
Abstract
Repeated mild traumatic brain injury (mTBI) can cause sustained cognitive and psychiatric changes, as well as neurodegeneration, but the underlying mechanisms remain unclear. We examined histologic, neurophysiological, and cognitive changes after single or repeated (three injuries) mTBI using the rat lateral fluid percussion (LFP) model. Repeated mTBI caused substantial neuronal cell loss and significantly increased numbers of activated microglia in both ipsilateral and contralateral hippocampus on post-injury day (PID) 28. Long-term potentiation (LTP) could not be induced on PID 28 after repeated mTBI in ex vivo hippocampal slices from either hemisphere. N-Methyl-D-aspartate (NMDA) receptor-mediated responses were significantly attenuated after repeated mTBI, with no significant changes in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor-mediated responses. Long-term potentiation was elicited in slices after single mTBI, with potentiation significantly increased in ipsilateral versus contralateral hippocampus. After repeated mTBI, rats displayed cognitive impairments in the Morris water maze (MWM) and novel object recognition (NOR) tests. Thus, repeated mTBI causes deficits in the hippocampal function and changes in excitatory synaptic neurotransmission, which are associated with chronic neuroinflammation and neurodegeneration.
INTRODUCTION
Mild traumatic brain injuries (mTBI) comprise more than 75% of the 1.7 million cases of head injury reported in the US annually. 1 There are growing concerns about the consequences of repeated mTBI, especially among athletes and military personnel.2,3 Repeated mTBI may be associated with cognitive and psychiatric alterations, such as impaired memory and disorders of behavior and mood, as well as neurodegeneration; more substantial disabilities and neurodegeneration have been reported in some individuals, a condition termed chronic traumatic encephalopathy.2,3 It is hypothesized that the first mTBI places the brain in a vulnerable state that is more susceptible to damage from subsequent injuries.46 However, the nature of the changes produced by a single mTBI remains insufficiently characterized.
Certain animal models of traumatic brain injury (TBI) produce cognitive impairments that share features with clinical head injury.7,8 For example, the lateral fluid percussion (LFP) moderate injury model produces ipsilateral cell loss in the hippocampus, altered hippocampal excitability, and deficits in long-term potentiation (LTP),9–12 suggesting that changes in synaptic plasticity contribute to long-term cognitive deficits. More recently, it has been observed that experimentally induced repeated mTBI produces ipsilateral cortical neuroinflammation and lasting deficits in learning and memory.4,5 Whether the contralateral hemisphere and subcortical structures are also affected remains unknown, as do the molecular and physiologic consequences.
We demonstrated previously that moderate LFP in rats induces secondary injury mechanisms causing neuronal cell loss and microglial activation that continue over weeks to months after the injury in the ipsilateral cortex and hippocampus.13,14 Limiting posttraumatic microglial activation in animal studies reduces not only the progressive neurodegeneration, but also associated cognitive deficits. 13 Whether mTBI also produces contralateral and subcortical neuroinflammation is unknown.
Based on our previous characterization studies 15 using the rat LFP model, we determined a level of mTBI that causes modest behavioral impairment, when compared with moderate TBI, which causes significant behavioral dysfunction coupled with progressive neuronal cell loss, and chronic microglial and astrocyte activation.13,15 And others have shown significant TBI-induced changes in neurotransmission and synaptic plasticity across hippocampal subregions using experimental models of more severe TBI.9–11 In the present study, we demonstrated that repeated mTBI using the rat LFP model produced similar functional and histologic changes as a single moderate injury—including impaired cognitive performance, neuroinflammation, and neurodegeneration. Bilateral changes were found in both hippocampal excitatory synaptic transmission and LTP after mTBI. Moreover, we demonstrated that repeated mTBI causes significantly more neuroinflammation than a single mTBI, and is associated with significantly greater hippocampal neurodegeneration and cognitive deficits. Together, these observations suggest that repeated mTBI may result in part from increased secondary brain injury and associated neurologic deficits by increasing delayed inflammatory responses.
MATERIALS AND METHODS
Lateral Fluid Percussion Injury
All surgical procedures and experiments were approved by and carried out in accordance with protocols approved by the University of Maryland Animal Care and Use Committee. Adult male Sprague–Dawley rats (10 to 12 weeks old; 310 to 330 g) (Harlan Laboratories, Indianapolis, IN, USA) were used. Based on previous studies, we determined that mild injury corresponded to a pressure pulse of 1.1 to 1.4 atmospheres (atm), producing modest behavioral impairment in contrast to moderate TBI using the LFP model, which causes significant behavioral impairments.13,15 Rats were subjected to single mild, repeated mild, or sham surgeries. Repeated mild groups received injury on days 0, 2, and 4. Single mild and sham groups were injured on day 4. Rats were anesthetized with isoflurane (4% induction; 2% to 2.5% maintenance; supply gas mixture: 70% compressed air + 30% oxygen) and immobilized in a stereotaxic frame. The scalp was incised, temporal muscles were reflected, and a small craniotomy (4 mm) was made midway between lambda and bregma sutures over the left parietal cortex. An adapter was cemented over the craniotomy and attached to our custom-built microprocessor-driven fluid percussion device, which produces a predetermined and adjusted pressure pulse causing deformation of the underlying brain. Sham animals received craniotomy and anesthesia but no injury. 15 Rats that received repeated mild LFP on days 2 and 4 were anesthetized as described above, the adapter was reattached to the previously craniotomized site, and the injury was induced at a pressure of 1.1 to 1.4 atm.
Composite Neurologic Scores
Chronic sensorimotor recovery was assessed using the composite neurologic scoring paradigm (n = 10 to 11/group), previously described. 15 The composite neuroscore reflects a combination of individually scored tests, using an ordinal scale ranging from zero (severe impairment) to five (normal function). The total composite neurologic score (0 to 35) was obtained by combining the scores of the following tests: (i) lateral pulsion (left and right), (ii) forelimb flexion (left and right), and (iii) inclined plane (two vertical and two horizontal positions for assessing maximum angle at which the animal can stand for 5 seconds; scoring: >50° = 5, 45° to 50° = 4,40° to 45° = 3, 35° to 40° = 2, 30° to 35° = 1, and <29° = 0). 15 Rats were tested on post-injury days (PIDs) 1, 7, 14, 21, and 28 days.
Morris Water Maze
Morris water maze was recorded by Any-Maze automated video tracking system (Stoelting, Wood Dale, IL, USA). The circular tank (180 cm in diameter) was filled with water (23 ± 2°C) made opaque with white Crayola non-toxic paint and surrounded by various extra-maze cues on the wall of the room. A transparent platform (10 cm in diameter) was submerged 0.5 cm below the surface of the opaque water. Spatial learning and memory were assessed using the acquisition paradigm of Morris water maze (MWM) on PID 14, 15, 16, and 17, as previously described.13,15,16 Rats underwent four trials per day and were allowed a maximum of 90 seconds(s) to find the hidden platform. Retention memory was assessed by a probe trial without the platform on PID 18, with time spent in each quadrant recorded over 60 seconds. Visual acuity was assessed on PID 18 after the probe test by placing a flag on the platform, with latency recorded over a 30-second period. Water maze search strategy analysis was performed as previously described. 17 Briefly, three strategies were identified using a categorization scheme: spatial strategies were defined as swimming directly to the platform with no more than one loop, or swimming directly to the correct target quadrant and searching; systematic strategies were defined as searching interior portion or entire tank without spatial bias and searching incorrect target quadrant; looping strategies were defined as circular swimming around the tank, swimming in a tight circle, and swimming around the wall of the tank. The search strategies were analyzed on all acquisition trials on PID 17 and the percentage of each strategy in each group was calculated.
Novel Object Recognition
Recognition memory was assessed by novel object recognition (NOR). The apparatus is a rectangular open field (60 × 24 × 46 cm) with two adjacent circular zones, as previously described.13,18 Zones were designated as ‘familiar object’ and ‘novel object’ zones using Any-Maze video tracking system. On PID 20, all animals were placed in the apparatus for 5 minutes each without objects present for habituation. On PID 21, two 5-minute trials were performed: the first trial had two objects for them to become familiar with (one object in each zone, training phase) and the second trial had one familiar object and one novel object present in their respective zones (testing phase). During the 60-minute intertriai interval, rats were placed back into their cages. Cognitive outcomes were quantified as the time spent in the novel and familiar object zones and a ‘% discrimination index’ for the second trial was calculated as:

Ex Vivo Slice Preparation and Electrophysiological Recordings
At PID 28, sham-operated and injured rats were anesthetized, decapitated, and brain tissue removed in ice cold oxygenated artificial cerebrospinal fluid (ACSF), composed of (in mmol/L): 125 NaCl, 2 KCl, 26 NaHCO3,3 CaCl2, 1 MgCl2, 20 glucose, titrated to pH 7.4 by bubbling with 95% O2−5% CO2. The contralateral and ipsilateral hippocampi were dissected free and 400 μm-thick sections cut on a vibratome. Slices were incubated in oxygenated immersion chamber in ACSF for 1 to 3 hours at room temperature before being transferred to a recording chamber. Recordings were done with slices continuously submerged in room temperature ACSF saturated with 95% O2−5% CO2 in a perfusion chamber under a stereoscope. Recordings were obtained using ACSF-filled glass micropipettes (tip resistance ca. 1 MQ) placed in stratum pyramidale for recording population spikes (PS) or str. radiatum for recording field excitatory post-synaptic potentials (fEPSP). Voltage signals were amplified 100-fold (npi Electronics, Tamm, Germany), low pass filtered at 1 kHz, and digitized and analyzed using pClamp software (Molecular Devices, Sunnyvale, CA, USA). Synaptic responses were evoked via a concentric bipolar tungsten stimulating electrode placed in str. radiatum >500 μm from recording electrodes. A constant voltage stimulus (amplitude 1 to 20V, duration 100 μs) was applied at 0.1 Hz. Stimulus intensity was chosen so as to elicit responses at about half of the maximal PS or fEPSP. For LTP experiments, baseline responses were recorded for 20 minutes, LTP was induced by high frequency stimulation (4 × 100 Hz trains, 15 seconds intertrain intervals), then followed by stimulation at 0.1 Hz for 1 hour. For α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)- and NMDA receptor-mediated responses, an input/output curve was generated by increasing the stimulus intensity in 0.5 V steps. Responses were first obtained in ACSF with OmM mg2+ and the GABAAR blocker, Picrotoxin (100 μmol/L). AMPARs were then blocked by washing in the antagonist 6,7-dinitroqui-noxaline-2,3-dione (DNQX) (50 μmol/L), leaving isolated N-methyl-D-aspartate receptor (NMDAR)-mediated responses, and a second input/output curve was generated. Three consecutive responses at each stimulus intensity were averaged and fEPSP slope was calculated. AMPAR-mediated responses were measured from the initial rising phase of the response, 2 to 4 ms after its initiation. Slopes for NMDAR-mediated responses were calculated over a 2 to 4-ms window in the rising phase of the response after 15 minutes of DNQX application. To calculate the AMPA-to-NMDA response ratio, the values collected at the middle three to five stimulus intensities were averaged for each response type in each slice. To avoid introducing sampling bias, data from different slices of one animal (ranging from 1 to 5) were averaged before the final ratios were calculated and sample size was reported as number of rats from each group. 19 Electrophysiological recordings were done within a 6-hour window from the time of decapitation to ensure tissue viability.
Immunohistochemistry
Anesthetized rats were transcardially perfused with saline (phosphate-buffered saline) followed by 4% buffered paraformaldehyde solution (Fisher Scientific, Pittsburg, PA, USA) for 15 minutes. The brains were removed and post-fixed in 4% paraformaldehyde for 24 hours and incubated in 30% sucrose for 48 hours at 4°C. Coronal sections (60 μm and 20 μm) were cut on a cryostat (Leica Biosystems, Buffalo Grove, IL, USA) and mounted onto glass slides. Lesion volume estimations were made on 60 μm sections from brains harvested at PID 28, stained with Cresyl Violet (FD NeuroTechnologies, Baltimore, MD, USA), dehydrated, and mounted for analysis. Lesion volume was quantified based on the Cavalieri method of unbiased stereology using Stereoinvestigator software (MBF Biosciences, Williston, VT, USA) by outlining the missing tissue on the injured hemisphere using the Cavalieri estimator with a grid spacing of 150 μm. Stereoinvestigator software was used to assess neuronal cell loss in the hippocampal subregions CA1, CA3, and dentate gyrus (DG) at PID 28, using the optical fractionator method of stereology. Every fourth Cresyl Violet-stained 60-μm section between −1.22 and −2.54 mm from bregma was analyzed beginning from a random start point. Sections were analyzed using a Leica DM4000B microscope with an optical dissector size of 50 μm by 50 μm in the × and y-axis, respectively and a dissector height of 10 μm and guard-zone height of 4 μm from the top of the section. The sampled region for each hippocampal subfield was demarcated and Cresyl Violet neuronal cell bodies were counted. Grid spacing of 300 μm in the x-axis and 100 μm in the y-axis was used for CA1 and CA3, and 300 μm in the x-axis and 150 μm in the y-axis was used for DG. Hippocampal subfield volumes were measured using the Cavalieri estimator method with a grid spacing of 100 μm. The estimated number of surviving neurons in each field was divided by the volume of the region of interest to obtain the cellular density expressed in counts/mm 3 .
For stereological assessment of microglial activation, sections were stained and processed as described previously;13,18 briefly, 60-μm sections were stained with lba-1 (1:1,000; #019–19741 Wako Chemicals, Richmond, VA, USA) overnight, incubated with biotinylated anti-rabbit immunoglobulin G antibody (Vector Laboratories, Burlingame, CA, USA) for 2 hours. Sections processed with avidin-biotin-horseradish peroxidase for 1 hour (Vectastain elite ABC kit, Vector Laboratories), and then reacted with 3,30-diaminobenzidine (Vector Laboratories) for color development. Stereoinvestigator software was used to count and classify the number of microglia in each of the three microglial morphologic phenotypes (ramified, hypertrophic, and bushy) at PID 28 days using the optical fractionator method of unbiased stereology as previously described.13,18 Sections were analyzed using a Leica DM4000B microscope with an optical dissector size of 50 μm by 50 μm in the × and y-axis, respectively and a dissector height of 10 μm and guard-zone height of 4 μm from the top of the section. Microglial phenotypic classification was based on the length and thickness of the projections, the number of branches and the size of the cell body as previously described.13,14,20 Hippocampal subregions were counted as a whole and the volume of the region of interest was measured using the Cavalieri estimator method with a grid spacing of 100 μm for the hippocampus. The estimated number of microglia in each phenotypic class was divided by the volume of the hippocampus to obtain the cellular density expressed in counts/mm 3 .
Statistical Analysis
The number of animals per group for each assessment was based on our prior studies using the LFP model 15 and satisfied scientific and statistical power requirements. Lesion volume, functional data, and the electrophysiological and stereological analyses were performed by an investigator masked to the groups. Quantitative data are expressed as mean±s.e.m. Morris water maze acquisition data were analyzed by repeated measures one-way analysis of variance (ANOVA), followed by multiple pairwise comparisons using Tukey's post hoc test. Data from the unbiased stereological assessments were analyzed by one-way ANOVA, followed by multiple pairwise comparisons using Tukey's post hoc test. Lesion volume was analyzed using a two-tailed unpaired Student's t-test. Electrophysiological data were analyzed using one-way ANOVA followed by multiple pairwise comparisons using Tukey's post hoc test. Search strategy for MWM was assessed using λ 2 analysis. The data were analyzed using SigmaPlot 12 (Systat Software, San Jose, CA, USA) or GraphPad Prism Version 4.0 (GraphPad Software, San Diego, CA, USA). A P<0.05 was considered statistically significant.
Results
Repeated Mild Traumatic Brain Injury Increased Cortical Lesion Volume and Caused Reactive Microgliosis and Progressive Neuronal Cell Loss
In order to study the consequences of mild brain injury, we used our previously described rat LFP brain injury model at a pressure used to generate a milder injury based on behavioral assessments. 15 Repeated mTBI resulted in a significantly larger lesion than single mTBI (18.2 ± 3.1 mm 3 versus 7.7 ± 2.2 mm 3 ; Figure 1A, P<0.05, Student t-test), and was comparable with moderate injury. 13 There was no measureable lesion in sham-operated rats.
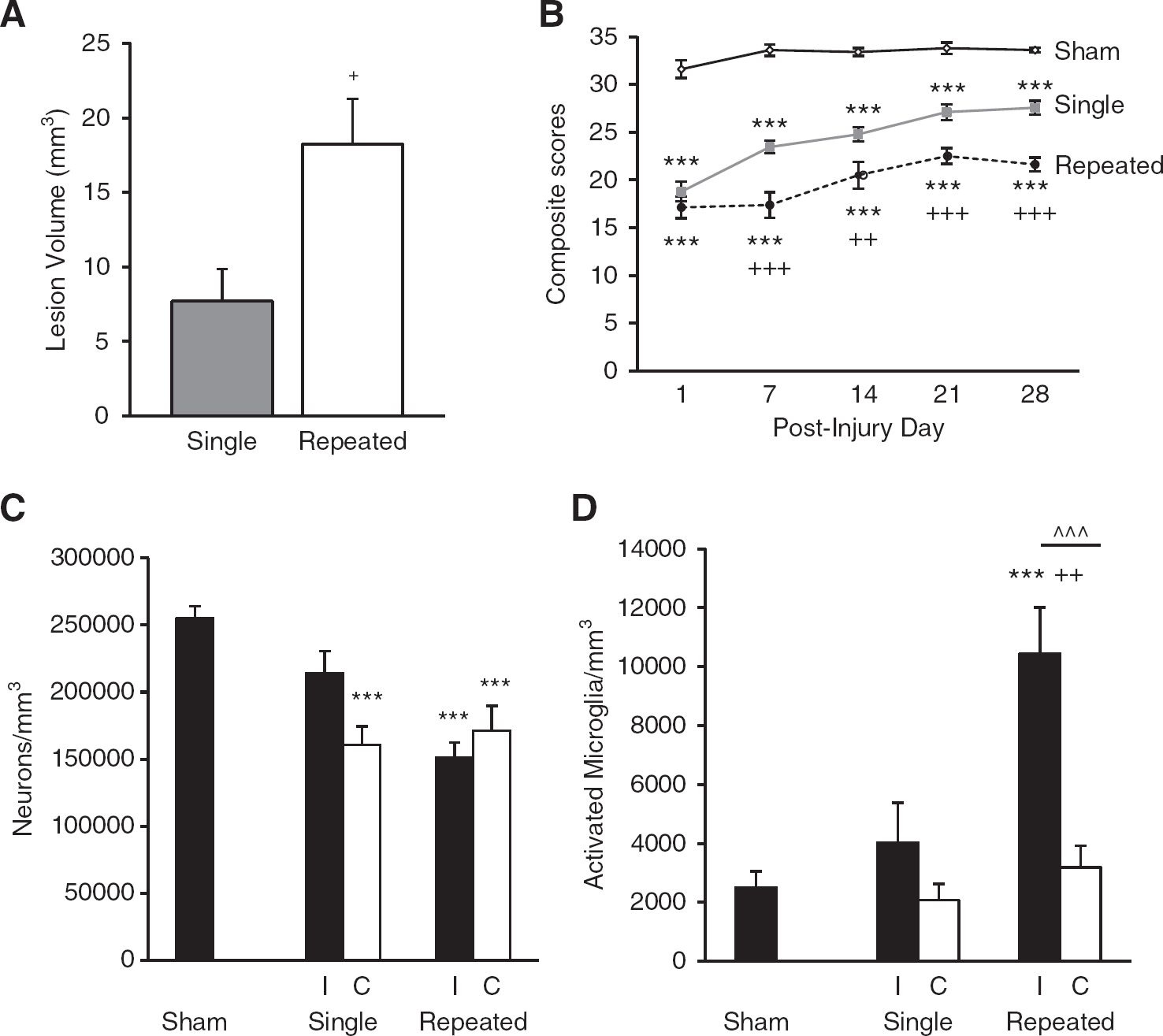
Cortical lesion volume, sensorimotor function, and bilateral cortical neurodegeneration and microglial activation. Quantitative analysis of lesion volume and unbiased stereological quantification of surviving neurons and activated microglial phenotypes were analyzed on PID 28. (
Assessment of fine sensorimotor function was performed using the composite neurologic scoring paradigm 15 and results were statistically analyzed by one-way (groups) repeated measures (scores/time) ANOVA followed by Tukey test. The interaction of ‘PIDs X groups’ (F(8,109) = 2.52, P = 0.016) was significant, and the factors of ‘PIDs’ (F(4/109) = 15.23, P<0.001) and ‘groups’ (F(2,109) = 1381.46, P<0.001) were statistically significant. Repeated mTBI induced significant sensorimotor impairments when compared with single mTBI on day 7 (P<0.001), day 14 (P<0.01), day 21 (P<0.001), and day 28 (P<0.001) and when compared with sham rats on all days analyzed (P<0.001, for days 1, 7, 14, 21, and 28; Figure 1B). Single mTBI induced significant motor impairments on all days compared with sham rats (P<0.001, for days 1, 7, 14, 21, and 28; Figure 1B).
To determine if mTBI produces chronic neurodegeneration and neuroinflammation, quantitative stereological assessment of surviving neurons and activated microglial phenotypes was performed bilaterally in the cortex. Repeated mTBI resulted in significant neurodegeneration in both ipsilateral and contralateral cortex compared with sham tissue (in neurons/mm 3 , ipsilateral repeated mTBI, 150,969.80 ± 11,450.68, P<0.001 versus sham; contralateral repeated mTBI, 171,094.05 ± 18,574.67, P<0.001 versus sham; sham, 254,953.15 ± 8,919.46; Figure 1C). Neurodegeneration did not significantly differ between single and repeated mTBI. There was significant neurodegeneration in single mTBI contralateral cortex compared with sham tissue (contralateral single mTBI, 160,818.85 ± 13,661.65, P<0.001 versus sham) but not in ipsilateral single mTBI tissue (214,172.56 ± 16,461.67). Activated microglial phenotypic classification was based on the length and thickness of the projections, the number of branches and the size of the cell body as previously described.13,14,20 Repeated mTBI caused a significant increase in activated microglial phenotypes in the ipsilateral cortex as compared with sham and single mTBI tissue (in microglia/mm 3 , repeated ipsilateral mTBI, 10,445.99 ± 1568.15, P<0.001 versus sham, P<0.01 versus single; sham, 2506.44 ± 541.00; single ipsilateral mTBI, 4024.54 ± 1352.86) as well as a significant bilateral difference within the repeated mTBI group (P<0.001 versus contralateral repeated mTBI, 3183.93 ± 741.58; Figure 1D). These data indicate an injury-dependent effect on chronic neurodegeneration throughout the cortex and specific injury-induced neuroinflammation isolated to the injured cortex.
Repeated Mild Traumatic Brain Injury Caused Progressive Neuronal Cell Loss and Reactive Microgliosis in the Hippocampus
To evaluate the effects of mTBI on subcortical regions, stereological assessment of surviving neurons was performed bilaterally in the hippocampal subregions (CA1, CA3, and DG). Repeated mTBI resulted in significant neurodegeneration in all the hippocampal subregions in both ipsilateral and contralateral tissue as compared with sham tissue (Figures 2A–C) and caused significant loss in contralateral DG compared with single mTBI. Contralateral CA1 neuronal numbers after single mTBI were not significantly different from shams (in neurons/mm 3 , shams, 395,602.43 ± 26,866.82; contralateral single mTBI, 344,452.95 ± 37,653.82); however, there was significant neurodegeneration in ipsilateral CA1 compared with sham tissue (ipsilateral single mTBI, 293,252.14 ± 14,507.37, P<0.05). Repeated mTBI caused significant neuronal degeneration bilaterally in CA1 compared with sham tissue (contralateral repeated mTBI, 251,196.13 ± 19,844.70, P<0.001; ipsilateral repeated mTBI, 244,192.70 ± 17,850.30, P<0.01). Both ipsilateral and contralateral CA2/3 neuronal cell numbers in single mTBI were significantly less than shams (ipsilateral single mTBI, 273,475.56 ± 14,902.63, P<0.05, contralateral single mTBI, 228,690.48 ± 29,363.26, P<0.01; shams, 386,245.13 ± 21,755.92) as were CA2/3 neuronal cell numbers after repeated mTBI (ipsilateral repeated mTBI, 232,907.98 ± 17,441.18, P<0.001; contralateral repeated mTBI, 195,128.35 ± 25,009.05, P<0.001). In the DG, repeated mTBI caused significant neurodegeneration in contralateral hippocampus compared with sham or single mTBI (sham, 570,507.20 ± 32,997.73; contralateral single mTBI, 467,497.28 ± 47,602.86; contralateral repeated mTBI, 287,399.83 ± 17,900.36, P<0.001 versus sham, P<0.01 versus single). These data indicate regional hippocampal vulnerability to neurodegeneration after mTBI, with CA2/3 seemingly the most vulnerable after a mild injury, and the DG showing injury severity dependence.

Bilateral hippocampal neurodegeneration and microglial activation. Unbiased stereological quantification of surviving neurons and activated microglial phenotypes was performed on post-injury day (PID) 28. (
To determine if mTBI produces neuroinflammation in subcortical regions, quantitative assessment of activated microglial phenotypes was performed in the contralateral and ipsilateral hippocampus on PID 28. Hippocampal subfields were combined and each hippocampal hemisphere was counted as a whole. Repeated mTBI resulted in significant increase in activated microglial phenotypes of hypertrophic and bushy in both the ipsilateral and contralateral hippocampus (Figure 2D) compared with shams (in microglia/mm 3 ; shams, 1843.40 ± 319.59; repeated ipsilateral, 4987.33 ± 782.39, P<0.01; repeated contralateral 4788.32 ± 939.40, P<0.01). Sham tissue and single mTBI tissue did not differ significantly with regard to reactive microglial phenotypes in either hemisphere.
Repeated Mild Traumatic Brain Injury Caused Impairments in Learning and Memory
To evaluate the effects of mTBI on cognitive function, we used the MWM and NOR, as previously described.13,15 Cognitive performance in the MWM in part reflects the hippocampal integrity.13,21 Consistent with the hippocampal neurodegeneration and increased neuroinflammation, there was a significant impairment in spatial learning in injured rats, and it was more severe in those subjected to repeated mTBI. While analysis by repeated measures ANOVA indicated that there was no significant interaction between injury groups × days (F(6/76) = 1.681, P = 0.137), there was significance among injury groups (F(2,76) = 8.793, P<0.001) and among days (F(3,76) = 22.661, P<0.001). During the acquisition phase of MWM, latency to the platform (seconds) on PID 16 was significantly greater for rats from the repeated mTBI group compared with the sham group (P<0.05), and the latency on PID 17 was significantly greater when compared with sham and single mTBI groups (P<0.001 versus sham; P<0.05, versus single mTBI, Figure 3A), indicating deficits in spatial learning and memory acquisition. Assessment of reference memory using the MWM probe trial13,15 showed that repeated mTBI resulted in significant cognitive impairments when compared with sham or single mTBI rats (P<0.001 versus sham, P<0.001 versus single, Figure 3B). Water maze search strategy analysis was performed as previously described. 17 The search strategies were analyzed on all acquisition trials on PID 17 and percentage of each strategy in each group was calculated. Repeated mTBI rats exhibited significantly higher reliance on systematic and looping strategies than spatial behavior (Figure 3C; P<0.001, χ 2 = 40.05), while single mTBI relied mostly on spatial and systematic, and shams relied only on spatial. All rats performed well in the visual cue test, and swim speeds did not differ across groups (data not shown).

Repeated mild traumatic brain injuries (mTBI) impairs performance on cognitive tasks. (
We next used the object exploration and discrimination trials of NOR task to assess recognition memory function13,18,22 and found that repeated mTBI resulted in significant cognitive impairments when compared with sham rats (P<0.01) and single mTBI rats (P<0.05, Figure 3D). Rats with a single mTBI had a significantly impaired discrimination index compared with sham rats (P<0.05).
Repeated Mild Traumatic Brain Injury Caused Bilateral Deficits in Hippocampal Long-Term Potentiation and N-methyl-D-aspartate Receptor-Mediated Synaptic Excitation
Previous rodent moderate and severe TBI studies demonstrated an impairment in expression of the hippocampal LTP at the SC-CA1 synapses.9,11,12 To investigate the potential cumulative effects of repeated mTBI on the hippocampal LTP, we recorded fEPSPs in stratum radiatum of area CA1 in response to stimulation of the SC pathway in acutely prepared slices from contralateral and ipsilateral hippocampus of single mild, repeated mild, or sham-operated rats on PID 28. Consistent with the cognitive outcomes, large and persistent LTP was induced and maintained for 60 minutes in response to high frequency tetanization in shams and single mTBI rat slices from both the contralateral and ipsilateral hippocampus. In slices taken from animals subjected to repeated mTBI, in contrast, no potentiation could be elicited in either hemisphere (Figures 4A–E). In contralateral hippocampus, we found no difference in magnitude of LTP in the hippocampal slices from single mTBI and sham-operated animals (Figure 4A; P = 0.912); however, there was a significant difference in the magnitude in single mTBI slices from the ipsilateral hippocampus compared with the shams (Figure 4B). The magnitude of potentiation in single mTBI slices from the ipsilateral hippocampus was also significantly higher than in the contralateral hippocampus from the same injury group (Figure 4D). There was significantly less potentiation in slices from repeated mTBI animals compared with both single mTBI and sham animals (Figures 4A and B). Long-term potentiation in the ipsilateral and contralateral slices from shams or repeated mTBI did not differ significantly within groups (Figures 4C and E). These results indicate that repeated mTBI causes deficits in the ipsilateral hippocampal LTP similar to those reported using moderate TBI, and that repeated injury affects the contralateral hippocampus as well, indicating the diffuse nature of the injury.
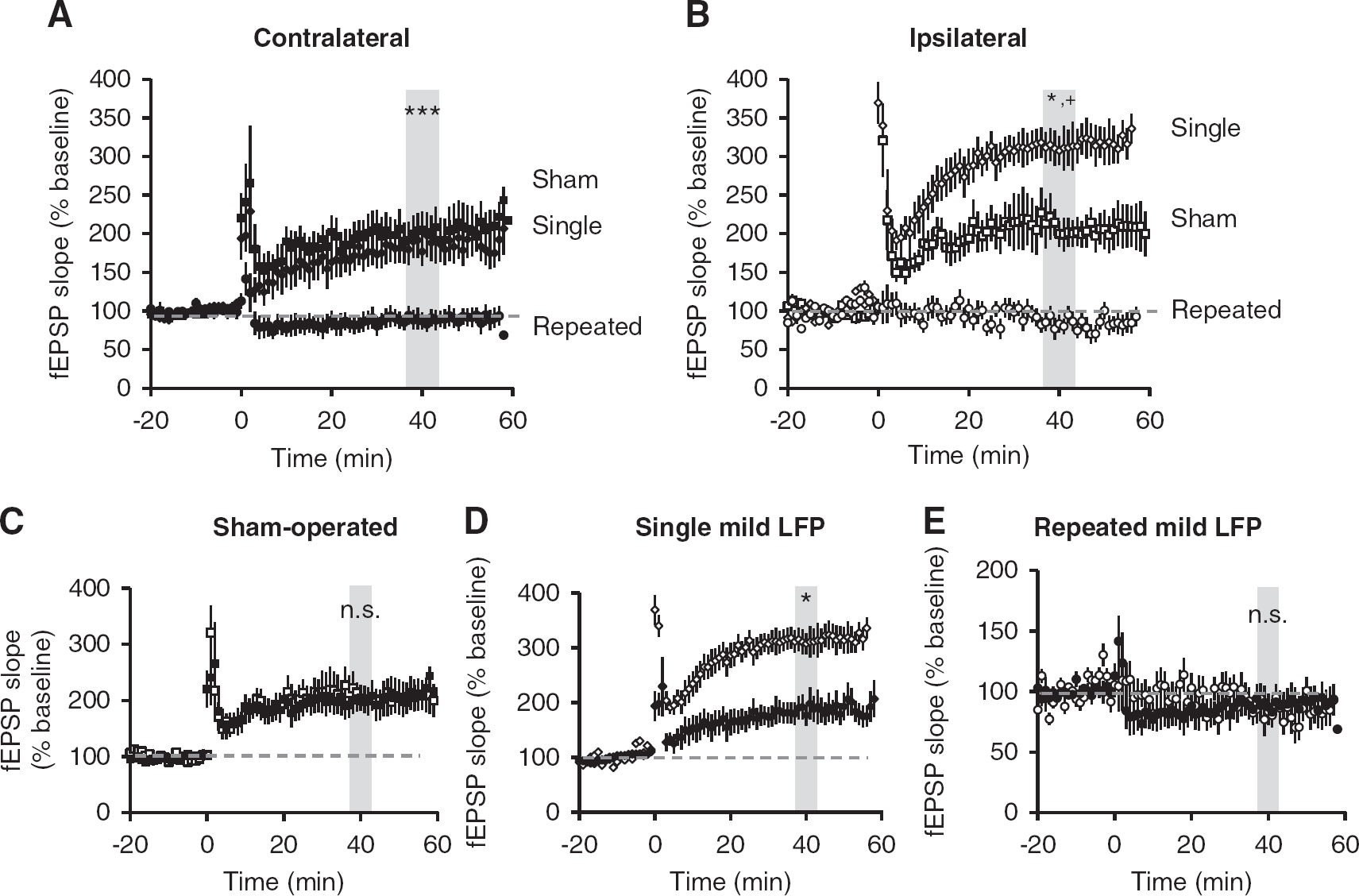
Mild traumatic brain injury (mTBI) alters potentiation at SC-CA1 synapses. Field excitatory post-synaptic potentials were recorded from stratum radiatum in area CA1 from acute ipsilateral and contralateral hippocampal slices. Long-term potentiation was induced after 20 minutes of stable baseline using high frequency stimulation. (
Simultaneous recording of PS in str. pyramidale of CA1 revealed that when there was no potentiation of fEPSPs (see Figure 4), there was potentiation of PS. Specifically, although there was no observed fEPSP potentiation in slices taken from ipsilateral or contralateral hippocampus of rats subjected to repeated mTBI, there was PS potentiation (Figure 5). The amount of potentiation in slices from repeated mTBI animals was not statistically different from that in sham slices from either ipsilateral or contralateral hippocampus. However, the level of potentiation of PS in the ipsilateral single mTBI slices was statistically higher than from repeated mTBI and sham slices (Figure 5B). There was no difference in potentiation between contralateral and ipsilateral hemispheres in sham-operated or in repeated mTBI slices (Figures 5C and E). However, the amount of potentiation achieved in the ipsilateral hippocampus was significantly higher than contralateral hippocampus after single mTBI (Figure 5D).
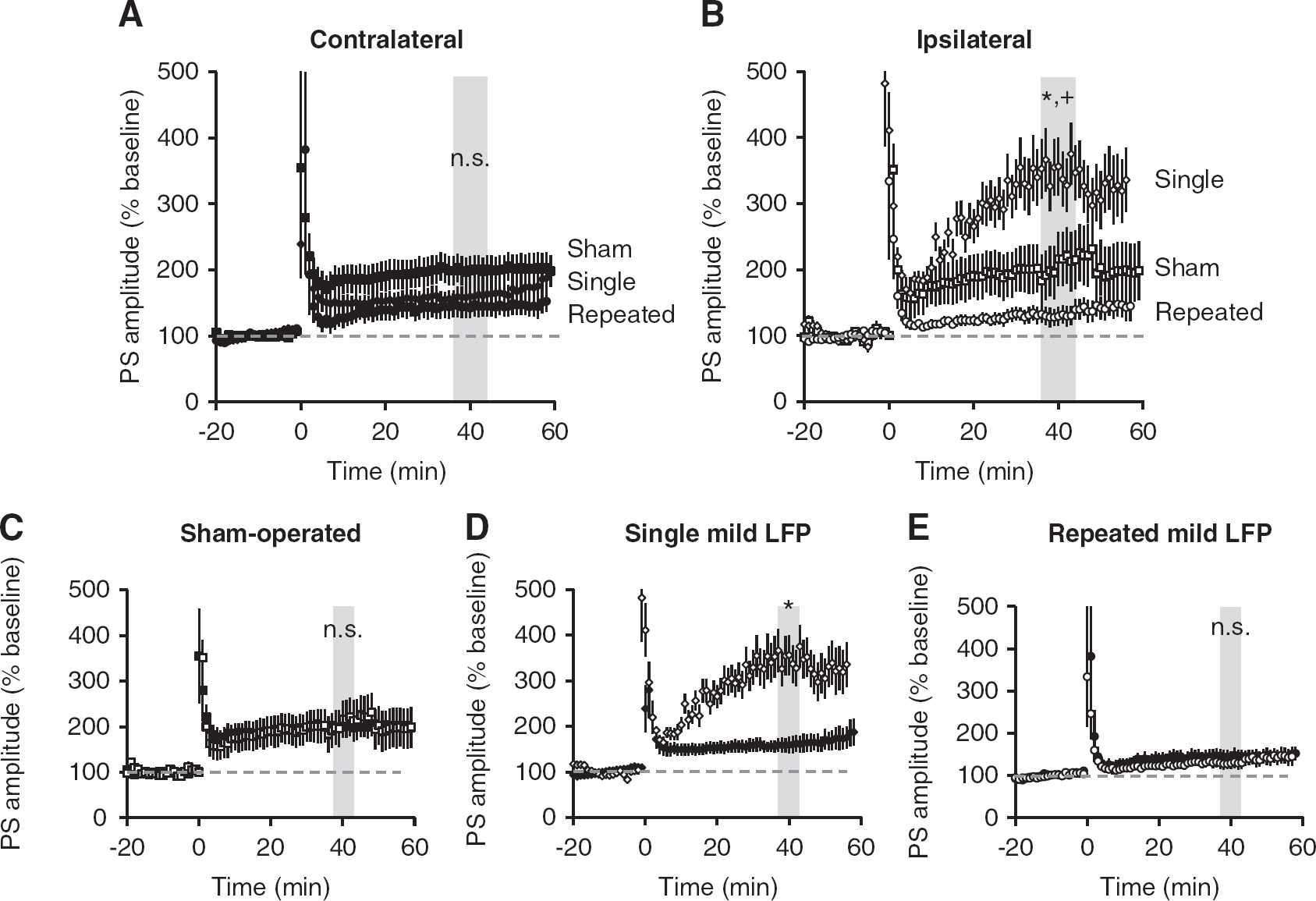
Mild traumatic brain injury (mTBI) alters potentiation in CA1 pyramidal cells. Population spikes (PS) were recorded from stratum pyramidale in area CA1 from acute ipsilateral and contralateral hippocampal slices. Long-term potentiation was induced after 20 minutes of stable baseline using high frequency stimulation (4 × 100 Hz). (
Changes in LTP after mTBI may be owing to changes in the baseline strength of excitatory synaptic transmission mediated by AMPA or NMDA receptors. Therefore, we used pharmacological approaches to determine their contribution to fEPSPs in str. radiatum (Figure 6A). To determine AMPA receptor-mediated responses, an input/output curve was generated by increasing the stimulus intensity in 0.5 mV steps starting at OmV in Mg 2 + -free ACSF containing 0.1 mmol/L Picrotoxin. Responses were then normalized across the slices and treatments by plotting the initial fEPSP slope as a function of the fiber volley amplitude, an indicator of the number of axons stimulated. AMPAR-mediated responses did not differ between injury groups in either ipsilateral or contralateral hippocampus (Figure 6B). NMDAR-mediated responses were then assayed after application of 50 μmol/L DNQX. Under these conditions, 80 μmol/L D-AP5 was washed into the bath for 5 minutes to abolish NMDAR-mediated responses. Although comparable NMDAR-mediated responses were readily obtained from slices from rats subjected to single mTBI and shams, DNQX essentially abolished fEPSP responses in slices from rats with repeated mild injuries, consistent with a dramatic decrease in NMDAR-mediated responses (Figure 6C). AMPA/NMDA ratios were calculated from responses before and after DNQX application from stimulation intensities eliciting fiber volleys of ca. 0.2 mV amplitude. There was a significant increase in AMPA/NMDA ratio after single mild compared with shams (P<0.05 versus sham) and repeated mild compared with sham and single mild (P<0.05 versus sham, P<0.05 versus single mild) (Figure 6D).
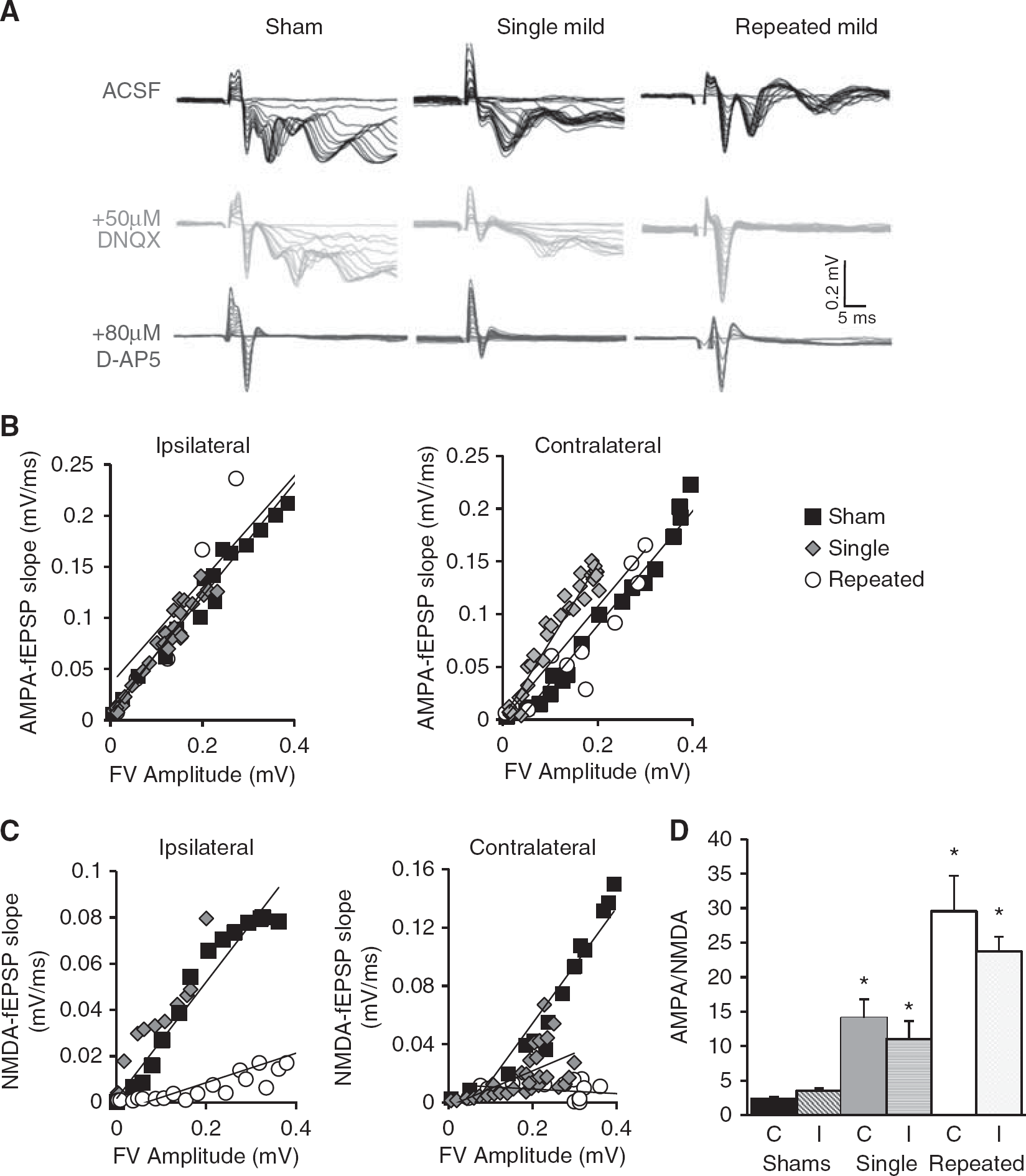
NMDAR-mediated synaptic responses in SC-CA1. Recordings from stratum radiatum were done in Ommol/L Mg
2
+ artificial cerebrospinal fluid with 0.1 mmol/L Picrotoxin. (
DISCUSSION
Chronic cognitive deficits are common to human TBI and experimental TBI models.4,8,23 There are no current pharmacological therapies that significantly reduce cognitive deficits after TBI in humans, 23 in part reflecting an insufficient understanding of underlying molecular mechanisms. It has been increasingly recognized that experimental TBI causes long-sustained neuroinflammation that may be associated with chronic neurodegeneration and cognitive impairments. 4 Moreover, we have shown that delayed therapeutic strategies to reduce such chronic inflammation can markedly improve cognitive recovery in experimental TBI models. 14 Whether similar changes underlie the consequences of repeated mild concussive brain injury is unknown. Repeated concussions can be associated with impaired memory and disorders of behavior and mood.2,3 However, how single concussion increases susceptibility to damage from subsequent injuries is unclear. Therefore, it is important to elucidate the underlying secondary injury mechanisms that sensitize the brain to subsequent mild head injuries. In the present study, we compared the effects of single or multiple mTBI on late hippocampal synaptic physiology and histopathology, inflammation and cognitive function.
Lateral fluid percussion injury is an experimental TBI model that simulates many pathobiological features of human contusive head trauma.15,24,25 The LFP model produces both focal and diffuse injury with vascular disruption, neuronal cell death, and glial proliferation.15,24,25 In order to examine the correlation between the functional and physiologic consequences of mTBI that contribute to significant cognitive dysfunction, we recorded bilaterally from hippocampal brain slices. Previous reports for moderate TBI in the ipsilateral hippocampus found LTP deficits in CA1 synapses at 4 hours, 24 hours, and 48 hours,9,12 7 days, 10 and 2 weeks. 23 Interestingly, we found that repeated mTBI via LFP, in both ipsilateral and contralateral SC-CA1 pathway, led to a failure to potentiate in response to high frequency tetanus at 28 days post injury, making repeated mTBI seemingly comparable with moderate TBI in its deleterious electrophysiological consequences. In contrast, LTP was observed in the SC-CA1 pathway bilaterally after single mTBI, and there was a significant increase in the level of potentiation in the synaptic and cell body layer in the injured hemisphere compared with the sham animals consistent with another published study. 26 However, the significance and mechanisms behind the altered excitatory neurotransmission in the ipsilateral hippocampus after a single mTBI remain to be determined.
Induction and expression of hippocampal LTP involves several critical steps. SC-CA1 LTP requires activation of post-synaptic NMDARs.27,28 Therefore, we hypothesized that the lack of LTP after repeated mTBI might be due to changes in NMDAR-mediated synaptic transmission. Indeed, we found that repeated mTBI caused virtual elimination of the NMDAR-mediated responses at SC-CA1 synapses. Notably, this lack of NMDAR-mediated responses correlated closely with the lack of LTP after repeated mTBI, and is therefore the likely explanation for lack of plasticity. Loss of NMDAR-mediated responses in the ipsilateral hippocampus has been reported previously in several moderate to severe TBI models, 10 including a closed head TBI model showing profound and long-lasting loss of NMDAR function associated with cognitive impairments.29–31 More importantly, this study extends the findings by revealing that NMDARs and LTP in the hippocampus are impaired bilaterally. We observed no difference in AMPAR-mediated transmission between injury groups. While there was a lack of fEPSP potentiation after repeated mTBI in the SC-CA1 synapses, PS in the pyramidal cell layer were potentiated in both ipsilateral and contralateral hippocampus. Population spike potentiation in the absence of fEPSP potentiation is unprecedented under any conditions, to our knowledge, although it appears to reflect a process independent of pyramidal cell NMDAR-activation. Plasticity of feed-forward GABAergic inhibition is one possibility.32,33 These injury-induced changes in synaptic transmission are likely to result in the hippocampal dysfunction and chronic deficits in the hippocampal-dependent tasks after repeated concussive injuries.
The physiologic alterations in the hippocampus after repeated mTBI were correlated with dysfunction of hippocampal-dependent learning and memory tasks, including impairment in spatial learning, reference memory, and retention memory functions. Hippocampal integrity, particularly in the CA3 and DG subregions, is essential for performance in the MWM.16,34 Impaired cognitive performance after repeated mTBI was associated with bilateral hippocampal neuronal loss. Furthermore, cognitive performance across the acquisition trials of the MWM can be assessed by swimming strategies or paths utilized by the rats to locate the platform. 17 Impaired cognitive performance in search of strategic analysis is indicated by a switch from spatial to looping swimming patterns across trials. Repeated mTBI-injured rats showed greater reliance on looping strategies than single mTBI and sham groups, indicating impairments in spatial learning and memory. The trials of NOR test, based on object exploration, attention and discrimination, all together assess the retention memory function.13,22 Here we show that single or repeated mTBI impair discrimination index in the NOR task. The loss of neurons in the hippocampus and/or the cortex after repeated mTBI may explain poor performance in the NOR task. However, the subtle hippocampal and cortical neurodegeneration after single mTBI cannot explain the observed NOR task abnormalities after single mTBI, but may be more owing to its dependence on the functional interactions between the hippocampus and cortex. 35 The role the hippocampus plays in recognition memory is controversial; some studies show that damage to the hippocampus has little effect,36,37 whereas others show a correlation with impaired performance.35,38 Barker and Warburton 35 reported that recognition memory tasks depend on functional interactions between the hippocampus and the cortex. 35 While the loss of neurons in the hippocampus after repeated mTBI might explain poor performance in NOR, the abnormalities after single mTBI cannot readily be attributed to the minimal neuronal loss in the hippocampus. However, we also show neuronal cell loss in the cortical regions after mTBI, likely contributing to poorer performance in NOR after single mTBI. Chronic neuroinflammation after TBI may provide a mechanistic connection between the resulting behavioral dysfunction and altered hippocampal physiology. Although both neuroprotective and neurotoxic microglial phenotypes have been described, 20 microglial activation and the release of associated inflammatory agents have been proposed as important contributing factors in chronic neurodegenerative disorders. 39 Previous studies have indicated that sustained microglial activation after moderate central nervous system trauma may have a role in the progressive neurodegeneration. 40 In addition, recent studies on mild injury indicate that repeated mTBI also causes neuroinflammation and concurrent cognitive deficits and neuronal cell loss. 5 We demonstrate that repeated mTBI, but not single injury, caused an increase in activated microglial phenotypes (hypertrophic/bushy) bilaterally in the hippocampus, as seen previously in the injured cortex after moderate TBI. 13
In the past decade, there have been several reports on the effects of TBI in humans on neuronal cell loss, synaptic plasticity, and cognition, but it has been limited to more moderate and severe injuries along with most of the focus on the injured side of the brain. Recently, there are growing concerns about the pathophysiological effects of repeated mTBI and their relationship to long-term functional impairments. Here we report chronic neuronal cell loss, deficits in synaptic plasticity, microglial activation, and the associated cognitive deficits after repeated mTBI that involve subcortical structures remote from the site of injury, including the contralateral hemisphere. Clinically diagnosed mTBI, which may occur without observable structural changes in the brain tissue, can be associated with chronic cognitive deficits, sleep disturbances, and other behavioral changes.7,8 Our study underscores the increased vulnerability of the brain to the damage caused by repeated mild trauma, and confirms that the consequences may be greater and more sustained than after a single mTBI. Repeated mTBI using the rat LFP model led to significant alterations in the hippocampal excitatory synaptic transmission and plasticity, associated with greater neurodegeneration and neuroinflammation, and cognitive impairments. These changes and behavioral deficits were similar to those observed after single moderate TBI. Our findings highlight the correlation between the physiologic changes, and histologic and functional impairments resulting from repeated mTBI, thus providing a potential mechanistic explanation for the underlying pathophysiology.
Footnotes
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors thank Katherine Cardiff and Juliane Faden for expert technical assistance.