
Abstract
Purpose:
Upon identifying the need for an alternative treatment option in the management of hepatitis C to decrease viral load and improve health parameters, the investigator has developed the hepatitis C virus (HCV) nosode.
Methods:
An open-label observational study in 24 HCV-positive individuals was conducted by using the HCV nosode at 30C and 50C potencies.
Results:
In this clinical trial, the HCV nosode was administered to HCV-positive participants. From week 12 to week 24, the mean viral load decreased; the median viral load decreased by half, from 1,557,567.50 IU/mL to 789,265.50 IU/mL. However, at 24 weeks, the average viral load increased significantly (p = 0.2206) in the participants completing the trial. The study has shown a double population: a large set of responders with marked improvement (week 12 [p = 0.0120] and week 24 [p = 0.0304] and from week 12 to week 24 [p = 0.0028]) and a small set of nonresponders with increasing viral load (week 12 [p = 0.0120] and week 24 [p = 0.0304] and from week 12 to week 24 [p = 0.0028]). Most participants in this study showed improvement in appetite and weight gain. The treatment using the nosode was found to be safe in the tested population.
Conclusion:
The HCV viral load was affected by using ultra-diluted preparation sourced from HCV, as per the Law of Similars, in responders. Further studies of longer duration in patients with uniform baseline characteristics and those that adjust the potency to the individual participant's requirement are recommended.
Introduction
H
Therapy for hepatitis C should be affordable for low-income countries; affordable treatment would also prevent transmission of the virus. Alternative medications with disease-modifying activity may be of benefit. 4 Many people who have hepatitis C choose to follow an alternative method of treatment because of adverse effects (AEs), high cost, varying success rates, and dissatisfaction with current medical therapy. Many patients with hepatitis C do not begin treatment but rather wait for newer, cheaper, and more effective drugs. Eradication of the virus, its reduction, and slow disease progression are the areas of interest for research in hepatitis C. This is especially the case for a growing number of people for whom treatment has not been successful and for those with a lower chance of achieving an SVR. More effective and better-tolerated treatments are urgently needed. 6
After identifying the need for alternative treatment to address the immune system and slow disease progression by reducing viral counts, the investigator developed a new homeopathy HCV nosode 7 and evaluated its effect on HCV-positive participants in a clinical trial. The preparation, standardization, safety, and evaluation of efficacy of the hepatitis C nosode were part of this project.
Materials and Methods
HCV nosode preparation
An institutional ethics committee approved the project for HCV nosode preparation and the informed consent forms. Blood samples from HCV-positive consented donors were collected for preparing the nosode. Samples containing HCV type I and III (and negative for other coinfections) were mixed and potentized as per the potentization method described in the Homeopathic Pharmacopoeia of India (HPI). 8 The author has developed 15 steps, standardized according to a scientific method of nosode preparation, for the HCV nosode. The source material, preparation process, and standardization with respect to viral count by polymerase chain reaction, polyacrylamide gel electrophoresis, pH, and the process of potentization (electromechanical device that calculates the impact of stroke) were documented. The HCV nosode in 30C and 50C potencies was prepared. 9
The extremely diluted (potentized) homeopathic medicines are generally administered in 30C or 200C (or more) potencies. A potency of 1C is achieved by making a solution of one part of drug substance mixed with 99 parts of vehicle (10−2) (generally alcohol, as per HPI), which undergoes rigorous succussions. One part of 1C is again mixed with 99 parts of vehicle to repeat the process, to arrive to 2C (10−4) 3C, 4C, and so forth to 30C (10−60) are prepared. 7
Clinical trial with HCV nosode
An open-label observational study in 26 HCV-positive individuals was conducted (Table 1 shows demographic characteristics). The trial was conducted at Life Force research center, which has a research team composed of five staff members (four physicians and one clinical trial expert). Individuals were enrolled in the clinical trial as per inclusion and exclusion criteria described in the protocol. The first participant was enrolled on October 13, 2010, and the last participant completed the last visit on November 1, 2012. The prescription were prescribed six pills for the HCV nosode (pallet size, 30) three times daily: 30C for the first 3 months and 50C for the next 3 months. This predefined protocol for potency (30C and 50C) and repetition (initially and 3 months later) was based on a similar study of an HIV nosode. 10 Because there was no precedence of any organized study of treatment of any chronic viral infection, such as hepatitis C, the investigator developed the protocol for potency and repetition for the study.
Values expressed with a plus/minus sign are the mean ± standard deviation.
Repetition of the daily dose was chosen on the basis of the aggressive nature of this infection, which tends to keep multiplying naturally; infrequent repetition was not considered adequate because of the author's personal clinical experience with the treatment of hepatitis and HIV. Future studies can be done to examine the effect of higher potencies. Study personnel took care of all documentation, such as participants' informed consent forms, laboratory investigations, and safety and ethical measures. Participants were monitored for viral count at scheduled visits at screening, week 12, and week 24. HCV RNA was quantified by standard procedure on COBAS AmpliPrep and COBAS TaqMan HCV test (HCV loadtestings were done by Metropolis Laboratory Mumbai, India, which used Roche Molecular System's analyzer [Pleasanton, CA]). The participants were followed critically for 6 months and the collected data were analyzed. Viral load reduction as primary outcome was examined at the end of week 12, at the end of week 24, and for the change from week 12 to week 24. Responder's viral load reduction, nosode safety, and general health parameter in terms of appetite, weakness, and weight gain were accessed as secondary outcome measurements. At each visit, the symptoms related to illness and associated conditions were recorded in case record form and a database. From these data, individual participant narratives were prepared and analyzed.
Patient population
Inclusion criteria
Participants satisfying all of the following criteria were included in the trial: (1) HCV viral load greater than 100,000 IU/mL, (2) no previous receipt of any experimental HCV vaccines, (3) male or female patients age 18–65 years inclusive at the time of signing the informed consent, (4) ability to receive an optimized regimen, (5) free from any acute infection or serious medical illness within 14 days before study entry, (6) ambulatory status and ability and willingness to give informed consent, (7) patients without actual or suspected allergies to any component of investigational product, (8) female patients of childbearing potential having negative pregnancy test before intraperitoneal administration, and (9) men and women who agreed to use effective birth control methods during the study.
Exclusion criteria
Participants satisfying any of the following criteria were excluded: (1) leukocyte count less than 3000 cells/mm3, platelet counts less than 80,000 cells/mm3, and bilirubin level greater than 1.5 mg/mL, (2) any other chronic coinfection (hepatitis B virus or HIV), (3) patients treated with conventional anti-HCV (interferon) treatment during the past year, (4) cancer of liver (detected on ultrasonography [USG]) or any form of malignancy, (5) patients with symptoms of esophageal varices, (6) renal failure, (7) coexistent serious cardiac and respiratory diseases, (8) neuropsychiatric disorders, (9) lactating mothers, (10) alcohol abuse, and (11) insulin-resistant diabetes (patients with chronic hepatitis C are at risk of developing type 2 diabetes mellitus and impaired fasting glucose, and this risk may increase among HCV–infected patients not responding to antiviral therapy.)
Guidelines, ethics, compliance, and approvals
The project was reviewed and approved on September 16, 2010, by the institutional ethics committee, as per the Indian Council of Medical Research guidelines. 11 The requirements for the obligations of investigators as per the “Guidance on Good Clinical Practice”/International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use Independent Ethics Committee (GCP-ICH) 12 were met. The clinical trial has been registered (CTRI/2011/12/002261) with the Clinical Trials Registry-India. 13 The study was conducted as per the GCP-ICH and available regulatory guidance for homeopathic medicines.
Statistical analysis
An independent agency (contract research organization) carried out data entry and data analysis (after the database was locked). Continuous data are presented with descriptive statistics: number, mean, standard deviation, median, and minimum and maximum values. Categorical data are presented as frequency and percentage. All p-values are reported on the basis of a two-sided significance test, and all statistical tests were interpreted at a 5% level of significance. Analysis was carried out using SAS software (SAS Institute, Inc., Cary, NC). Individual-patient narrative was prepared from the subjective qualitative data collected at each visit and was analyzed at the end of the study.
Results
Of 26 randomly assigned participants, 21 were male and five were female; the mean age was 42 years, and the mean weight was 63 kg. Twenty of 24 participants completed the study. One participant voluntarily withdrew consent, and five participants were lost to follow-up. Of the five participants lost to follow-up, four had completed the study until visit 2 and hence were considered as evaluable (total of 24 participants were evaluable) (Fig. 1).

Flowchart. HCV, hepatitis C virus. *Participants who completed visit (V) 2 were considered evaluable for analysis.
At 24 weeks, the average viral load increased significantly in the participants completing the trial. However, from week 12 to week 24, the mean viral load decreased; in this period the median viral load decreased by half, from 1,557,567.50 to 789,265.50 IU/mL. The highest viral load was 7,136,934 IU/mL, and the lowest was 136,907 IU/mL. The study showed a double population: a large set of responders with marked improvement and a small set of nonresponders with increasing viral load (Table 2). Of 24 participants, 17 (71%) had responded (viral load reduction) to the nosode at week 12 or week 24 or from week 12 to week 24. Seven participants (29%) did not respond (no viral load reduction) to the treatment.
Unless otherwise noted, values are expressed as IU/mL. SAS software: Table 14.2.1.sas; analysis dataset: INFSTA.sas7bdat; table generation: 04JUN13:10:47:38. Change from baseline is calculated as screening visit − postbaseline visit. For any missing value in the data, last available postbaseline value is carried forward using last-observation-carried-forward method. p-Value was calculated using paired t-test at 5% level of significance.
SD, standard deviation; min, minimum; max, maximum.
The participants who responded to the treatment showed an average 56.52% reduction in viral load. The reduction in viral load was statistically significant in responders from baseline to week 12 (p = 0.0120) and week 24 (p = 0.0304) and also from week 12 to week 24 (p = 0.0028) (Table 3).
Unless otherwise noted, values are expressed as IU/mL or percentages. SAS software: Table 14.2.3.sas; analysis dataset: INFSTA.sas7bdat; table generation: 04JUN13:12:30:07; data source: listing 15.2.1 Percentage change from baseline is calculated as ((baseline – postbaseline value)/baseline) × 100. p-Value was calculated using paired t-test at 5% level of significance. Analysis has been done for average percentage improvement in viral load only in responders.
Six participants (25% participants) showed an average 56.52% improvement (decrease in viral load) at the end of 12 weeks (28.90% to 96.34%) (Fig. 2). Eleven participants (45.83%) showed an average 51.81% improvement (decrease in viral load) at the end of 24 weeks of treatment. Thirteen participants (51.16%) showed an average 52.63% improvement (decrease in viral load) between 12 and 24 weeks (2% to 96%) (Figs. 3 and 4).
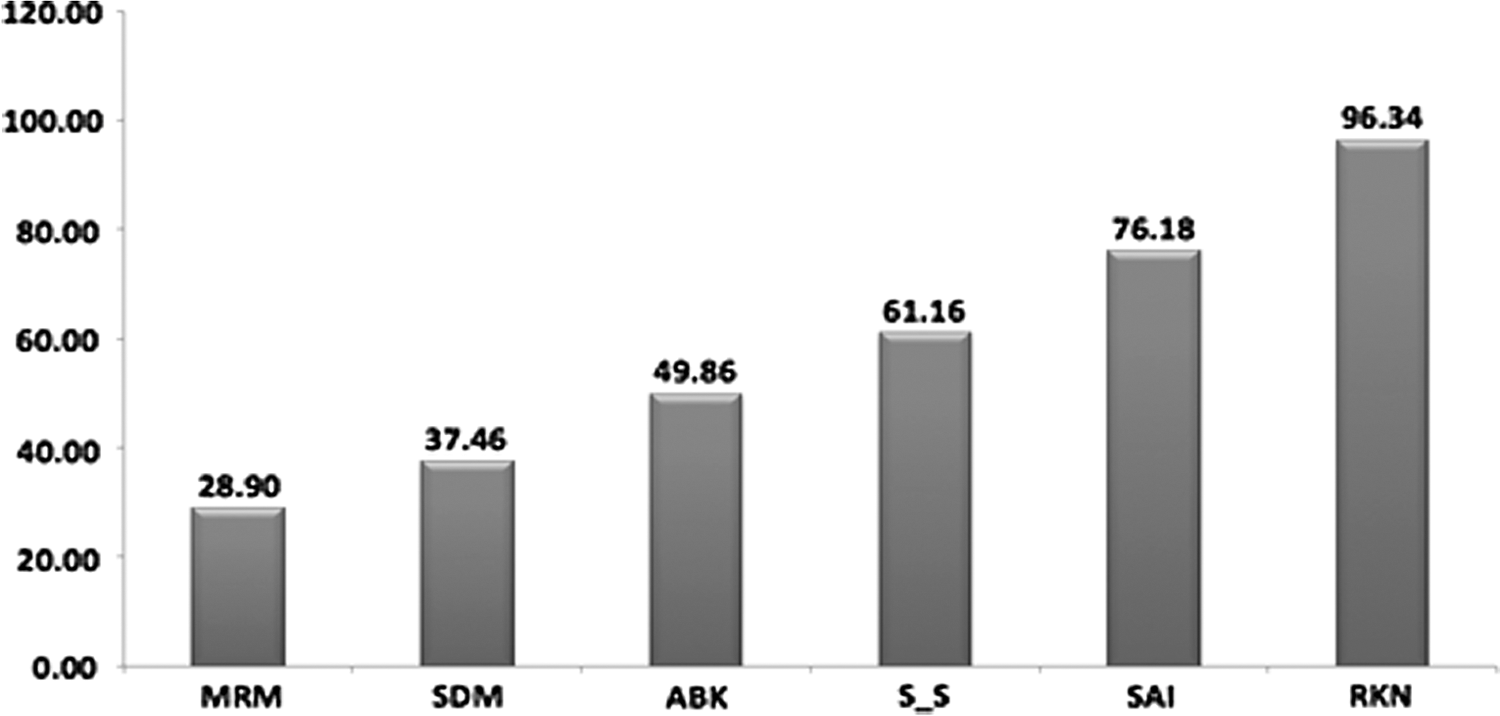
Percentage improvement in viral load reduction in responders at the end of 3 months. Letters are from alphanumeric codes used to identify patients.
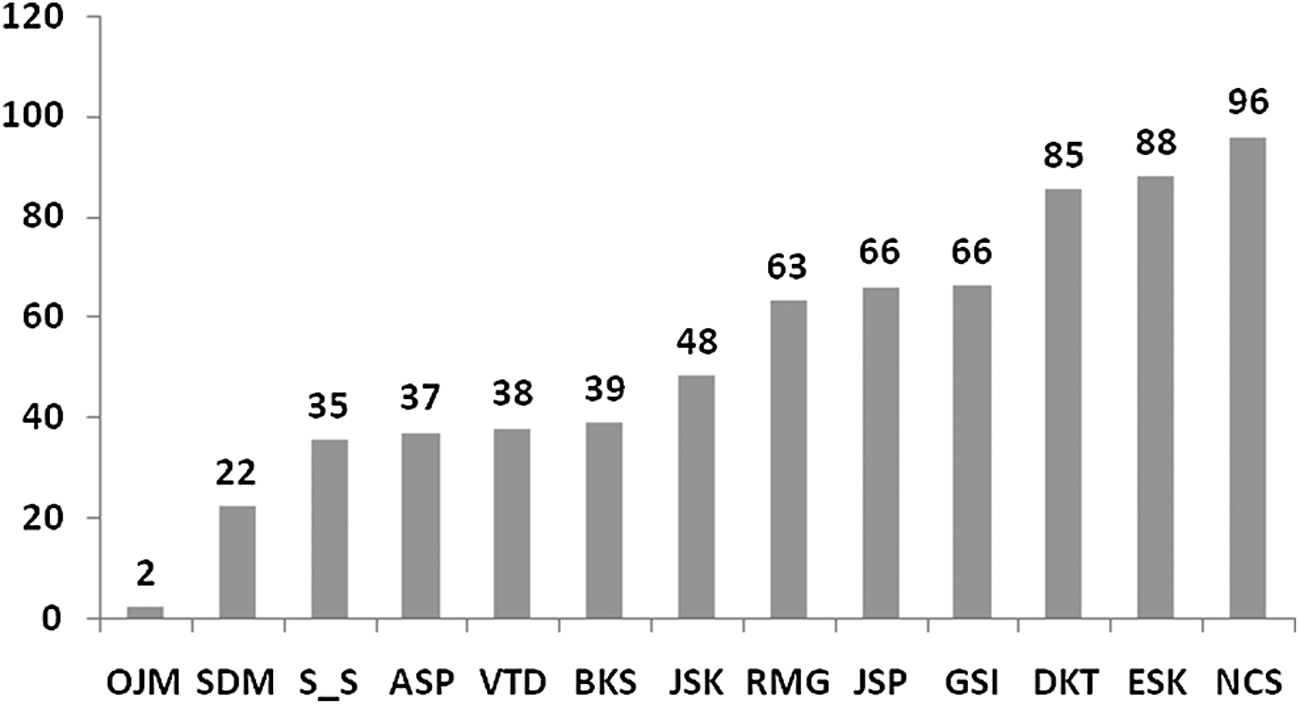
Percentage improvement in viral load reduction in responders at the end of 6 months. Letters are from alphanumeric codes used to identify patients.

Visit-wise viral load in responders as compared with baseline at the end of 6 months. Letters are from alphanumeric codes used to identify patients.
In the nonresponder group, viral load was increased at week 12 (p = 0.0603) and week 24 (p 0.1045) and from week 12 to week 24 (p = 0.2938). However, the increase in viral load (p = 0.2206) was not statistically significant (Table 4 and Fig. 5).
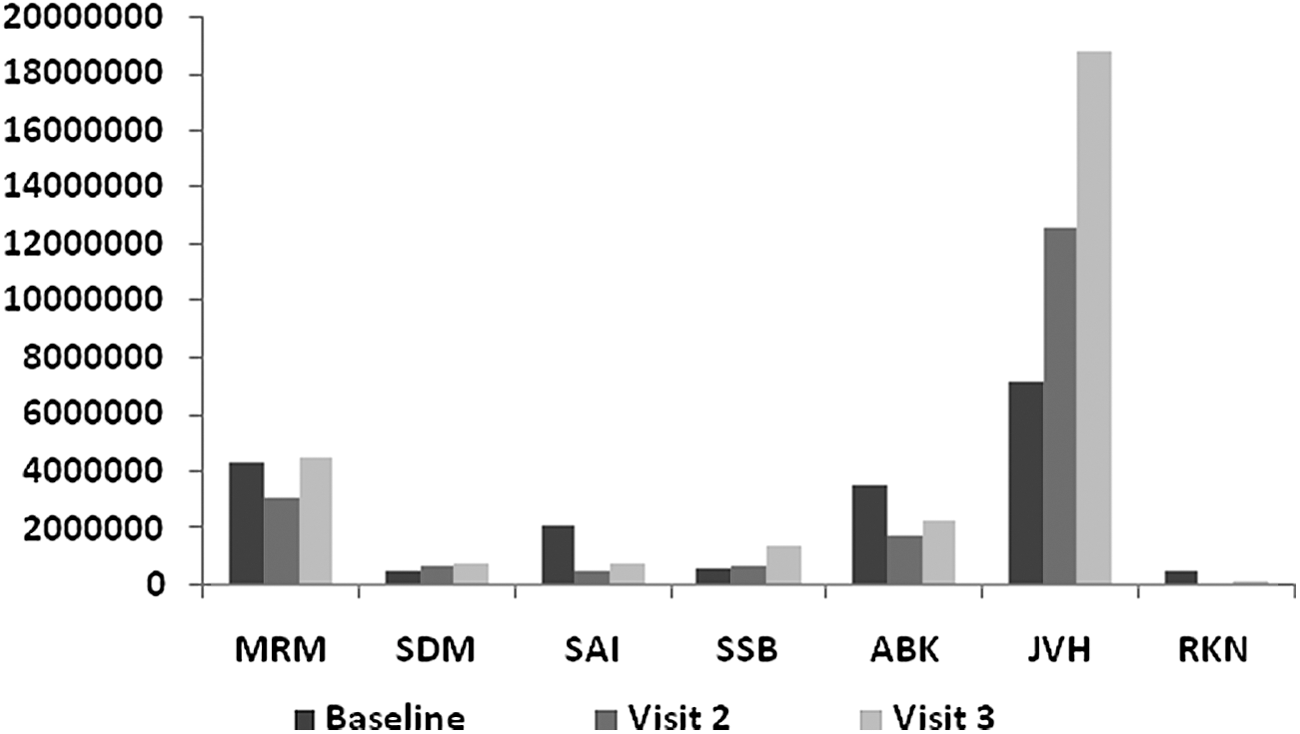
Visit-wise viral load in nonresponders as compared with baseline at the end of 6 months. Letters are from alphanumeric codes used to identify patients.
Unless otherwise noted, values are expressed as IU/mL or percentages. SAS software: Table 14.2.3.sas; analysis dataset: INFSTA.sas7bdat; data source: Listing 15.2.1. Percentage change from baseline is calculated as ((baseline – postbaseline value)/baseline) × 100. p-Value was calculated using paired t-test at 5% level of significance. Analysis has been done for average percentage improvement in viral load only in nonresponders.
No significant change was observed in the percentage of responders for sustained viral reduction from week 12 to week 24 in the population studied. However, Table 3 shows that the mean viral load for responders was 1,151,274.83 IU/mL at week 12 and 623,940 IU/mL at week 24. This reduction in viral load may be due to the contribution of great improvement in a few participants at week 24. Only two responders who showed improvement from baseline to week 12 continued the improvement from week 12 to week 24. There were different groups of participants who showed improvement at week 12 and week 24 and from week 12 to 24. Seven (27%) of 26 participants experienced adverse events (AEs). None of the AEs were related to the study drug. No serious AEs were reported during the study.
Most of the AEs (72.73%) experienced by the participants belonged to the system organ class of “infections and infestations” (11.54%) and “respiratory, thoracic and mediastinal disorders” (11.54%). The most common AEs reported were cough (7.69%) and productive cough (7.69%), followed by pyrexia (31.82%). All the participants were treated with concomitant medications, except one for whom no action was taken. Concomitant medication is a drug or biological product, other than the investigational product, not targeted for the treatment of hepatitis C. During the study, the participants used appropriate concomitant medication (e.g., Omez-D [Dr. Reddy's Laboratories, Hyderabad, India], Cordarone [Pfizer, New York, NY], antibiotics, calcium supplement) were used by the participants.
No participants discontinued treatment because of AEs. No AEs were suspected to be due to the study drug. All the AEs reported during the study were mild to moderate in intensity.
Discussion
Hepatitis C has been identified as a serious infection in humans, presenting a challenge for its treatment and calling for examining different possible methods. Homeopathy, which originated in Germany in 1796, is based on The Law of Similars, which states that any substance that has a capacity to produce symptoms in humans also has the capacity to relieve similar symptoms if administered in very small doses. 14 Nosodes are homeopathic preparations sourced from live organisms (e.g., Mycobacterium tuberculosis, Neisseria gonorrhea) used in potentized forms since 1878. 15,16 The author has prepared a new nosode in potentized form, prepared from HCV, with documented safety in healthy human volunteers. 7 In addition, the homeopathic pathogenetic trial, a placebo-controlled study in healthy human volunteers, has demonstrated effects of an ultra-diluted, potentized nosode at 30C potency by producing a set of symptoms similar to those during HCV infection. 7 As a next step, the current clinical trial was conducted in participants with HCV infection to examine the fundamental Law of Similars.
This exploratory study was conducted to examine the role of homeopathy in the light of laboratory-based immunologic parameters. Participants were prescribed the HCV nosode at 30C potency for the first 12 weeks and at 50C potency for the next 12 weeks. This potency regimen was based on the investigator's experience with the nosodes. Because of the active presence of the virus in HCV-infected individuals, a regimen of frequent repetition of the nosode was chosen rather than an infrequent repetition, which is a common practice when nosodes are used in homeopathy for conditions other than infection with the same organisms. In a similar study of treatment of HIV-infected individuals with the HIV nosode, an identical potency and repetition protocol were followed. In addition, the investigator has substantial experience with the use of frequent repetitions in the treatment of HIV, 10 hepatitis C, and other infections.
The reduction in viral load was statistically significant in responders from baseline to week 12 (p = 0.0120) and week 24 (p = 0.0304) and from week 12 to week 24 (p = 0.0028). The clinical trial demonstrated the effect of the HCV nosode in reducing the viral load to a certain extent without adverse effects, opening new avenues for further studies. Viral load, measured in terms of log change, is considered a standard measure in hepatitis treatment with interferon (or antiviral agents), and the response to the treatment is thus based on logarithms. This study enrolled participants whose viral load exceeded 100,000 IU/mL, without setting any upper limit. As a result, the intensity of the disease at baseline was not uniform.
The objective of the study was to examine the effect of the nosode on viral load. This was an observational study, and viral load change was calculated in numbers of viral copies. This study demonstrated that a potentized HCV preparation could reduce the viral load.
The participants were administered the HCV nosode as a blanket prescription without consideration of the constitutional symptoms. Studies may be carried out to examine whether this nosode could produce superior results if supported by concurrent use of constitutional medicine based on individualized symptoms.
The individual participant narratives reveal that most participants showed some improvement in their general health. Most participants reported improvement in symptoms, such as appetite, general well-being, and weight gain, and felt energetic. This finding indicates that the HCV nosode could help improve the overall health of participants with chronic hepatitis. Although the viral load did not decrease significantly in nonresponders, the improvement in the general condition of the participants may mark the beginning of a response to treatment. A mixed response was observed at week 24, wherein some participants improved and some had a relapse in their viral load.
Six genotypes have been identified to date. The polyvalent nosode was intended to include as many genotypes as possible. The investigator could find donors for genotype I and III that met the inclusion criteria. The HCV nosode could be updated with more genotypes to evaluate the response accordingly, and further research could be undertaken.
The HCV nosode was observed to be safe in the treatment of participants with hepatitis C, and none of the AEs reported during the study were related to the study drug.
Response to standard therapy is usually based on SVR and persistence of SVR and, when supported by other histologic findings, can be considered to represent eradication. The study was conducted for 24 weeks to explore the possibility of a response. In light of a few beneficial effects observed during the trial, a follow-up study of a longer duration and with a larger sample size and different potencies is warranted. Clear and superior results could be expected with a longer duration of observation and uniform baseline characteristics and after adjustment of the potency per the susceptibility of individual participants. More studies with various nosodes could also be explored for different diseases.
Footnotes
Acknowledgments
The author thanks members of the institutional ethics committee; subject experts for their technical, ethical, legal, and medical input; and the volunteers for their participation in the study.
The HCV nosode trial project was funded by Homeopathy India Pvt Ltd. The sponsors had no role in data collection, analysis, and interpretation.
Author Disclosure Statement
No competing financial interests exist.