vivo. We observed that mice exposed to middle cerebral artery occlusion displayed two distinct patterns of injury, small striatal or large corticostriatal lesions and when lesions were divided into the two subtypes, low doses of memantine reduced the amount of lesion volume present whereas high doses increased it. These results suggest that memantine is an effective neuroprotectant at clinically useful doses while the toxicity found at high concentrations may explain previous variability in the degree of protection reported.
To investigate the effect of memantine in vitro, we developed a HDCs model which allowed acute cell death, [Ca2+]i and extracellular glutamate release to be monitored in real time simultaneously in astrocytes, neurons, and mixed cultures. Memantine applied to normal-density culturs at high concentrations (100 to 300 μM) produced a toxic effect under control artificial cerebrospinal fluid conditions and potentiated the injury produced by OGD. An absence of protection against ischemic conditions at this concentration is found in experiments using isolated rat optic nerve, 10 an effect that is counteracted by coperfusion with a non-NMDA glutamate receptor blocker; while negative effects of higher memantine doses have been noted previously in vivo.31, 32, 33 The toxic effect of memantine may be because of the interaction of the antagonist with the NMDA receptor or at a site distinct from the receptor. To test this, memantine was applied under depolarizing conditions, which did not potentiate toxicity, indicating the toxic effect is not because of the interaction of memantine at the NMDA receptor since actions at this site are highly voltage-dependent. 18 High concentrations of MK-801 (100 μmol/l) were not toxic, confirming that there is no specific toxicity after NMDA receptor blockade in this preparation. Memantine has been shown to interact with various other receptors including the 5-HT and α-nicotinic receptors,34, 35, 36, 37 and toxicity may arise from nonselective effects of this type.
Although higher concentrations of memantine potentiated cell death during 90 minutes of OGD in coculture, significant protection was seen in the presence of lower concentrations (0.5 to 2 μmol/l). The current well-tolerated therapeutic dose for Alzheimer's treatment is 5 to 10 mg/day, with 5 mg/day producing a CSF concentration in patients of ∼0.05 μmol/l, 38 although the effective concentration at the receptor mouth in vivo is estimated to be an order of magnitude higher (Xia et al 2010). Results here show that NMDA receptor inhibition at these memantine concentrations attenuates both astrocyte and neuronal cell death in coculture, while ischemic conditions evoked glutamate release from astrocyte but not neuronal monocultures. Caution must be used when interpreting a negative result of this type, however, it is possible that the lower density of neuronal monocultures resulted in lower levels of glutamate release compared with the denser glial monoculture. Neuronal cell death was potentiated by coculture with astrocytes and was preceded by biphasic cytotoxic [Ca2+]i rises, while exogenous application of glutamate agonists evoked [Ca2+]i rises in neurons with little effect in astrocytes. The findings are consistent with ischemic glutamate release from astrocytes evoking neuronal [Ca2+]i rises and cell death via a glutamate receptor dependent form of excitotoxicity, a cascade interrupted by memantine leading to cell protection. Despite the low incidence and amplitude of NMDA receptor responses in astrocytes, low memantine concentrations protected both cell types; an effect indicative of bidirectional glial-neuronal feedback during ischemic conditions, where neuronal injury may potentiate astrocyte injury, while astrocyte glutamate release triggers neuron cell death.
Antiexcitotoxic drugs were first identified as potential stroke therapies over 30 years ago, but have failed to translate into clinical practice because of dose-dependent complications. 39 Memantine is a moderate affinity noncompetitive NMDA receptor antagonist, binding directly within the pore of the channel in its open configuration.35, 36, 40 Although it displays fast on/off kinetics, it also shows partial trapping on agonist removal. 41 These properties make memantine effective in models of stroke at high (20 mg/kg) concentrations when administered in the acute treatment window;42, 43 20 mg/kg/day infusion produces a CSF concentration in rodents of 0.5 to 1 μmol/l, 18 an order of magnitude higher than that reported in patients receiving a standard 5 mg/day regime. 38 Although there are no data indicating rodent-CSF levels at lower infusion doses, the 0.2 mg/kg/day dose found to be protective in the current experiments is two orders of magnitude lower than the standard 20 mg/kg/day protocol use elsewhere and is likely to correlate to the 0.05 μmol/l levels reported in patients taking 5 to 10 mg/day. That protection was found at doses lower than previously reported and which are likely to correlate to well-tolerated current clinical practice suggests that prophylactic treatment for patients at risk of stroke is feasible. A discrepancy between the low protective concentrations found in vitro (0.5 to 1 μmol/l) and those likely to be present in the CSF after infusion of 0.2 mg/kg/day in vivo may result from the known potentiation of the blocking effects of low memantine concentrations when applied for a long period. 10 The standard 20 mg/kg/day memantine dose used largely for postlesion studies of focal ischemia was found to exacerbate the amount of ischemic damage produced when administered as a pretreatment. A similar reversal of memantine protection at higher concentrations has been reported in models of Alzheimer's disease.44, 45
In the current study, as others have also reported, we observed two distinct patterns of ischemic injury; small striatal lesions and large corticostriatal lesions. Why a standard protocol of focal ischemia should produce one of two distinct injury patterns may relate to variability in vascular anatomy or genetic makeup, or may arise from a normal distribution in the vascular field size producing lesions that encroach from the striatum into the cortex in a proportion of cases, 46 where a separate injury cascade may spread the lesion in an all or none manner throughout a large cortical area. Initiation of spreading depression, for example, may act to propagate injury from a relatively small cortical border zone through the cortical hemisphere.47, 48
This study has showed both the protective and toxic effects of memantine after in vitro and in vivo ischemia. Such dichotomous effects of memantine were dose-dependent. Memantine is licensed and approved for treatment of Alzheimer's disease at low systemic concentrations, which produce few side effects. 45 Because of the strong correlation of increased stroke risk with patient factors including previous stroke or transient ischemic attack, hypertension, age and atrial fibrillation,49, 50, 51, 52 patients at high risk of stroke can be identified. The potentially neuroprotective effect of prophylactically administered low-dose memantine may thus represent a pharmacological intervention of potentially immediate clinical utility for a significant number of patients.
Footnotes
The authors declare no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.