
Abstract
Therapeutic hypothermia can partially reduce long-term death and disability in neonates after hypoxic-ischemic encephalopathy. The aim of this study was to determine whether prolonging the duration of cooling from 3 days to 5 days could further improve outcomes of cerebral ischemia in near-term fetal sheep. Fetal sheep (0.85 gestation) received 30 minutes bilateral carotid artery occlusion followed by 3 days of normothermia (n = 8), 3 days of hypothermia (n = 8), or 5 days of hypothermia (n = 8) started 3 hours after ischemia. Sham controls received sham ischemia followed by normothermia (n = 8). Cerebral ischemia was associated with profound loss of electroencephalography power and spectral edge, with greater and more rapid recovery in both hypothermia groups (P < 0.05). Ischemia was associated with severe loss of neurons in the cortex, hippocampus and thalamus (P < 0.05), with a significant improvement in both hypothermia groups. However, the ischemia-3-day hypothermia group showed greater neuronal survival in the cortex and dentate gyrus compared with ischemia-5-day hypothermia (P < 0.05). Ischemia was associated with induction of iba1-positive microglia, which was attenuated in both hypothermia groups (P < 0.05). Extending the duration of delayed therapeutic hypothermia from 3 to 5 days did not improve outcomes after severe ischemia, and was associated with reduced neuronal survival in some regions.
INTRODUCTION
There is now compelling clinical and experimental evidence that therapeutic hypothermia can reduce neuronal loss and improve neurological outcome after a severe hypoxic–ischemic insult in term infants. 1 Follow-up studies of large randomized trials have shown that infants with hypoxic-ischemic encephalopathy treated with therapeutic hypothermia showed a reduction in death, cerebral palsy and disability, and improved neurocognitive functioning that persisted into middle childhood.1–4 The major challenge for the future is that current protocols for therapeutic hypothermia are partially neuroprotective, with a number needed to treat of eight. 1
In part, partial protection is likely related to the formidable clinical difficulties involved in starting hypothermia within the optimal window of opportunity. 5 A recent study found that asphyxiated infants cooled within 3 hours of birth had better motor outcomes than when hypothermia was started between 3 and 6 hours. 6 However, in a controlled trial, hypothermia was only started in 12% of infants within 4 hours of birth. 7 Preclinical studies suggest that some of the loss of efficacy associated with delayed onset of hypothermia can be salvaged by more prolonged cooling. For example, in adult gerbils, 12 hours of hypothermia initiated 1 hour after global ischemia effectively reduced hippocampal injury after 3 minutes, but not 5 minutes, of global ischemia. 8 However, if the duration of hypothermia was extended for 24 hours, near total preservation of CA1 neurons was seen after 5 minutes of global ischemia. 8 In the near-term fetal sheep, delayed cerebral hypothermia started before the onset of post-ischemic seizures is partially protective, and is associated with moderate secondary microgliosis.9,10
The aim of this study was to determine whether continuing mild cerebral hypothermia for 5 days compared with 3 days, could further improve gray matter injury and recovery of brain activity and correspondingly suppress secondary neural inflammation after global cerebral ischemia in near-term fetal sheep. Hypothermia was started 3 hours after the end of ischemia to represent a realistic clinical delay before treatment.
MATERIALS AND METHODS
Fetal Surgery
All procedures were approved by the Animal Ethics Committee of The University of Auckland under the New Zealand Animal Welfare Act, and the Code of Ethical Conduct for animals in research established by the Ministry of Primary Industries, Government of New Zealand. In brief, 32 time-mated Romney/Suffolk fetal sheep were instrumented using sterile techniques at 118 to 124 days of gestation (term is 145). Food, but not water was withdrawn 18 hours before surgery. Ewes were given long acting oxytetracycline (20 mg/kg, Phoenix Pharm, Auckland, New Zealand) intramuscularly 30 minutes before the start of surgery. Anesthesia was induced by intravenous injection of propofol (5 mg/kg; AstraZeneca, Auckland, New Zealand) and maintained using 2–3% isoflurane in O2. The depth of anesthesia, maternal heart rate, and respiration were constantly monitored by trained anesthetic staff. Ewes received a constant infusion isotonic saline drip (at an infusion rate of ~250 mL/h) to maintain fluid balance.
After a maternal midline abdominal incision, the fetus was exposed and both fetal brachial arteries were catheterized with polyvinyl catheters to measure mean arterial blood pressure. An amniotic catheter was secured to the fetal shoulder. Electrocardiography electrodes (Cooner Wire, Chatsworth, CA, USA) were sewn across the fetal chest to record fetal heart rate. The vertebral–occipital anastomoses were ligated and inflatable carotid occluder cuffs were placed around both carotid arteries.9,11 A 3S Transonic ultrasonic flow probe (Transonic systems, Ithaca, NY, USA) was placed around the right carotid artery. Using a 7 stranded stainless steel wire (AS633–7S's SF; Cooner Wire), two pairs of electroencephalography (EEG) electrodes were placed on the dura over the parasagittal parietal cortex (10 and 20 mm anterior to bregma and 10 mm lateral) and secured with cyanoacrylate glue. A reference electrode was sewn over the occiput. Two electrodes were sewn in the nuchal muscle to record electromyographic activity as a measure of fetal movement. A thermistor was placed over the parasagittal dura 30 mm anterior to bregma to measure extradural temperature and a second thermistor was inserted into the esophagus to measure body temperature. A cooling cap made from silicon tubing (3 × 6 mm, Degania Silicone, Kibbutz Degania Bet, Israel) was secured to the fetal head. The uterus was then closed and antibiotics (80 mg Gentamicin, Pharmacia, and Upjohn; Rydalmere, NSW, Australia) were administered into the amniotic sac. The maternal laparotomy skin incision was infiltrated with 10 mL 0.5% bupivacaine plus adrenaline (AstraZeneca). All fetal catheters and leads were exteriorized through the maternal flank. The maternal long saphenous vein was catheterized to provide access for post-operative maternal care and euthanasia.
Post-Operative Care
Sheep were housed together in separate metabolic cages with access to food and water ad libitum. They were kept in a temperature-controlled room (16°C ± 1°C, humidity 50% ± 10%), in a 12 hour light/dark cycle. Antibiotics were administered daily for 4 days intravenously to the ewe (600 mg benzylpencillin sodium, Novartis, Auckland, New Zealand, and 80 mg gentamicin). Fetal catheters were maintained patent by continuous infusion of heparinized saline (20 U/mL at 0.15 mL/h) and the maternal catheter maintained by daily flushing.
Data Recording
Data recordings began 24 hours before the start of the experiment and continued for the remainder of the experiment. Data were recorded and saved continuously to disk for off-line analysis using custom data acquisition programs (LabView for Windows, National Instruments, Austin, TX, USA). Arterial blood samples were taken for pre-ductal pH, blood gas, base excess (Ciba-Corning Diagnostics 845 blood gas analyzer and co-oximeter, Massachusetts, USA), glucose, and lactate measurements (YSI model 2300, Yellow Springs, OH, USA). All fetuses had normal biochemical variables for their gestational ages.12,13
Experimental Protocols
At 128 ± 1 day of gestation, ischemia was induced by reversible inflation of the carotid occluder cuffs with sterile saline for 30 minutes. Successful occlusion was confirmed by the onset of an isoelectric EEG signal within 30 seconds of inflation. The carotid occluder cuffs were not inflated in sham control experiments. Fetal blood samples were drawn just before the occlusion and 2, 4, and 6 hours after occlusion followed by daily sampling for the remainder of the experiment.
Fetuses were randomized to ischemia–normothermia (n = 8), ischemia-3-day hypothermia (n = 8), ischemia-5-day hypothermia (n = 8), or sham control (n = 8) groups. Cooling was started 3 hours after reperfusion and continued until 72 or 120 hours after the onset of ischemia in the ischemia-3-day hypothermia and ischemia-5-day hypothermia groups, respectively. Cooling was performed by linking the cooling coil over the fetal scalp with a pump in a cooled water bath and circulating cold water through the cooling coil. The target extradural temperature was between 31°C and 33°C. In the ischemia–normothermia and sham control groups, the water was not circulated and the cooling coil remained in equilibrium with fetal temperature.
Data Analysis
The total EEG power was log transformed [dB, 20 times log (power)], as this transformation gives a better approximation of the normal distribution, and then normalized about the 24-hour baseline period. Spectral edge is a measure of the relative frequency of the EEG, and was defined as the frequency below which 90% of total EEG power was present. Impedance increases as the temperature of the medium, through which the signal passes, falls. Therefore, in each fetus, the slope of impedance change at the onset of hypothermia was used to correct the impedance signal: corrected impedance = impedance – (slope × Δtemperature).9,14 Sleep state cycling was defined on the 1-minute-averaged EEG data as a repetitive alternating pattern of high and low voltage activity, with each phase lasting ~20 minutes. The day on which sleep state cycling resumed was determined visually by a masked observer and analyzed by Fisher exact test. Animals that did not resume sleep state cycling by the end of the experiment were assigned a value of 7 days. As these data were not normally distributed, a non-parametric independent samples median test was used for statistical analysis. Other data were analyzed using analysis of variance (ANOVA) or repeated measures ANOVA, followed by the Tukey post-hoc test when a significant difference was found. Statistical significance was accepted when P < 0.05.
Immunohistochemistry
Fetal brains were perfusion fixed with 10% phosphate-buffered formalin. Coronal slices (10 μm thick) were cut using a microtome (Leica Jung RM2035, Leica Microsystems Pty Ltd, North Ryde, NSW, Australia) starting at the level of the dorsal hippocampus. Slides were dewaxed in xylene and rehydrated in decreasing concentrations of ethanol, then washed in 0.1 mol/L phosphate-buffered saline. Antigen retrieval was performed using the citrate buffer boil method followed by incubation in 1% H2O2 in methanol to block endogenous peroxidase activity. Blocking was performed in 3% normal goat serum for NeuN and normal horse serum) with 0.1% Triton-X100 (Scharlau Chemie, Sentmenat, Spain) for Iba1 for 1 hour at room temperature. Sections were labeled with 1:400 mouse antineuronal nuclei monoclonal antibody (NeuN, Chemicon International, Temecula, CA, USA) and 1:400 or 1:200 anti-Iba1 (Abcam, Cambridge, England) overnight at 4°C. Sections were incubated for 3 hours in biotinconjugated 1:200 anti-mouse (NeuN) or 1:200 anti-goat (Iba1, Vector Laboratories, Burlingame, CA, USA) in 3% normal goat serum for NeuN and 3% normal horse serum for Iba1. Slides were then incubated in ExtrAvidin (1:200, Sigma-Aldrich, St Louis, MO, USA) in phosphate-buffered saline for 2 hours at room temperature and then reacted in diaminobenzidine tetrachloride (Sigma-Aldrich). The reaction was stopped by washing in phosphate-buffered saline and the sections dehydrated and mounted.
Neurons in the cortex of the first and second parasagittal gyri, CA1, CA3, CA4, and dentate gyrus of the hippocampus, thalamus, caudate nucleus, and putamen, respectively, were imaged using light microscopy (Nikon eclipse 80i, Scitech, Preston, VIC, Australia) at ×20 magnification by an investigator masked to the treatment group by separate coding of the slides. Normal-appearing NeuN-positive neurons were identified morphologically by the presence of typical nuclei; cells showing condensed nuclei or fragmented appearance were not counted. 15 Microglial number was assessed using light microscopy at ×40 magnification in the cortex of the first parasagittal gyrus. Microglia showing either an amoeboid or ramified morphology were included.
RESULTS
Blood Gas, Glucose, and Lactate Measurements
There were no significant differences in baseline blood gas, pH, glucose, or lactate measurements between groups (Table 1). There was a significant increase in lactate concentration after ischemia in all groups.
Blood gas, pH, glucose, and lactate data before and after 30 minutes of cerebral ischemia
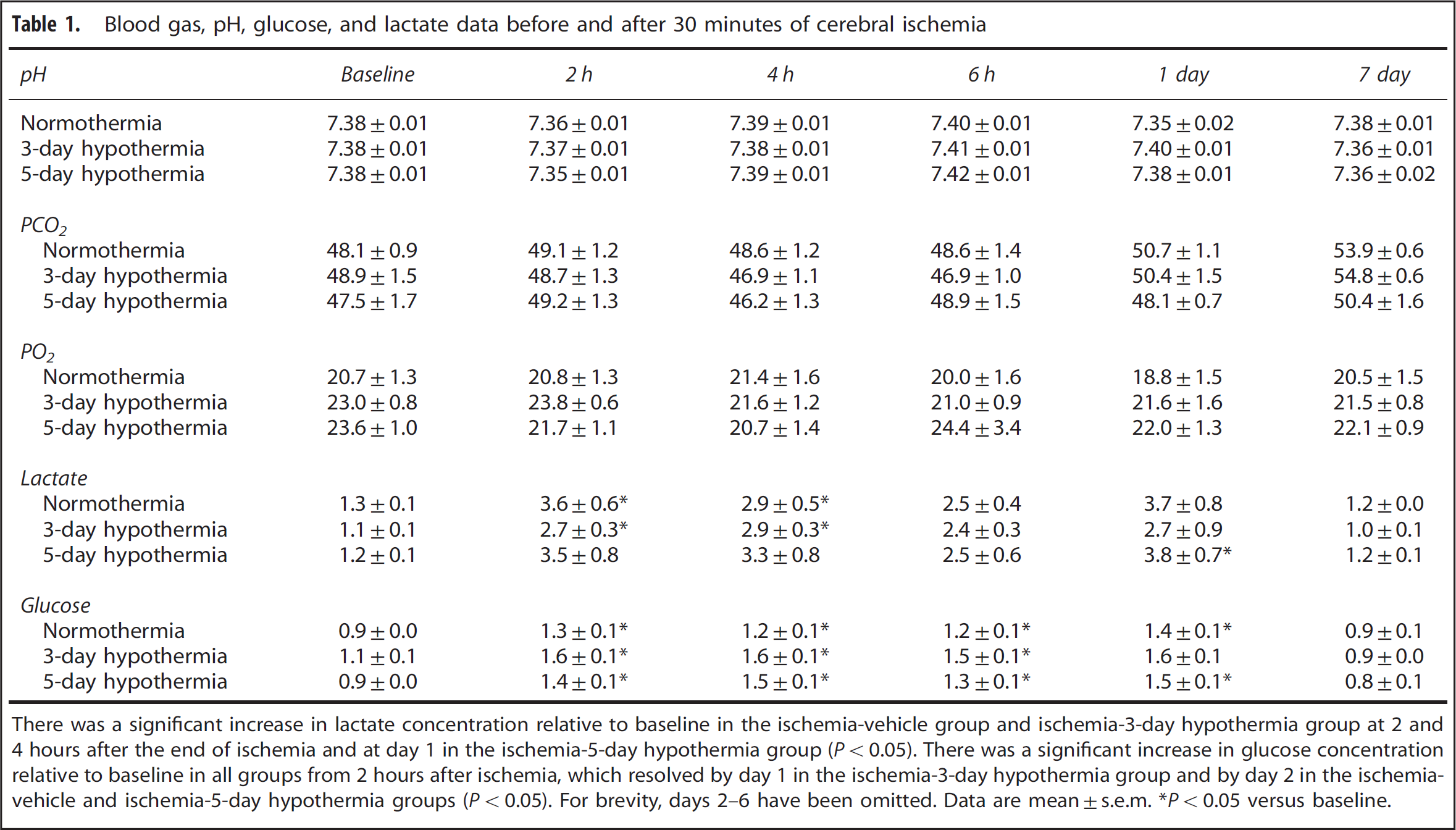
There was a significant increase in lactate concentration relative to baseline in the ischemia-vehicle group and ischemia-3-day hypothermia group at 2 and 4 hours after the end of ischemia and at day 1 in the ischemia-5-day hypothermia group (P<0.05). There was a significant increase in glucose concentration relative to baseline in all groups from 2 hours after ischemia, which resolved by day 1 in the ischemia-3-day hypothermia group and by day 2 in the ischemia-vehicle and ischemia-5-day hypothermia groups (P<0.05). For brevity, days 2–6 have been omitted. Data are mean±s.e.m. ∗P<0.05 versus baseline.
Cardiovascular and Temperature Changes
At the onset of hypothermia, extradural temperature fell to 31.3° C ± 0.2°C in the ischemia-3-day hypothermia group and 31.8° C ± 0.3° C in the ischemia-5-day hypothermia group compared with 39.5° C ± 0.1° C in the ischemia–normothermia group (P < 0.05, Figure 1). Esophageal temperature fell to between 37° C and 38° C during the treatment period (P < 0.05, Figure 1).

Changes in mean arterial blood pressure (MAP), carotid blood flow (CBF), and extradural and esophageal temperature before, during and after 30 minutes of global cerebral ischemia in the ischemia-normothermia, ischemia-3-day, and ischemia-5-day hypothermia groups. There were no significant differences in MAP at any time between groups (P > 0.05). A significant increase in CBF was seen in the ischemia–normothermia group between 6 and 48 hours after ischemia compared with either hypothermia group (P < 0.05). At the onset of hypothermia (3 hours after ischemia), extradural temperature was significantly reduced in the ischemia-3-day hypothermia group and the ischemia-5-day hypothermia group compared with the ischemia–normothermia group (P < 0.05). Extradural temperature returned to baseline after the end of the period of hypothermia at 72 hours or 120 hours in the ischemia-3-day hypothermia and ischemia-5-day hypothermia groups, respectively. A significant reduction in esophageal temperature was seen with the onset of hypothermia, with temperature remaining stable during the treatment period (P < 0.05). Data are represented as mean ± s.e.m., *P < 0.05 ischemia-3-day hypothermia versus ischemia–normothermia group. #P < 0.05 ischemia-5-day hypothermia versus ischemia-normothermia.
Brain Activity
Ischemia was associated with rapid, profound suppression of EEG power (Figure 2). After release of the occlusion, activity remained suppressed until the onset of a period of intense seizure activity from ~ 8 to 48 hours after ischemia. After seizures resolved, total EEG power fell to below baseline levels in the ischemia–normothermia group. In contrast, both the 3-day and 5-day hypothermia groups showed substantially greater recovery of EEG power than ischemia–normothermia from 30 hours until the end of the experiment (P < 0.05). Ischemia was also associated with rapid suppression of spectral edge, which remained below baseline until the end of the experiment in the ischemia– normothermia group. In contrast, both the ischemia-3-day and ischemia-5-day hypothermia groups showed significantly greater spectral edge from 64 hours until the end of the experiment (Figure 2, P < 0.05).
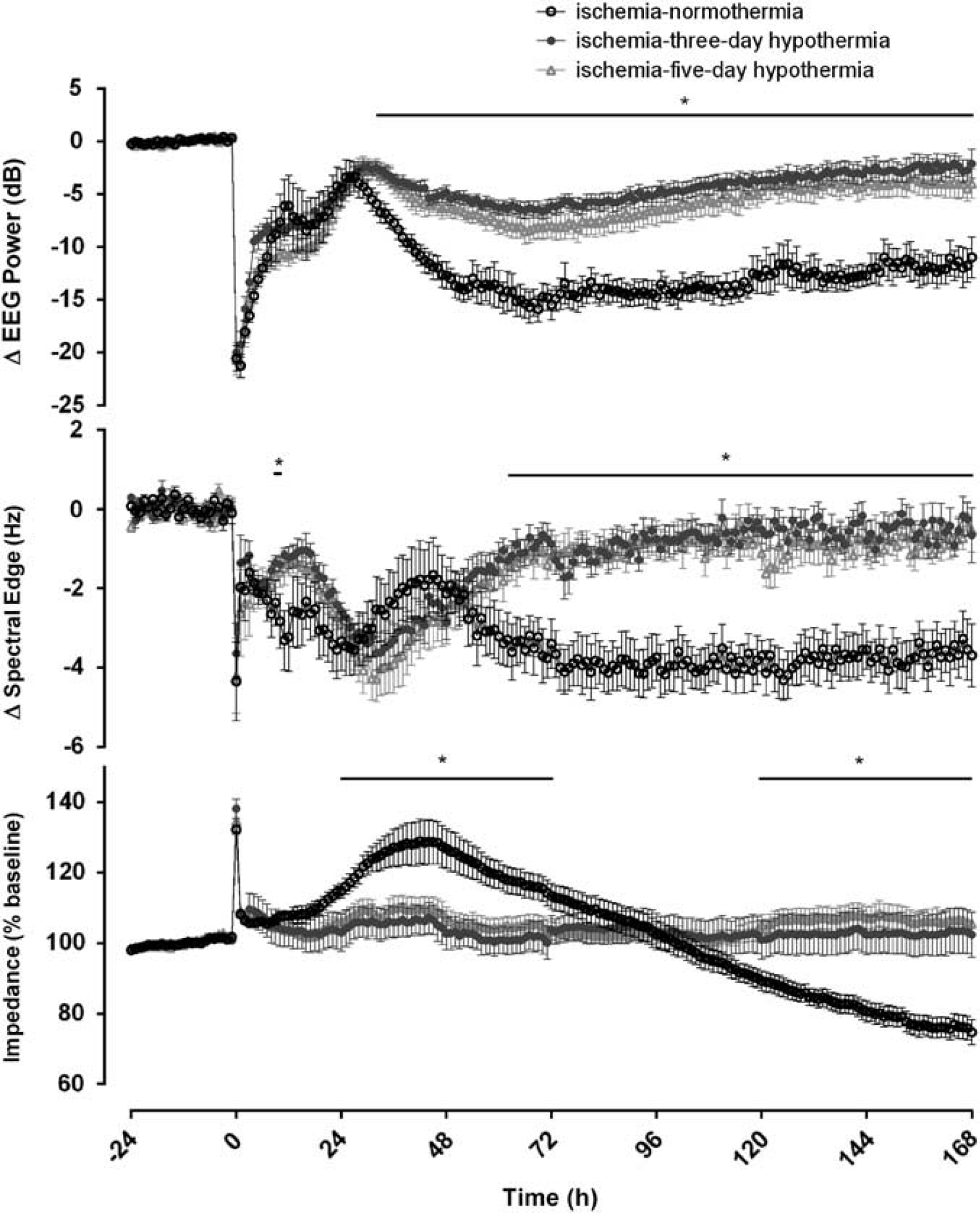
Changes in electroencephalography (EEG) activity, spectral edge frequency and impedance before, during and after 30 minutes of global cerebral ischemia in the ischemia–normothermia, ischemia-3-day and ischemia-5-day hypothermia groups. Time point zero denotes the start of ischemia. Electroencephalography activity was suppressed in all groups during ischemia. A rebound in EEG activity was seen during the seizure period between 8 and 48 hours. Electroencephalography activity in the ischemia–normothermia group was reduced for the remainder of the experiment, while both hypothermia groups showed a significant recovery of EEG power from 60 hours onwards (P < 0.05). Spectral edge was suppressed in all groups during ischemia and remained suppressed until the end of the experiment in the ischemia–normothermia group. A significant increase in spectral edge was seen in both hypothermia groups between 8 and 12 hours and from 64 hours onwards compared with the ischemia–normothermia group (P < 0.05). Ischemia was associated with an increase in impedance in all groups, which resolved after the end of ischemia. A significant rise in impedance was seen between 24 and 72 hours in the ischemia–normothermia group compared with the hypothermia groups followed by a significant reduction in impedance in the ischemia–normothermia group from 120 hours until the end of the experiment (P < 0.05). Data are represented as mean ± s.e.m., *P < 0.05 versus ischemia–normothermia group. *P < 0.05 vs sham control. #P < 0.05 vs ischemia-3-day hypothermia. +P < 0.05 vs ischemia-5-day hypothermia.
The ischemia–normothermia group showed a secondary rise in cortical impedance from 24 and 72 hours after ischemia compared with the ischemia-3-day and ischemia-5-day hypothermia groups (P < 0.05). Impedance fell below baseline levels from 72 hours onwards in the ischemia–normothermia group, and was significantly lower than either hypothermia group from 120 hours until the end of the experiment (P < 0.05).
Clearly defined sleep state cycling resumed by day 7 in four or eight animals in the ischemia–normothermia group, whereas it recovered in all animals in both hypothermia groups (P < 0.05, Fisher exact test). Within fetuses that resumed sleep state cycling, the average time until return of sleep state cycling was longer (5.6 ± 0.5 days (P < 0.05)) in the ischemia– normothermia group than either ischemia-3-day hypothermia (2.8 ± 0.5 days) or ischemia-5-day hypothermia (3.1 ± 0.3 days) groups.
Physiological Parameters
A significant increase in carotid artery blood flow was seen in the ischemia–normothermia group between 6 and 48 hours after ischemia, which was suppressed in both hypothermia groups (Figure 1, P < 0.05). There were no significant differences in mean arterial pressure (Figure 1), fetal heart rate, or nuchal electromyography between groups (data not shown).
Immunohistochemistry
Neuronal survival was decreased in the cortex, CA1, CA3, CA4, and dentate gyrus of the hippocampus as well as the thalamus, but not the caudate nucleus and putamen, in the ischemia–normothermia group compared with sham controls (P < 0.05 Figures 3, 4 and 5). Neuronal survival was significantly improved overall in both hypothermia groups. However, neuronal survival was significantly less in the cortex and dentate gyrus in the ischemia-5-day hypothermia group than after ischemia-3-day hypothermia (P < 0.05).

Neuronal cell count in the cortex, CA1, CA3, CA4, and dentate gyrus, thalamus, caudate nucleus, and putamen in the ischemia–normothermia, ischemia-3-day hypothermia, and ischemia-5-day hypothermia groups 7 days after 30 minutes of global cerebral ischemia. A significant decrease in neuronal number was seen in all regions except the caudate nucleus and putamen, in the ischemia–normothermia group compared with sham control (P < 0.05). Neuronal number was significantly higher in the cortex, CA1, CA3, CA4, and dentate gyrus, caudate nucleus and thalamus in the ischemia-3-day hypothermia group compared with the ischemia–normothermia group (P < 0.05). Neuronal number was significantly higher in the cortex, CA4, and thalamus in the ischemia-5-day hypothermia group compared with the ischemia–normothermia group (P < 0.05). Neuronal number was significantly lower in the cortex, CA1, CA3 and dentate gyrus in the ischemia-5-day hypothermia group and in the CA1 in the ischemia-3-day hypothermia group compared with sham control (P < 0.05). Neuronal number was significantly lower in the cortex and dentate gyrus in the ischemia-5-day hypothermia group compared with the ischemia-3-day hypothermia group (P < 0.05). *P < 0.05 vs sham control. #P < 0.05 vs ischemia-3-day hypothermia. +P < 0.05 vs ischemia-5-day hypothermia.
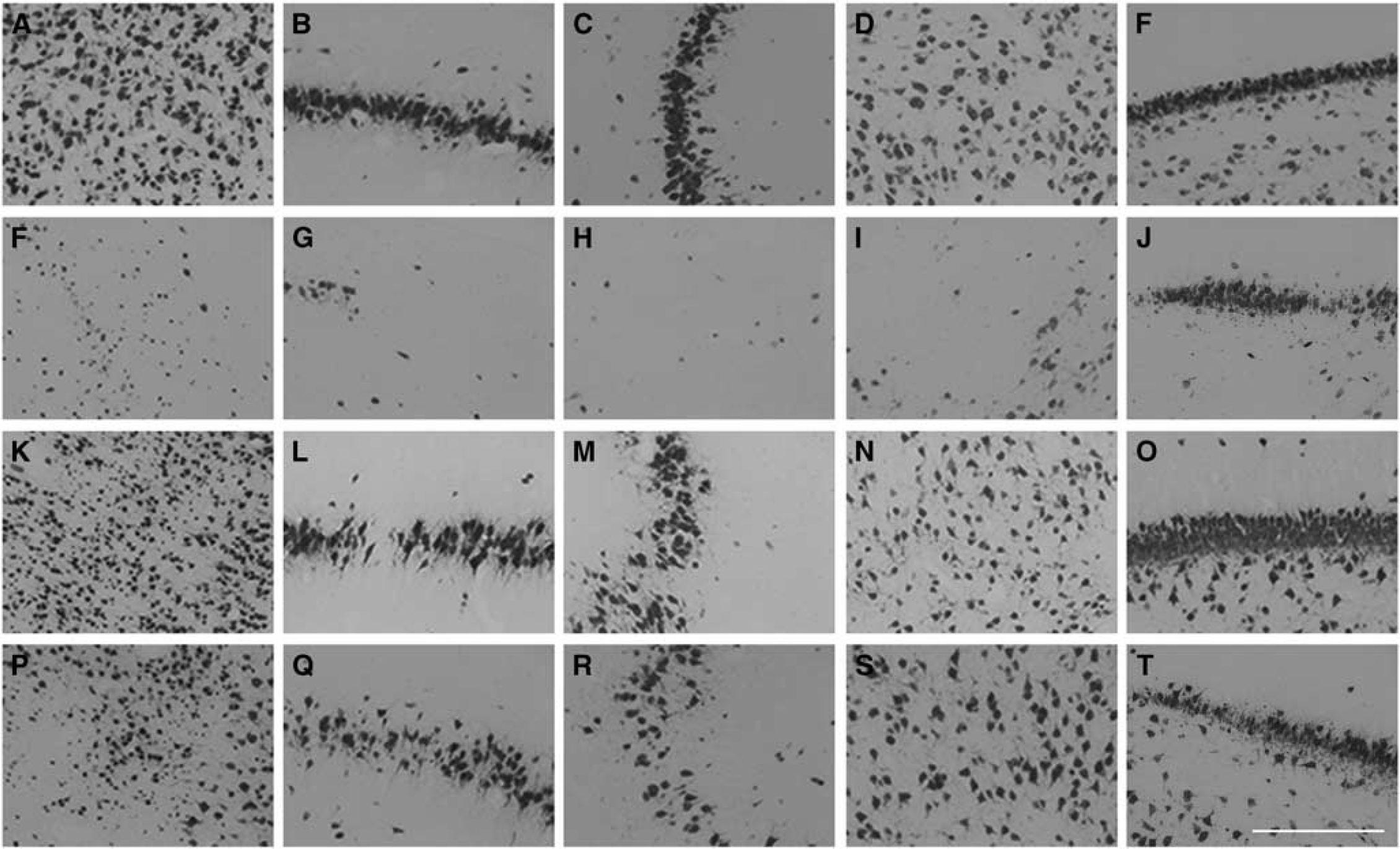
Photomicrograph of neuronal survival in the cortex (
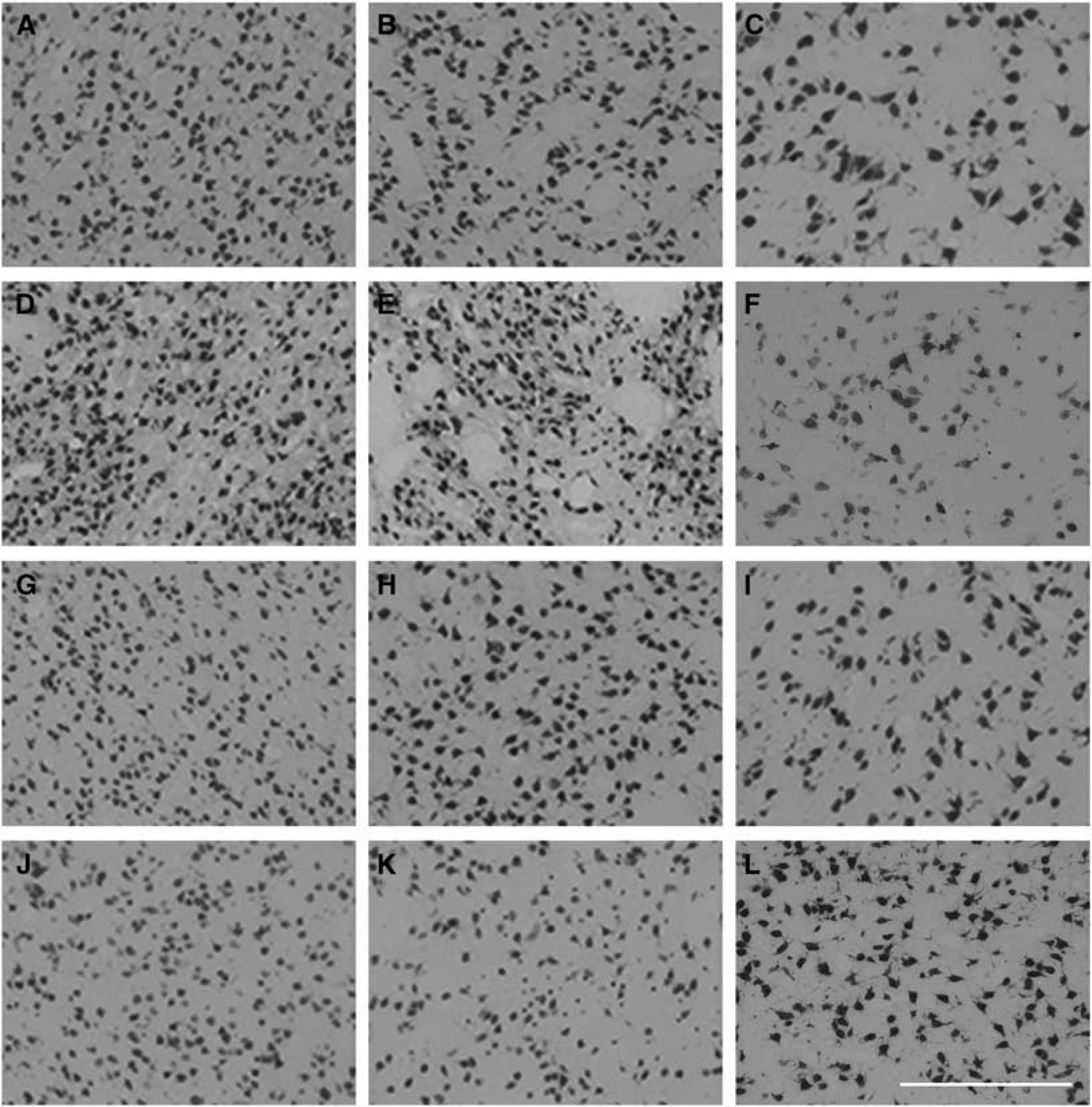
Photomicrograph of neuronal survival in the caudate nucleus (
A significant increase in patchy Iba1-positive microglial staining was seen in the cortex of the first parasagittal gyrus in the ischemia-normothermia group compared with sham control (P < 0.05, Figure 6). This was significantly reduced in both the ischemia-3-day and 5-day hypothermia groups (P < 0.05), which were not significantly different to sham control.

Change in Iba1-positive microglial number in the cortex of the first parasagittal gyrus in the sham control (
DISCUSSION
This study has demonstrated that therapeutic hypothermia started 3 hours after the end of 30 minutes of global cerebral ischemia was associated with significantly improved neuronal survival, reduced microglial activation and improved recovery of EEG power, and sleep state cycling, after a 7-day recovery from ischemia. Prolonging the period of hypothermia from 72 to 120 hours had no additional beneficial effect in any region, and indeed was associated with reduced neuronal survival in the cortex and dentate gyrus and no greater suppression of microglial activation. These findings are consistent with a recent clinical trial of optimizing cooling strategies that found that cooling for 120 hours was unlikely to be superior to cooling for 72 hours at 33.5°C, and might be deleterious, and so this trial was stopped early. 16
Recovery of EEG activity and spectral edge was significantly improved in the ischemia-3-day and ischemia-5-day hypothermia groups compared with the ischemia–normothermia group. Hypothermia was associated with improved EEG power from 30 hours onwards, and greater spectral edge frequency from 64 hours after ischemia. These quantitative changes were in turn associated with significantly earlier and more consistent recovery of sleep state cycling compared with normothermia animals. This is highly consistent with clinical evidence that in infants with moderate to severe HIE, more rapid recovery of sleep wake cycling within the first 36 to 48 hours of life is associated with a significantly better outcome.17,18 Interestingly, in contrast with the present study, there is evidence that hypothermia may delay but did not prevent recovery of sleep state cycling in infants with favorable outcomes. 19 Speculatively, this apparent difference may reflect the severity of cortical neuronal loss in our paradigm or the use of sedatives and anticonvulsants in clinical practice.
Further, hypothermia almost completely abolished the secondary rise in impedance, reflective of cell swelling. 20 Of interest, hypothermia also attenuated the late fall in impedance after 72 hours in normothermia. It is likely that this late reduction of impedance below baseline levels reflects cell loss, reducing impedance to flow of current through the extracellular space, consistent with the greater cell loss seen in the ischemia–normothermia group. 11
In contrast, in the present study, neuronal survival was less in the 5-day hypothermia group than the 3-day group in the parasagittal cortex and the dentate gyrus of the hippocampus. The specific mechanisms are unknown but speculatively may involve the delayed restoration of the normal cellular environment and homeostasis. These mechanisms do not seem to involve suppression of prolonged inflammation, as cortical microglial activation at 7 days was similarly attenuated in both hypothermia-treated groups compared with the ischemia-normothermia group.
Finally, it is unlikely that the suboptimal effect of cooling for 5 days reflects direct changes in neuronal activity, given the similar recovery of EEG activity during both 3 and 5 days of hypothermia in the present study. Consistent with this, there is reassuring evidence in adult rats exposed to ischemia induced by occlusion of two vessels, that even prolonged hypothermia applied between 2 and 7 days was not associated with impaired neurogenesis or synapse formation. 21
This study has shown that extending the duration of delayed cooling after cerebral ischemia, from 3 to 5 days, was not associated with any additional benefit, and indeed reduced neuronal survival in some regions. It may be that the immediate focus for improving the outcome of clinical treatment should be to refine clinical protocols to encourage earlier initiation of treatment.
AUTHOR CONTRIBUTIONS
JOD, LB, and AJG conceptualized and designed the study. JOD and GW undertook experiments and analyzed the data. CAY and FGZ undertook immunohistochemistry, cell quantification, analysis, and preparation of figures. AJG provided overall oversight of the research. All authors critically reviewed the manuscript and approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.