
Abstract
The prevalence of dementia is increasing in our aging population at an alarming rate. Because of the heterogeneity of clinical presentation and complexity of disease neuropathology, dementia classifications remain controversial. Recently, the National Plan to address Alzheimer’s Disease prioritized Alzheimer’s disease-related dementias to include: Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia, vascular dementia, and mixed dementias. While each of these dementing conditions has their unique pathologic signature, one common etiology shared among all these conditions is cerebrovascular dysfunction at some point during the disease process. The goal of this comprehensive review is to summarize the current findings in the field and address the important contributions of cerebrovascular, physiologic, and cellular alterations to cognitive impairment in these human dementias. Specifically, evidence will be presented in support of small-vessel disease as an underlying neuropathologic hallmark of various dementias, while controversial findings will also be highlighted. Finally, the molecular mechanisms shared among all dementia types including hypoxia, oxidative stress, mitochondrial bioenergetics, neuroinflammation, neurodegeneration, and blood–brain barrier permeability responsible for disease etiology and progression will be discussed.
Introduction
In recent years, dementia due to various causes is becoming common in the elderly. Notably, abnormal protein deposition has been shown to co-exist with damaged neurovasculature in dementias at various disease stages. The relationship between vascular dysfunction and dementia has been described several decades ago. During the nineteenth century, pathologists have noted small blood vessel narrowing in the brain and described its association with hypoperfusion leading to brain damage. This became known as arteriosclerotic dementia—the hardening of arteries.1,2 In 1907, Alois Alzheimer reported senile plaques in the brain of patients, which have become the neuropathologic hallmark of Alzheimer’s disease (AD). Further, Alois Alzheimer posited that circulatory system impairment might serve as the trigger for AD pathology. Yet most pathologists at that time classified AD as purely degenerative, with uncertain etiology. In the mid-twentieth century, experts began to challenge the dichotomy between arteriosclerotic and degenerative dementia, questioning the etiology of AD. From that point on, the concept of vascular dementia (VaD) has evolved and transformed (for additional information, refer to ‘Classifications of dementias’ section). In 1974, Hachinski et al. 3 defined arteriosclerotic dementia to include multiple small-vessel infarcts, offering a new perspective on the cerebrovascular causes of dementia. With the advent of noninvasive computed tomography of the brain and, later, magnetic resonance imaging (MRI) technology, the clinical understanding of vascular disease was improved. This led to mounting evidence on a wide spectrum of ischemic changes and abnormal findings in apparent cognitively normal elders, which further perplexed investigators and clinicians alike. 4 Since the emergence of MRI as a unique tool coupled with advances in related diagnostic technologies, the diagnosis of dementia today is integrative and uses a variety of diagnostic tools to merge neuroimaging and immunohistopathologic analysis to assess the contributions of various neuropathologic mechanisms, including vascular factors, in dementia. 5
Cerebrovascular changes are common neuropathologic findings in aged subjects with dementia. 6 Vascular remodeling and pathologic changes to the macro- and microvasculature may disrupt blood vessel integrity. Notably, such remodeling leads to vascular disease and cerebral hypoperfusion associated with neuronal injury, structural and functional brain damage. 7 More specifically, neuroimaging findings indicate white matter hyperintensities, cerebrovascular lesions, and cerebral amyloid angiopathy (CAA). 8 Other ultrastructural abnormalities to the microvasculature associated with small-vessel disease, and exacerbated by aging, include capillary wall deterioration and the accumulation of erythrocytes, 9 basement membrane thickening, and pericyte degeneration, 10 resulting in blood–brain barrier (BBB) permeability 11 and vascular cognitive impairment (VCI). Vascular cognitive impairment is a broad term that encompasses cognitive deficits associated with vascular disease, ranging from mild-to-severe cognitive impairment including VaD. 12 A new and all-inclusive term was recently coined referred to as VCID (vascular contributions to cognitive impairment and dementia). It is now often used by many groups, including NIH, since it is not restrictive to a particular diagnosis but rather highlights the contributions of many dementias. Moreover, hemodynamic impairment of mean blood flow velocity, pulsatility index, and cerebrovascular reactivity were found in AD patients as compared with controls using transcranial Doppler to monitor the middle cerebral artery. 13 These results suggest hemodynamic impairment as a critical marker of cognitive decline and confirm its role as an early predictor of vascular damage in AD. Similarly, previous studies have observed cerebrovascular changes in DLB, partially (in 30% of the cases) characterized by cortical microbleeds and CAA. 14 Some cases of FTD associated with white matter alterations and small-vessel disease. 15 Interestingly, VaD incorporates certain key neurovascular features of AD-related dementias (i.e., cerebral microbleeds, infarcts, CAA), which significantly correlates with vascular risk factors in clinical studies. 16 However, it is unclear whether this relationship is purely coincidental and cumulative or, alternatively, casual and synergistic. Treatments aimed at reducing vascular risk factors, such as better blood pressure control, improved diet, and exercise have been shown to improve disease incidence and progression. 17
The brain histopathology and pathophysiology may not necessarily reflect clinical symptoms. The disease(s) may take years to manifest their cognitive, behavioral, and functional impairments. Several autopsy studies of nondemented, aged individuals confirm mixed cerebrovascular pathologies and neuropathologic findings, suggesting a complex and silent form of subclinical disease.18,19 Additional support for this claim is derived from the seminal nun studies. Autopsies of nearly 500 human brains found that the prevalence of dementia in persons beyond the age of 75 is very high. Moreover, only 10% of the nuns in this group exhibited ‘brain reserves’, or the ability to tolerate high-stage AD-related neuropathologic alterations without developing clinical symptoms of dementia. 20 In another related study, nuns diagnosed with dementia often showed signs of underlying cerebrovascular dysfunction as defined by the presence of lacunar infarcts and these participants tended to have less severe AD-related changes than participants with similar cognition but without overlapping vascular disease pathology. 21
With the wide spectrum of available literature on cerebrovascular alterations in different dementias, in the following sections, we will first provide current evidence linking cerebrovascular abnormalities to AD-related dementia. Next, a discussion of vascular impairments in various other forms of dementia will follow, along with controversial hypotheses regarding vascular contributions to cognitive impairment. Finally, we will highlight emerging vascular risks and molecular mechanisms predisposing individuals to dementia.
Classifications of dementias
The incidence of dementia is exponentially increasing and has become one of the major public health concerns in recent years. According to the World Health Organization census in 2010, there were approximately 35.6 million people worldwide living with dementia, a number that is expected to triple by 2050.
22
With the yearly diagnosis of 7.7 million new cases of dementia worldwide, the medical cost burden surpasses that of cancer and heart disease combined.
23
Furthermore, dementias may be difficult to clinically diagnose because of their multifactorial causes, overlapping symptoms, and variety of degenerative pathologies, resulting in inconsistent clinical presentation and diagnostic challenges.
24
For example, a retrospective study using the National Alzheimer’s Coordinating Center database was recently conducted on the accuracy of AD diagnosis. Autopsies of 533 AD patients demonstrated that 119 subjects did not meet the criteria for definite AD diagnosis. Among other neuropathologic findings, researchers found DLB (n = 35, 29%), VaD (n = 15, 13%), and FTD (n = 14, 12%). These observations suggest that a wide and diverse clinical spectrum of dementia exists in the population, which may be attributed to underlying vascular alterations as a part of the neuropathology. Therefore, careful examination and analysis of this relationship is necessary.
25
A schematic of different types of dementia and associated neuropathologic features is presented in Figure 1.
Classifications of dementia subtypes and associated neuropathologic features. A hierarchy listing the four major categories of dementia with corresponding neuropathologic findings. A mixed phenotype represented by double-sided arrows indicates a shared disease neuropathology between two dementia types. Aβ, amyloid-β; AD, Alzheimer’s disease; APP, amyloid precursor protein; α-synuclein, alpha-synuclein; CAA, cerebral amyloid angiopathy; DLB, Lewy body dementia; ET, endothelial; FTD, frontotemporal dementia; FUS, inclusions of fused in sarcoma; NFTs, neurofibrillary tangles; p-tau, hyperphosphorylated tau; TDP-43, transactive response DNA-binding protein-43; VaD, vascular dementia.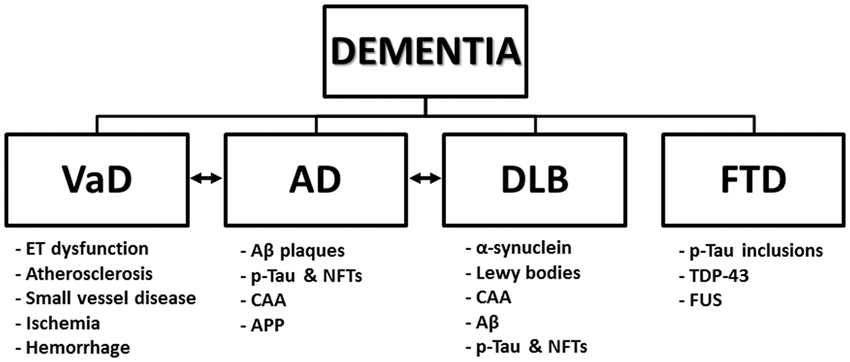
Alzheimer’s Disease
Alzheimer’s disease is the most common form of dementia, which often occurs with VCI. 26 The neuropathologic hallmarks of AD are the extracellular accumulation of senile plaques composed of Aβ peptide and intraneuronal accumulation of neurofibrillary tangles (NFTs) composed of hyperphosphorylated microtubule-binding protein-tau (p-tau). These pathologic markers positively correlate with neuronal degeneration, neuroinflammation, microglia activation, BBB dysfunction, and cognitive decline.27,28 Furthermore, NFT pathology in AD develops in three stages: transentorhinal, limbic, and isocortical, which is the basis of the Braak and Braak staging system of AD, 29 and serves as a better predictor of cognitive decline as compared with Aβ pathology. 30 As a detailed discussion of AD characterization and clinical diagnosis is beyond the scope of the review, the reader is referred to current comprehensive reviews on the topic.31,32
Cerebrovascular effects of amyloid
Recent findings indicate that vascular changes often coexist with changes linked to AD, suggesting a synergistic effect on cognitive decline. Pathologic studies of the Alzheimer’s brain microcirculation have shown alterations of blood vessel morphology and decreased vascular density, combined with increased vessel tortuosity. 33 One significant contributor to AD-mediated cerebrovascular injury is Aβ (a cleaved fragment of the amyloid precursor protein or APP), which is known to exert powerful vascular effects. The Aβ1–40 fragment has also been shown to be deposited in the cerebrovascular blood vessels in human autopsy studies.28,34
Additional data indicates Aβ deposition on cerebral blood vessel walls (CAA), 35 resulting in the loss of smooth muscle and narrowing of the lumen. Direct effects of soluble Aβ1–40 and Aβ1–42 on cerebrovascular vasomotor regulation have been observed in rodent penetrating arterioles, which are responsible for increased cerebrovascular resistance. 36 Even soluble Aβ1–40 has been shown to induce nicotinamide adenine dinucleotide phosphate oxidase-dependent reactive oxygen species production in the endothelial wall, thus resulting in Aβ-induced neurovascular dysfunction. 37 Additional information on BBB permeability in AD can be found in ‘Cerebrovascular risk factors and neuropathological correlates of dementia’ section. Effects of Aβ deposition on vessel structure and function also include thickening of the basement membrane, disruption of vascular smooth muscle cells, and impaired drainage. Weakened vasculature walls can lead to leaky vessels and/or hemorrhages in the brain. Cumulatively, these studies suggest that CAA and Aβ limit the ability of the blood vessels to adapt normally to physiologic stimuli and pose a potential risk for hemorrhages and BBB opening as a result of Aβ-induced weakness of the vessel wall. 38
Although the majority of AD is sporadic, there is a strong genetic component to the disease. Apolipoprotein E-ɛ4 allele has been classified as one of the important genetic risk factors for late-onset AD and drives the accumulation of parenchymal Aβ plaques. 39 This gene is viewed as a susceptibility gene, in contrast to deterministic genes, such as the APP or presenilin mutations. 40 Recent reports suggest that the apolipoprotein E-ɛ4 genotype correlates with CAA, with the greatest prevalence found in the occipital lobe. 41 These findings suggest that genetic risk may be involved in exacerbating cerebrovascular injury in late-onset AD.
Dementia with Lewy Bodies
Dementia with Lewy bodies is the second most common cause of neurodegenerative dementia in the elderly. 42 It shares clinical and pathologic characteristics of other dementias that may occur during the course of Parkinson's disease and other neurologic conditions.43,44 Pathologically, DLB is characterized by abnormal aggregation of the synaptic protein α-synuclein as ‘Lewy bodies’ in neurons associated with brain atrophy, which is not as robust as in AD. 45
Cerebrovascular dysfunction and neuroinflammation in dementia with Lewy bodies
Emerging evidence supports the contributions of hypoxia and vascular hemodynamic alterations in DLB. 46 Reductions in cerebral blood flow and decreased microvessel density associated with vascular endothelial growth factor deficiency, secondary to the accumulation of α-synuclein, were described in the occipital cortex of DLB patient brains as compared with controls. 47 A recent study suggested that α-synuclein levels increase in human SH-SY5Y neuroblastoma cells after oxygen-glucose deprivation, confirming that a state of hypoperfusion may promote protein aggregation. 47 Increasing evidence also suggests altered inflammatory responses during the neurodegenerative processes, which accompany DLB, particularly, in terms of glial activation. 48 The presence of autoantibodies against amyloid and glial-derived structures was determined in the cerebrospinal fluid (CSF) of demented patients. 49 Notably, serum autoantibody levels against α-synuclein, Aβ1–42 myelin oligodendrocyte glycoprotein and myelin basic protein were significantly higher in DLB compared with tau-associated dementias (AD, FTD), VaD, and controls. Consistently, reports of CSF and plasma concentrations of hyaluroinic acid, an adhesion molecule known to regulate both vascular and inflammatory processes, was found to be higher in patients with DLB as compared with cognitively normal and age-matched controls. 50 Taken together, these data suggest that DLB patients experience a robust inflammatory response and vascular abnormalities, which further exacerbate Lewy body-induced neuropathology.
Controversies of dementia with Lewy bodies as a cerebrovascular disease
Data regarding the prevalence of cerebrovascular pathologies in DLB patients remain scarce and contradictory. An interesting study by Ghebremedhin et al. 51 found that patients with co-existing vascular disease were likely to have less Lewy body pathology than patients with only Lewy body disease in autopsy samples of DLB brains, consistent with a cumulative pathology hypothesis. The extent of Lewy body pathology inversely and significantly correlated with the severity of vascular pathologies, such as atherosclerosis, infarcts, and small-vessel disease. In contrast, CAA and NFTs were positively and significantly correlated with DLB pathology. Consequently, the authors concluded that patients with advanced Lewy body pathology were less likely to present with vascular dysfunction and/or stroke.
Although the complexities of AD-related dementias and their clinical diagnosis remain a challenge, there is a clear gap in knowledge regarding vascular contributions in pure DLBs. 52
Frontotemporal Dementia and Related Tauopathies
Frontotemporal dementia is a heterogeneous neurodegenerative group of non-AD dementias with a variant clinical and pathological profile. Several genes have been identified that cause the autosomal dominant familial forms of FTD. However, our focus will be on sporadic FTD, which represents 60% of cases. 53 The sporadic form of the disease typically develops in the elderly during the sixth decade of their life. 54 The term frontotemporal lobar degeneration (FTLD) refers to the neuropathology of the FTDs. Pathologically, the FTLDs are characterized by atrophy of the frontal and temporal lobes (lobar atrophy), which contrasts the diffuse atrophy of AD, and by some or all of the following microscopic findings: neuronal loss and gliosis, vacuolization of the superficial cortex (spongiosis), and ballooned neurons. 55 Frontotemporal dementia can be further subdivided into three different categories: (1) FTD-tauopathies (examples: progressive supranuclear palsy, Pick’s disease, corticobasal degeneration, Argyrophilic grain disease, etc.); (2) FTD-ubiquitin with ubiquitin-positive tau-negative neuronal inclusions (examples: motor neuron disease and sporadic amyotrophic lateral sclerosis), and (3) FTD-without tau- or ubiquitin-positive inclusion (examples: amyotrophic lateral sclerosis, and in some cases of AD, Pick’s disease, and DLB). 56 These variants differ in terms of clinical presentation, cognitive impairment, and affected brain regions. 57 This categorization is reasonably well accepted.
Pathologic markers of frontotemporal dementia
The neuropathologic hallmarks of these diseases include gliosis, neuronal loss, superficial spongiform degeneration, and atrophy of the frontal and temporal lobes, often culminating with progressive aphasia, disintegration of personality and behavior, severe memory loss, and language impairments. 58 Neuroimaging modalities such as MRI are necessary to provide an accurate diagnosis. Distinctive MRI features often include frontal and anterior temporal lobe atrophy, altered white matter signals reflecting gliosis, and frontotemporal brain hypometabolism. Another common histopathologic finding associated with the FTD group of diseases is tissue deposition of abnormally aggregated proteins. Three major pathogenic proteins have been implicated in the autopsy studies of sporadic FTD: (1) FTD-Tau: cellular p-tau inclusions (in both sporadic tauopathies and familial FTD linked to parkinsonism on chromosome 17, FTDP-17T) 59 and (2) FTD-transactive response DNA-binding protein-43 (TDP-43): the main ubiquinated peptide in tau-negative FTD, 60 representing the majority of cases, 61 while (3) inclusions of fused in sarcoma (FUS) proteinopathy are a seldom occurrence associated with a clinical diagnosis of behavioral variant FTD. 55 The tau-protein is typically involved in the regulation of microtubule assembly and disassembly, and has been shown to have an important role in FTD cases of tau- or ubiquitin-positive inclusions. 62 These tau abnormalities may also lead to tau aggregation or microtubule dysfunction, which in turn, may affect axonal transport.
Vascular risk factors associated with frontotemporal dementia
Only a few risk factors have been recognized to contribute to FTD etiology and only a few studies have investigated the relationship between vascular impairment and FTD. One of these risk factors is family history of FTD. Recent autopsy data of FTLD brains with tau inclusions supports the role of small-vessel dysfunction in FTD disease progression. 15 Investigators reported the prevalence of white matter arterial changes within frontal and temporal lobe regions associated with demyelination. These effects correlated with older age and suggest the presence of vascular co-morbidity in FTD. However, there is evidence arguing against the role of cerebrovascular dysfunction in FTD, including a negative effect or no correlation altogether, consistent with a cumulative pathology hypothesis. One reason that conclusions may be particularly difficult to reach regarding the role of vascular disease in FTD is the diverse etiologies associated with each of the FTDs, including both genetic and environmental influences (i.e., FTD because of trauma manifests differently than FTD associated with TDP-43). A case–control study containing 80 cases of veterans with sporadic FTD found a significant association between FTD and head injury. In fact, the prevalence of traumatic brain injury was significantly greater in veterans with FTD versus those with non-FTD dementias. Surprisingly, the FTD group also experienced a lower prevalence of heart and cerebrovascular disease, although vascular risk factors such as hypertension were similar between both the studied groups. 63 Additional evidence demonstrating an inverse relationship between FTD and cerebrovascular dysfunction is present in autopsy studies of FTLD brains. Young patients with α-synucleinopathies, FTLD because of tau and TDP-43, and prion disease showed a lower prevalence of cerebrovascular disease than patients with AD. 64 Another possible explanation for the observed inverse correlation between FTD and vascular impairment may be offered by the spatio-temporal variations of pro-inflammatory cascades during the acute and chronic phases of brain injury. 65 Future research should address the mechanisms underlying FTD and the vascular contributions in these dementias.
Vascular Dementia
The term VCI comprises the heterogeneous group of cognitive disorders that share a presumed vascular cause and includes both dementia and cognitive impairment without dementia. 66 Vascular dysfunctions, including large vessel disease, cardioembolic disease, and small-vessel disease are considered causal in patients with VaD compared with its additive or synergistic effects in association with the other dementias explained above. On the basis of the vascular hypothesis, VaD is caused by diminished cerebral blood flow, leading to hypoxia and BBB permeability from prolonged vasculotoxic and neurotoxic effects promoting neurodegeneration and amyloid deposition. 67 Vascular dementia has been classified into the following six dementia subcategories: (1) multi-infarction dementia, (2) strategic infarction dementia, (3) hemorrhagic dementia, (4) mixed dementia, (5) subcortical ischemic vascular dementia (SIVD), and (6) other forms of VaD. 68 The sudden occurrence of infarction and hemorrhagic stroke dementia subtypes associated with acute cerebrovascular diseases may parallel specific cortical or subcortical symptoms related to stroke-affected brain regions. Beyond the major categories of large vessel disease, cardioembolic and small-vessel disease, the remaining VaD subtypes, represent heterogeneous etiologies, including vasculitis, CAA, and inherited disease such as CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy). 69
The following section will address an emergently important VaD subtype, which is known to be caused by small-vessel disease in the brain and has been implicated in VaD.
Subcortical ischemic vascular dementia
SIVD is characterized by small-vessel vascular disease and is classified as a VCI subtype. 70 Hypertension represents a primary risk factor in sporadic SIVD (refer to ‘Cerebrovascular risk factors and neuropathological correlates of dementia’ section). The neuropathology is driven by severe stenosis and microvessel occlusion, culminating as white matter ischemia and multiple lacunar infarctions in subcortical structures. 71 Clinical studies of SIVD patients using dynamic contrast-enhanced MRI show significantly elevated albumin index and increased BBB leakiness, indicating abnormal white matter damage in the SIVD group as compared with controls. 72 Moreover, SIVD may be further divided into several clinical subtypes. The first presents with arteriosclerotic leukoencephalopathy in the white matter and is known as Binswanger's disease. This type of ischemic change in the brain is formed by widespread incomplete infarctions or white matter hypoperfusion. 73 The second is a rare hereditary disease referred to as CADASIL. Case reports of CADASIL patients have noted elevated Aβ1–42 levels in the cerebral cortex and increased Aβ plaque load. It has been further postulated that Aβ deposition is derived as a consequence of elevated Aβ synthesis or dysfunctional clearance because of the arteriolar alterations associated with CADASIL. 74
Mixed Dementia
A mixed etiology of AD and VaD is thought to become more common with increasing age, particularly in individuals older than 85. 75 Data from epidemiologic studies indicate that approximately one-third of patients with AD present with vascular pathology, 76 suggesting that there is a strong vascular component promoting brain injury. 77 Clinicopathologic studies report a heterogeneous phenotype comprising AD (Aβ plaques and NFTs) and ischemic lesions or small-vessel disease. 78 Common pathways connecting AD to VaD have been implicated in the literature. 75 The association of the apolipoprotein E-ɛ4 genotype with an increased risk for both AD and VaD proposes a potential link between atherosclerosis, cerebrovascular disease, and AD. 79 Conversely, amyloid deposition in cerebral blood vessels because of AD increases the risk for hemorrhagic strokes and subsequent VaD. 80 In an interesting study exploring the pathologic substrates of mixed dementia using neuropathologic correlates of 156 autopsied AD brains, it was found that the clinical presentation of vascular neuropathologic findings (i.e., white matter lacunes, periventricular, and diffuse white matter demyelination and focal and diffuse cortical gliosis, presence of NFTs, and cortical microinfarcts) is dependent upon cerebral lesion type, localization, and the degree of AD pathology. 81 Nevertheless, studies on mixed dementia are scarce, with the majority of reports merging VaD and mixed dementia throughout the data analysis process, thus making data interpretation difficult.
Cerebrovascular risk factors and neuropathologic correlates of dementia
Diabetes, hypertension, hypercholesterolemia, smoking, and old age are some of the vascular risk factors known to increase the risk for AD-related dementias.
82
Among these, hypertension has emerged as the strongest predictor of cognitive impairment, serving as a common denominator for a variety of neurodegenerative conditions. The following sections will focus on the role of hypertension in dementia, a novel research area that is only now beginning to emerge and describe known mechanisms of neuroinflammation and neurodegeneration leading to BBB permeability and manifesting dementia subtypes. We will also review neuropathologic findings associated with dementia, which are known to contribute to cerebrovascular risk (Figure 2).
Neurovascular factors driving neuropathology and dementia. A schematic representing the mechanisms of cerebrovascular injury because of hypoxia/hypoperfusion, metabolic dysfunction, and altered cerebrovascular hemodynamics with corresponding neuropathologic hallmarks leading to dementia. Additional pathologic markers of dementias include amyloid-β, cerebral amyloid angiopathy, phosphorylated-tau, Lewy Bodies, transactive response DNA-binding protein-43, fused in sarcoma inclusions, and reactive oxygen species such as nitric oxide, superoxide, and nitrogen peroxide.
Hypertension
Hypertension is a shared risk factor of AD and VaD associated with significant memory impairments in late life. 83 Chronically elevated blood pressure promotes cerebrovascular remodeling, leading to a reduction in the lumen diameter by an increase in the wall-to-lumen ratio. 84 Additional consequences of prolonged systemic blood pressure include altered morphology of small cerebral arterioles that supply vulnerable brain regions necessary for cognitive function. 84
Epidemiologic and preclinical studies have recently begun to address the complex interactions between aging, hypertension, and AD pathology. The direct effects of hypertension on Aβ accumulation have been shown in the brain parenchyma. The Honolulu Asia Aging study prospectively investigated the correlation between midlife blood pressure, Aβ plaque formation, and the risk for late-onset AD in 667 Japanese American men. The authors observed that Aβ-related risk for AD positively correlated with blood pressure, suggesting that chronic hypertension may compromise vascular integrity and is a major contributor to CAA as well as impaired Aβ clearance. 85 In support of these observations, animal studies using the spontaneously hypertensive stroke-prone rats, an animal model of chronic arterial hypertension and cerebral small-vessel disease, have demonstrated an age-dependent parenchymal Aβ accumulation similar to that observed in AD models. 86 Additional studies in the spontaneously hypertensive stroke-prone rats rat reveal an age-specific increase in Aβ deposition at 12 weeks of life, followed by APP overexpression at 20 weeks and p-tau by 26 weeks in the cortex and hippocampus. 87 Remarkably, the administration of antihypertensive drugs in an AD mouse model has been shown to restore cerebrovascular dysfunction by a significant reduction in oxidative stress, thus resulting in global improvements in cognitive function. 88
Efforts to determine the mechanisms of Aβ accumulation and the role of oxidative stress and mitochondrial bioenergetics as related to vascular impairment are underway. The first study showing a connection between the cerebrovasculature and the RAGE (receptor for advanced glycation end products), which regulates Aβ transport at the BBB, was recently published in a mouse model of hypertension. 89 Chronic vascular insult induced RAGE upregulation in the blood vessels of the cortex and hippocampus. Furthermore, RAGE inhibition reversed the hypertension-induced AD neuropathology phenotype, while reducing parenchymal Aβ deposition and improving cognitive function. 89 Interestingly, novel animal models have been recently created, expressing dual AD (APP and presenilin-1) and hypertension (angiotensin II infusion) pathology. 90 Cerebrovascular alterations, including a reduction in cerebral microvessel density and increased CAA deposition were reported, 91 correlating with a decrease in vascular endothelial growth factor-A expression and a reduction in nitric oxide synthase levels in experimental mice as compared with controls. These results agree with the prior studies92,93 and suggest that the chronic loss of endothelial nitric oxide in the cerebrovasculature may be an important contributor to the pathogenesis of sporadic AD by the regulation of Aβ, leading to cognitive decline. To counteract these effects, new evidence shows that the upregulation of the deacetylase, sirtuin 1 can prevent cerebral hypoperfusion injury through the deacetylation and upregulation of endothelial nitric oxide synthase, thus offering a novel mechanism for the restoration of cerebral blood flow. 94 Furthermore, mitochondrial dysfunction is a well-established hallmark of Aβ-induced neuronal toxicity. 95 Notably, microarray analysis revealed alterations in the genes relevant for hypoxic tolerance, energy/lipid metabolism, and mitochondrial bioenergetics/oxidative stress in 2- and 9-month-old spontaneously hypertensive rat model of hypertension. 96 However, the molecular sequence of events leading to hypertension-induced AD neuropathology has not been well characterized and additional studies are needed to address this issue.
Only a few studies, thus far, have addressed the relationship between hypertension and other dementia types. Neuroimaging MRI studies quantified white matter lesions and brain atrophy in late-life dementia. Earlier studies suggested that deep white matter hyperintensities did not correlate with measures of brain atrophy, ventricular dilatation, or age, but were associated with a history of hypertension, thus supporting an association of deep white matter hyperintensities with ischemic and vascular risk factors. 97 In contrast, no correlation was found between cardiovascular risk factors and FTD. Clinical evaluation of 100 FTD elderly patients (mean age > 70) as compared with 200 age-matched controls found no differences with rate of hypertension, dyslipidemia, and obesity. 98 However, further studies are needed to confirm these results. A previous study from our group has investigated the mechanisms of VCI in a hypertensive model of small-vessel disease (spontaneously hypertensive stroke-prone rats with carotid occlusion and a high-salt diet), using EPR (electron paramagnetic resonance) technology. 99 An EPR lithium crystal was implanted in vivo and used to monitor interstitial tissue O2 levels in the spontaneously hypertensive stroke-prone rats model. Our results demonstrated a reduction in tissue oxygenation within the white matter, which was exacerbated by age and associated with neuroinflammation. 100 In summary, these findings suggest a strong mechanistic link between cerebrovascular disease caused by hypertension and cerebral hypoperfusion. 99
Neuroinflammation
Neuroinflammation is prevalent in AD and related dementias. A growing body of literature links active inflammatory responses to AD neuropathology. 101 Epidemiologic data suggest that neuroinflammation serves as an independent predictor of early death in AD. 102 Microglia, the resident macrophage of the brain, have been shown to associate with Aβ plaques in AD. 103 Clustering of activated microglia and astrocytes have been reported surrounding Aβ plaques and correlated with elevated inflammatory cytokines such as interleukin (IL)-1, IL-6, and tumor necrosis factor-α.104,105 Furthermore, peripheral monocytes/macrophages are capable of clearing vascular Aβ. 106 In an in vivo multi-photon imaging study, circulating monocytes have been shown to enter luminal walls of Aβ-positive veins and carry amyloid back into the bloodstream. 107 These Ly6C+ monocytes naturally target and eliminate Aβ within the lumen of vessels.
Mechanisms of hypoperfusion-induced neuroinflammation in Alzheimer’s disease
Recent evidence suggests that hypoxia and pro-inflammatory mediators of neuroinflammation may precede Aβ and p-tau neuropathology, 108 while others argue that neuroinflammation is found downstream of AD protein aggregation.109,110 Hypoxia-induced inflammation has been shown to cause tissue damage and neurologic deficits in the central nervous system, correlating with an upregulation of an oxygen (O2)-dependent nuclear transcription factor, hypoxia-inducible factor-1α. 111 Hypoxia-inducible factor-1α is known to be upregulated during hypoxic stress and to activate a plethora of cellular regulatory factors, including the expression of pro-inflammatory and metabolic genes. 112 Recently our group has found that elevation of hypoxia-inducible factor-1α precedes white matter injury and neuroinflammation, leading to edema and BBB disruption in an animal model of VCI. 113 Consistently, reports of hemoglobin (Hgb), a major component of red blood cells known to increase blood O2 carrying capacity by 70%, 114 was significantly decreased in the red blood cells of AD patients as compared with non-AD controls. 115 In the early stages of AD, reduced Hgb reactivity has been found in practically all the neurons containing p-tau, NFTs, and α-synuclein, while Hgb expression was preserved in neighboring healthy neurons. 116 Additional studies support the important contributions of Hgb to hypoxia in AD by demonstrating its interactions with Aβ and co-localization with vascular amyloid deposits in post-mortem AD brain tissues. 117 Recently, data from triple transgenic AD mice confirmed a 50%–70% reduction in total brain tissue Hgb concentrations between 3 and 20 months of age. 118 Moreover, this severe state of hypoxia corresponded with increased concentrations of Aβ1–40 and Aβ1–42, as well as cerebrovascular factors such as vascular endothelial growth factor and endothelial nitric oxide synthase in the brain. Yet the spatio-temporal alterations in Hgb expression and correlation to Aβ and p-tau pathologies as well as hypoxia-inducible factor-1α/Hgb role in non-AD dementias remain unknown.
The role of neurovascular inflammation in dementia subtypes
The effects of neurovascular inflammation injury to the blood vessel wall because of adhesion of inflammatory markers are currently being investigated using novel AD animal models and innovative techniques. These studies have led to the observation that such ‘neurovascular inflammation’ is mediated by the peripheral systemic immune response, and that the two immune systems may work synergistically, thus influencing AD neuropathology and cognition. 101 Although significant neurovascular inflammation has been reported, its role and contributions to the AD brain remains unclear.
Reports of neuroinflammation in other AD-related dementias further support its critical contributions to disease pathophysiology. For instance, in the early phases of DLB, patients showed extensive microglia activation in several associative cortices, as measured by the binding potential of [(11)C]-PK11195 with positron emission tomography. 119 On further examination, it was discovered that extracellular α-synuclein released from neuronal cells serves as an endogenous agonist for Toll-like receptor 2, which activates inflammatory responses in microglia. 120 The activation of the pro-inflammatory cascade was also reported in FTD patients with the P301S human tau-protein mutation. Activated microglia and infiltrating macrophages were detected in the cortex and hippocampus, along with IL-1β and cyclooxygenase-2 upregulation in neurons and glial cells, thus enhancing disease progression. 121 Moreover, the immune response was shown to be critical in cerebral small-vessel disease and VaD, displaying increased T-cell production in the microvasculature and decreased IL-10 levels in the CSF. 122
Neurodegeneration
One timely question in the field is whether neurodegeneration precedes Aβ and tau formation or whether neuroinflammation, cerebrovascular dysfunction, and BBB injury occur after neuronal cell death. Several labs have begun to address this question. The consensus among these studies, including those from our own lab (unpublished data), seem to indicate that cerebrovascular impairment, neuroinflammation, neurodegeneration, and tauopathies precede Aβ plaque formation and may occur independently of Aβ deposition, thus promoting VCI and the early opening of the BBB.123,124 However, the interaction between neurodegeneration and β-amyloidosis in AD and the pathologic sequence of events leading to cognitive impairment remains to be determined. In 2010, Jack et al. 125 proposed a hypothetical model integrating a biomarker timeline of neuropathologic events culminating in cognitive impairment. The hypothesis highlighted the preclinical stages of AD, stating that Aβ changes occur rapidly at first, followed by a slower progression of p-tau, alterations in brain structure, memory loss, and cognitive deficits, presented as a sigmoidal curve. In the current, revised 2013 version, referred to as the ‘integrative model’, tau pathology appears before Aβ, although the latter can be detected first in CSF during middle-age. 126 Additional neuroimaging studies from the same group exploring the link between amyloid accumulation and neurodegeneration in nondemented individuals are now emerging. A cross-sectional study in 50–89-year-old subjects revealed an age-dependent increase in cerebral β-amyloidosis and neurodegeneration. Investigators found that Aβ and neurodegeneration associated with old age, apolipoprotein E-ɛ4 carriers, and women. 127 This was confirmed by other neuroimaging studies achieving similar results, 128 suggesting that the accumulation of Aβ and neurodegenerative pathologic correlates, associated with an abnormal MRI, is inevitable by old age, albeit that the clinical presentation is of normal cognitive function. 127
Several potential cerebrovascular risk factors have been implicated in neurodegeneration. The first is fibrinogen, known to induce clot formation. The deposition of fibrinogen in the AD brain promotes neuronal cell death and correlates with disease pathology. Moreover, small decrease in fibrinogen levels were shown to promote neuronal health and reduce Aβ pathology in an AD mouse model. 129 Second, accumulating evidence suggests both Aβ and p-tau may trigger neurotoxicity.130,131 Third, clinicopathologic reports of VaD patients demonstrate regional pyramidal neuronal atrophy in the dorsolateral prefrontal cortex, corresponding with diminished executive function, suggesting a vascular basis for the highly specific pyramidal neuron atrophy in individuals with dementia. 132
Finally, previous studies report an association between loss of function in α-synuclein (for DLB) tau, TDP-43 and FUS (for FTD), and neuronal atrophy (implicated in familial DLB 133 and FTDs). 134 However, data supporting the mechanisms of neurodegeneration in familial DLB/FTDs and neurodegenerative changes in the sporadic form of the DLB/FTD are currently unavailable.
Blood–Brain Barrier Dysfunction
The neurovascular unit, composed of neurons, astrocytes, myocytes, pericytes, extracellular matrix components, and endothelial cells forming the tight junctions of the BBB, is known to create a protective biochemical barrier between the brain microenvironment and the peripheral circulation. 135 Pathophysiologic changes occurring throughout the aging process, and exacerbated by cerebrovascular and neurodegenerative disease, may result in blood vessel tortuosity and vessel-wall thickening, causing reduced vascular reactivity. Vascular risk factors such as hypertension and diabetes associated with small-vessel disease may lead to BBB abnormalities, 136 reflected in increased albumin levels in the CSF and matrix metalloproteinase upregulation, known to disrupt tight junction proteins and promote cerebral edema. 137
In the AD brain, neurovascular unit pathology has been described as a consequence of Aβ accumulation along blood vessel walls (CAA) and profound neurovascular inflammation. 138 Reduced Aβ clearance has been described in AD and is associated with reduced cerebral blood flow and impaired cognitive function. 139 Epidemiologic data support an important role for pericytes, as their number and density in the cortex and hippocampus of AD patients was significantly reduced as compared with nondemented controls. 140 Similar observations in Aβ overexpressing mice showed that pericyte deficiency causes an upregulation in Aβ and p-tau expression. 141 Although significant progress has been made in BBB research, our understanding of the neurovascular unit integrity is incomplete in neurodegenerative dementias. Future studies should target a molecular biomarker, which may provide a readout for BBB leakiness in living patients, thus helping us understand the relationship between BBB dysfunction and progression of AD dementia.
Furthermore, BBB damage has also been noted in the other dementias. Observations of significant white matter abnormalities and demyelination because of underlying microvascular dysfunction in FTD were reported. 15 A separate study describes the rare occurrence of cerebrovascular lesions, 142 whereas microbleeds were more prevalent in the cerebral cortex of DLB brains. 142 Additional analyses by the same group revealed the prevalence and severity of cerebrovascular lesions in post-mortem brains of patients with DLB as compared with age-matched controls. 14 The authors concluded that these histopathologic changes reflect the neurodegenerative process, which is associated with BBB injury. 14 Increased BBB permeability reported in VaD patients corresponds to a higher mean albumin ratio (CSF/serum) as compared with healthy controls. 143 As there was no correlation between the albumin ratio and clinical vascular risk factors, the authors concluded that the observed BBB disruption is most likely because of small-vessel disease rather than cerebral infarcts. These findings were also confirmed among elderly individuals diagnosed with VaD, thus providing evidence for BBB leakiness as an early marker of cerebrovascular change in the disease process before the onset of clinical dementia. 144 A possible cause for the observed BBB damage in VaD may be attributed to the presence of matrix metalloproteinases in the white matter of patients as a result of underlying microvascular disease. 145
Conclusions and future directions
In conclusion, this review highlights several important key points. First, the possible multifactorial nature of sporadic AD and related dementias is associated with vascular etiologies as a part of the disease process. Second, the review focuses on the molecular cascades and associated cerebrovascular factors contributing pathologically to different dementias. Third, although the timing of disease symptoms and progression is important, often patients present with significant tau and Aβ load by the time symptoms begin. Given the contributions of vascular impairments at certain points during the disease process, it is important to investigate the vascular pathology before the disease becomes too severe to reverse. Finally, it is clear that the majority of dementia types involve an underlying small-vessel disease or cerebrovascular dysfunction at some point during disease progression. However, the causality dilemma still exists in identifying the circular cause and consequences between vascular dysfunction and the clinical signatures of different dementias, which is an important exploratory topic of future research.
A recent report released by the Alzheimer’s Association, in collaboration with NINDS, argues for the need to create basic science animal models to address the neurobiologic mechanisms underlying sporadic AD and related dementias and to develop novel biomarkers for clinical trials. 146 More specifically, several top priority objectives were recognized in the field: (1) animal models to understand the mechanistic link between cerebrovascular dysfunction and cognitive decline and to investigate the neuropathologic time course of neuronal and white matter injury. Small-vessel disease models mimicking human AD-related and mixed dementias are particularly needed; (2) biomarkers capable of detecting preclinical dementia are necessary, as early detection is the key to future therapeutic interventions. If it fails, neurodegeneration will be impossible to reverse once pathologic cascades are initiated; (3) reducing Aβ levels may hold promise in asymptomatic persons to delay the onset of cognitive impairment; 147 (4) the establishment of pathologic boundaries between normal aging and AD-related dementias using better diagnostic, biomarker-based criteria is essential for improved clinical outcomes. 148
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by NIH, RO1 grant, R01NS083704 (PI: KB) and NIH/NINDS, RO1 NS045847-07A1 (PI: Gary Rosenberg).
Acknowledgement
The authors would like to thank our collaborators, Drs Karen Santa Cruz and Gary Rosenberg for their guidance and support throughout the review drafting and revisions process.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
All the authors contributed equally to the conceptualization, design, and outline of this manuscript. LR collected the literature, created the figures, and provided the initial draft of the review. JK offered valuable clinical insight to answer the reviewers’ comments and ensure clinical accuracy of the manuscript during the revisions process. KB provided feedback throughout the entire writing process and was instrumental during the editing process.