
Abstract
Many drugs and other small molecules used to modulate biological function are amphiphiles that adsorb at the bilayer/solution interface and thereby alter lipid bilayer properties. This is important because membrane proteins are energetically coupled to their host bilayer by hydrophobic interactions. Changes in bilayer properties thus alter membrane protein function, which provides a possible mechanism for “off-target” drug effects. We have previously shown that channels formed by the linear gramicidins are suitable probes for changes in lipid bilayer properties, as experienced by bilayer-spanning proteins. We now report a gramicidin-based fluorescence assay for changes in bilayer properties. The assay is based on measuring the time course of fluorescence quenching in fluorophore-loaded large unilamellar vesicles, due to entry of a gramicidin channel-permeable quencher. The method is scalable and suitable for both mechanistic studies and high-throughput screening for bilayer-perturbing, potential off-target effects, which we illustrate using capsaicin (Cap) and other compounds.
Introduction
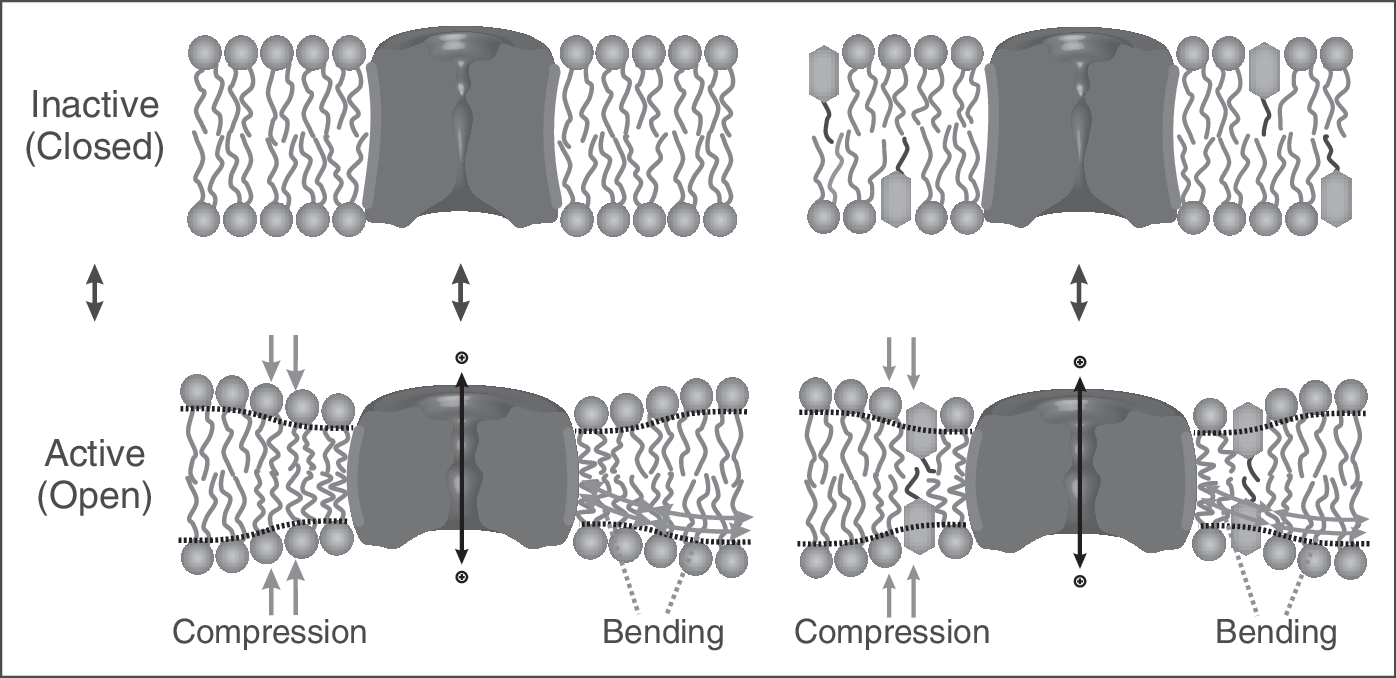
Amphiphiles can alter protein function without binding to the protein. The hydrophobic coupling between membrane proteins and their host bilayer provides for energetic coupling of the protein conformational preference to bilayer material properties, as the bilayer adjusts to the changes in protein conformation. The figure illustrates this for an ion channel, where the closed and open states have different hydrophobic lengths. The associated bilayer deforming energy will contribute to the equilibrium distribution between the states. Because amphiphiles can alter the bilayer material properties (compression and/or bending moduli), they can alter protein function without binding to the protein.
Drug adsorption to the bilayer/solution interface, taken together with the energetic coupling between membrane proteins and their host bilayer, thus provides a mechanism for nonspecific, off-target changes in membrane protein function by small molecules. 12 –17 Moreover, increased hydrophobicity leads to molecules with higher lipid bilayer partition coefficients, 18,19 and thus increased bilayer-modifying propensities. The hydrophobic coupling between lipid bilayers and embedded proteins 14,20 thus provides insights into why increased hydrophobicity is associated with increased attrition in drug development. 2,21 That is, whether or not the intended target of an amphiphilic drug or drug lead is membrane-embedded, these molecules may alter membrane protein function by adsorbing to the bilayer and thereby altering the energetics of membrane protein conformational changes. It thus becomes important to have methods to probe the bilayer-perturbing propensities of drug leads, and to determine at what concentrations they perturb lipid bilayer properties sufficiently to alter membrane protein function.
Many different methods are used to characterize how small molecules perturb lipid bilayer properties, for example, 18,19 but it remains unclear how changes in bilayer properties may alter the function of bilayer-embedded membrane proteins. It is therefore advantageous to have comprehensive methods that can detect whether amphiphiles alter lipid bilayer properties as sensed by bilayer-spanning proteins. One such method is to use gramicidin A (gA) channels as probes. 22 gA channels have been extensively employed as molecular force probes to monitor changes in collective bilayer properties using electrophysiological (single-channel) methods 8,12 –17,23 (see refs. 22 and 11 for recent reviews). The single-channel approach is powerful but difficult to scale. We therefore developed a gramicidin channel-based fluorescence assay, using large unilamellar vesicles (LUVs) filled with a fluorophore and doped with gramicidin (Fig. 2). Changes in the gramicidin monomer ↔ dimer equilibrium, caused by the adsorption of amphiphiles, can be measured by adding a gramicidin channel-permeable fluorescence quencher to the extravesicular solution and measure the time course of fluorescence quenching in the absence and presence of the amphiphiles. The method will work with many different combinations of fluorescent indicators and gramicidin channel-permeable ions; we chose the fluorescence indicator/quencher pair 8-aminonaphthalene-1,3,6-trisulfonate (ANTS)/Tl+, which has been successfully used previously. 24,25
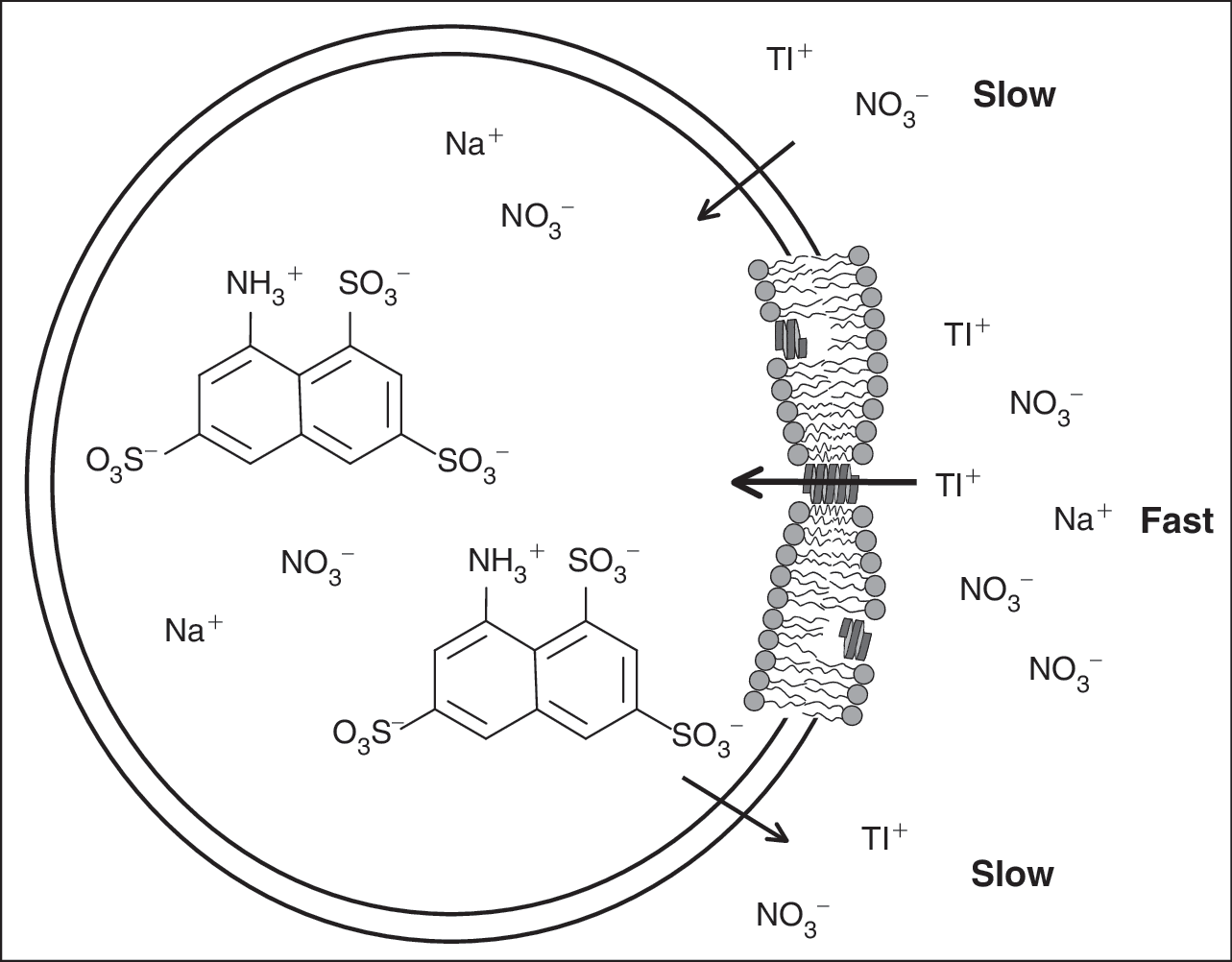
A fluorophore-loaded large unilamellar vesicle (LUV) doped with gramicidin A (gA) and exposed to quencher. Illustrated here with the 8-aminonaphthalene-1,3,6-trisulfonate (ANTS)/Tl+ fluorophore/quencher pair. Tl+ and ANTS cross the lipid bilayer poorly, whereas gA channels are Tl+ permeable. The rate of Tl+ influx into the vesicle, and the rate of fluorescence quenching, is proportional to the number of (bilayer-spanning) conducting gA channels in the LUV. The “expanded” view to the right shows a lipid bilayer segment with the 2 major gA forms: nonconducting monomer and conducting dimers.
Materials and Methods
Fluorescent Liposomes
The fluorophore 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) disodium salt, from Invitrogen (Eugene, OR), was loaded into LUVs using hydration/mini-extrusion. 26 1,2-Dierucoyl-sn-glycero-3-phosphocholine in chloroform (Avanti Polar Lipids, Alabaster, AL) was used without further purification. For each batch of LUVs, the lipid solution was dried under nitrogen and further dried in a dessicator under vacuum overnight, to fully remove chloroform. The dried lipid film was rehydrated in 100 mM NaNO3, 25 mM ANTS, 10 mM HEPES (pH 7.0) at room temperature overnight; the electrolyte volume was adjusted to give a 10 mM lipid suspension. The suspension was sonicated at low power for 1–2 min, subjected to 5–6 freeze-thaw cycles, and extruded 21 times, at room temperature, using an Avanti mini-extruder and a 0.1 μm polycarbonate membrane filter. Extravesicular ANTS was removed using a PD-10 Desalting column (GE Healthcare, Piscataway, NJ), and the ANTS-LUV stock solution was stored at 12°C in the dark for a maximum of 7 days. Final phospholipid concentrations were assayed as described in ref. 27.
For the fluorescence experiments, the ANTS-LUV stock was diluted 1:20 with 140 mM NaNO3 plus 10 mM HEPES (pH 7.0). Gramicidin-doped vesicles were prepared by adding the desired amount of gramicidin from Bacillus brevis (Sigma Chemical Co, St. Louis, MO), which is about 80% gA, diluted in dimethylsulfoxide (DMSO), and incubating for 24 h at 12°C in the dark.
All compounds tested were of the highest possible purity, used without further purification, and diluted in DMSO. Capsazepine, epicatechin (EC), (−)-epigallocatechin gallate (EGCG), GdCl3, genistein, genistin, and mefloquine were from Sigma; capsaicin (Cap) from ICN Biomedicals (Plainview, NY); and Gd3+, (β-OG, and Triton X-100 from Calbiochem (San Diego, CA). The compounds were incubated with the ANTS-LUV solution for 10 min at 25°C in the dark, and in each experiment the final [DMSO] was kept constant (<0.5%) except when DMSO was tested. ANTS has no effect on bilayer properties, as assayed using single-channel experiments and by comparing the fluorescence time course in LUVs prepared with different [ANTS] (results not shown).
Fluorescence Spectroscopy
The ANTS fluorescence emission was measured at 25°C using a SX.20 Stopped-Flow Spectrometer (Applied Photophysics, Leatherhead, UK), with a 150-W xenon lamp and a 2 sample rapid mixing unit with machine dead time ∼1.2 ms and integrated water bath. The excitation was at 352 nm and the fluorescence was recorded above 455 nm using a high-pass filter with Pro-Data control software from Applied Photophysics and a sampling rate of 5,000 points/s. For each sample, at least 5 repeated mixing trials were measured, from 2 or more vesicle preparations. Each repeat was separately analyzed using MATLAB v7.4 (The MathWorks, Inc., Natick, MA). The time course of fluorescence quenching was normalized to the average fluorescence without quencher, for that sample, and the first 2–100 ms fitted by a stretched exponential. 28 Because the Tl+ quenching is a near-linear function of [Tl+] at [Tl+] < 25 mM (Appendix, Fig. 1), the initial influx rate can be estimated from the rate of the stretched exponential at 2 ms. For additional details, see Appendix, Methods.
Results
Fluorescence Quenching Depends on Gramicidin Activity
Gramicidin channel activity is monitored using stopped-flow spectrofluorometry by measuring the rapid, Tl+-induced loss of fluorescence. The fluorescence decay is due to the increased intravesicular [Tl+], which is determined by the rate of Tl+ influx (Fig. 2). Tl+ permeates lipid bilayers slowly 29 but passes readily through conducting (bilayer-spanning) gramicidin channels. 30,31 The rate of fluorescence quenching is therefore proportional to the gramicidin channel activity (time-averaged number of conducting channels in the vesicle membrane).
In the absence of quencher (Tl+), the fluorophore (ANTS)-filled LUVs fluoresce steadily without photobleaching over the relevant time scale (Fig. 3A, top curve). In the presence of Tl+, but without any gramicidin (Fig. 3A, second curve from top), there is a small instantaneous drop in fluorescence due to the quenching of extravesicular ANTS. There is also a slow fluorescence decay due to leakage of Tl+ across the vesicle membrane. Doping the LUVs with increasing amounts of gramicidin (Fig. 3A, curves 3–5 from top) increases the rate of fluorescence decay due to Tl+ influx through an increasing number of conducting gramicidin channels. The distribution of LUV sizes in the samples (see Appendix, Methods) means that the time course of fluorescence quenching is described by a stretched exponential, 28 rather than by a single exponential; Figure 3B shows fits of a stretched exponentials to the influx rates (see Appendix, Methods).

Fluorescence quenching in the absence and presence of gramicidin A (gA). Relative fluorescence signal obtained with 8-aminoaphthalene-1,3,6-trisulfonate (ANTS)-filled vesicles shown (from top to bottom): without quencher, with quencher, and with quencher and doped with 87, 260, and 780 nM gA. (
Effects of the Bilayer Modifier Capsaicin
To illustrate the effects of bilayer-modifying amphiphiles we use capsaicin (Cap), the active component in chili pepper, as an example. Cap activates transient receptor potential vanilloid (TRPV1) channels at high nM concentrations 32 but is a promiscuous ion channel modulator at low μM concentrations. 14 Cap alters lipid bilayer material properties, as sensed by gA channels, at the concentrations where it becomes a nonspecific modulator of membrane protein function. 14 Figure 4 shows the effect of Cap in the gramicidin-based fluorescence assay. At micromolar concentrations, Cap by itself has negligible effects on Tl+ permeation (Fig. 4, curves 2 and 3 from top); the time course of fluorescence quenching is comparable in the absence and presence of 90 μM Cap, meaning that Cap does not significantly increase the amount of external ANTS (rupture vesicles) or promote Tl+ leak across the vesicle membrane. (Because Cap will distribute between the aqueous and the membrane phase, the actual Cap concentration in the aqueous phase will be less than the nominal concentration based on the amount added, see the on-line Appendix.) In the presence of gramicidin, Cap increases the rate of fluorescence quenching (Tl+ influx) in a concentration-dependent manner (Fig. 4, curves 4–7 from top). That is, when Cap partitions into the lipid bilayer, it shifts the gramicidin monomer ↔ dimer equilibrium toward the ion permeable dimers. By changing the bilayer material properties, Cap, thus lowers the energetic cost of the bilayer hydrophobic adaptation to the gramicidin channel (see Fig. 1).
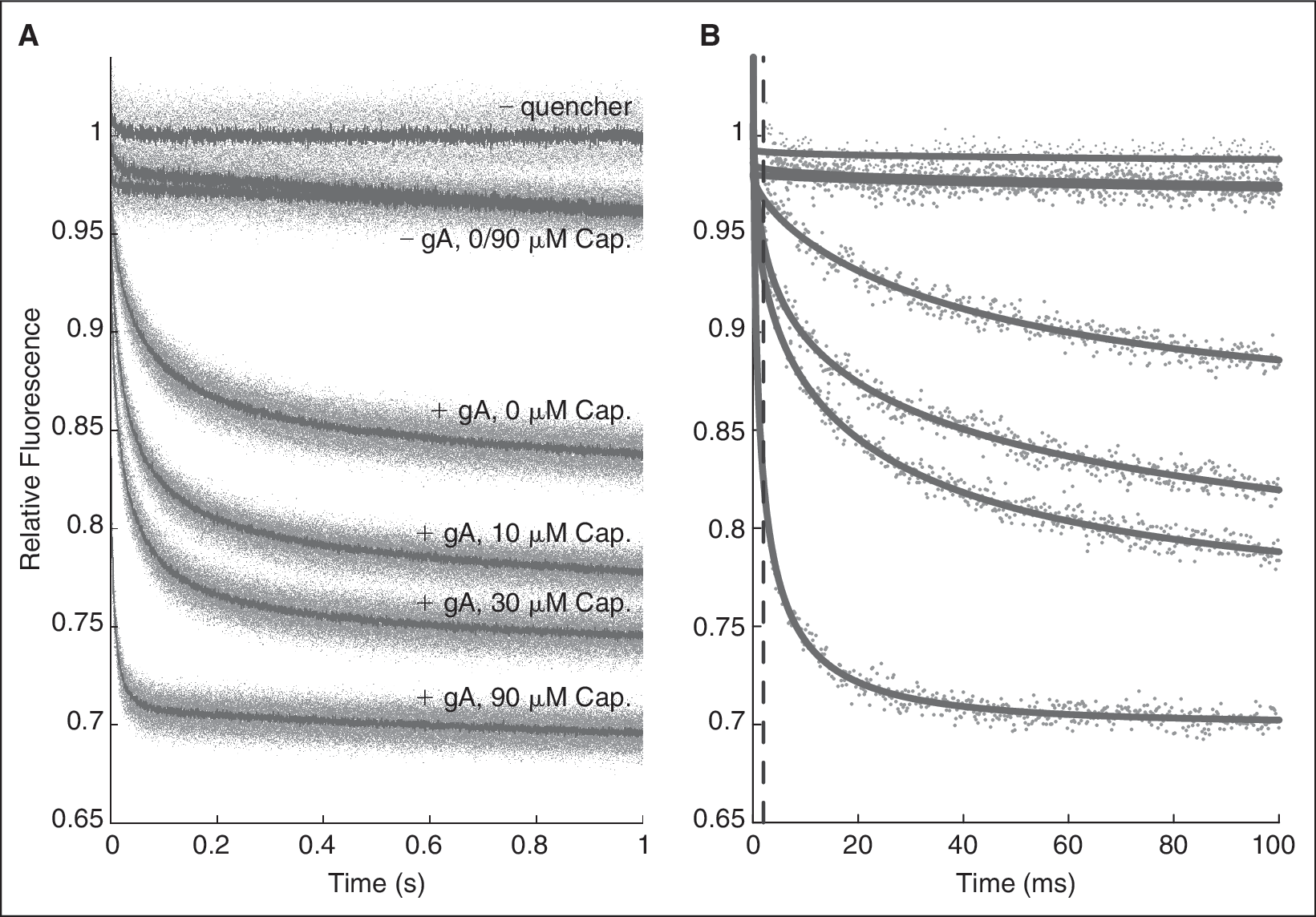
Effect of capsaicin (Cap) on the time course of 8-aminonaphthalene-1,3,6-trisulfonate (ANTS) fluorescence quenching. (
Comparison to Single-Channel Method
To validate the method, we tested additional compounds, including drugs and phytochemicals, which have been characterized in single-channel experiments (Fig. 5). Figure 5A shows the changes in gA channel lifetimes, as measured in single-channel experiments; Figure 5B shows the changes in gramicidin activity, as measured by the relative change in the rate of fluorescence quenching. For all compounds, the highest concentration used was also tested in the absence of gramicidin, where they had negligible effects on the “background” Tl+ permeability. Given the difference in experimental approach, one focusing on the channel dissociation rate constant (lifetime) and the other on the dimerization constant (time-averaged number of conducting channels), the agreement is good.
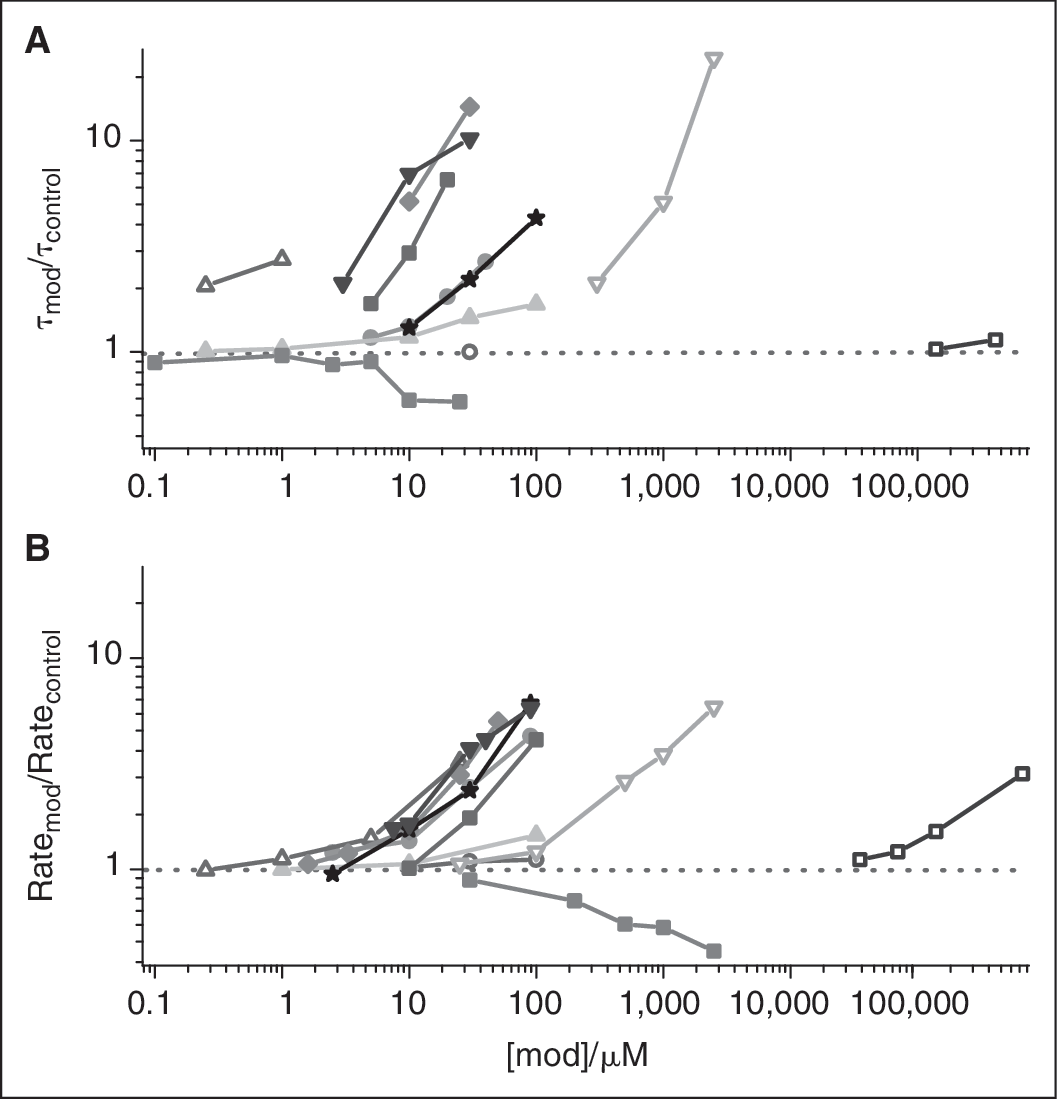
Comparison to single-channel method. (
The LUVs used in Figure 5B are formed in the absence of organic solvents, whereas the planar lipid bilayers in the single-channel experiments (Fig. 5A) are formed in the presence of n-decane, indicating that solvent-free and hydrocarbon-containing bilayers respond similarly to the adsorption of amphiphiles. Importantly, Cap and Triton X-100 have similar effects in both systems, even though they have opposite effects on the intrinsic lipid curvature. 14,33
A more detailed description of the compounds' structure and the concentrations at which they alter bilayer material properties are given in Appendix Table 1.
Discussion
We have developed a fluorescence-based assay to determine whether small amphiphiles, such as drugs and drug leads, alter membrane protein function through drug-induced changes in membrane properties. The assay exploits the power of gramicidin channels as probes for changes in bilayer properties, 11 as sensed by a bilayer-spanning channel (the energetic cost of a channel-bilayer hydrophobic mismatch). The results obtained using the fluorescence-based assay are in good agreement with results from single-channel gA experiments (Fig. 5 and Appendix Table 1), indicating that this method can be used for mechanistic studies as well as for screening compound libraries. Using the present configuration of the assay, we can test dozens of samples a day; which is 1 to 2 orders of magnitude higher throughput than possible using the single-channel approach. There are no fundamental rate-limiting steps in the fluorescence-based assay, meaning that it can be extended to run in true high-throughput mode. Given the time course of fluorescence quenching (10–100 ms), due to the vesicles' small volume and the fast quencher influx through the gA channels, such implementations of the assay will require a rapid mixing system. This may be difficult to implement in a conventional 96- or 384-well plate format, but can be achieved by automating the loading of the stopped-flow system.
It remains unclear whether there is a causal relationship between increasing lipophilicity of drug leads and increasing attrition in drug development 2 –4 and, if so, what are the underlying mechanism(s)? Nevertheless, because membrane proteins tend to be regulated by changes in their membrane environment, 10 it would be prudent to test whether amphiphilic drugs and drug leads alter pertinent lipid bilayer properties and, if so, at what concentrations? Because amphiphile partitioning into the lipid bilayer will deplete the aqueous phase; the relevant concentration is the free concentration in the aqueous phase—which may be orders of magnitude less than the nominal concentration in the system, for example. 16,17,33 Determining drug concentrations in the aqueous and lipid phases may be problematical due to lipid composition-dependent partitioning and drug absorption to the experimental setup. When comparing to in vivo plasma concentrations, it further becomes important to consider drug binding to plasma proteins. It is therefore important that one can vary the vesicle lipid composition, lipid:water ratio, and presence of plasma proteins. Addition of 600 μM serum albumin, for example, right-shifts the dose-response curve for Cap 3- to 5-fold (results not shown), presumably reflecting Cap binding to albumin. 35
If a molecule's desired (biological) effects occur at concentrations where it alters bilayer properties, it becomes important to distinguish between the “non-specific,” bilayer-mediated changes in membrane protein function, as opposed to direct effects due to (high-affinity) binding to one or more target proteins. Knowing the bilayer-modifying propensity of a drug lead, discovered in conventional high-throughput screening, therefore is likely to be important for decisions regarding its further development.
Any amphiphile—at some concentration—will alter some bilayer property. Key concerns therefore become: at what concentration and are the changes in bilayer properties sensed by (bilayer-spanning) membrane proteins? Here, we exploit the ability of gramicidin to form channels by transbilayer dimerization. 36 This makes them useful probes for the energetic coupling between lipid bilayers and bilayer-embedded proteins, and for exploring whether small molecules alter bilayer properties that are sensed by membrane proteins. The assay is fast, reliable, and scalable, and therefore suitable for both biophysical studies and for the screening compound libraries for drugs with potential bilayer-perturbing effects.
Footnotes
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01GM021342 to OSA), the Josiah Macy, Jr. Foundation (OSA), and the Tri-Institutional Program in Computational Biology and Medicine (HII). We thank Tashalee R. Brown, Shemille A. Collingwood, and E. Ashley Hobart for the unpublished results with Gd3+, DMSO, and Mefloquine, the Weill Cornell Medical College electron microscopy core for assistance with the electron micrography of the LUVs, and Michael J. Bruno, Ruchi Kapoor, Radda Rusinova, and Jon T. Sack for stimulating discussions.
Author Disclosure Statement
A provisional patent application that covers the methods described in the manuscript has been filed. The co-inventors are HII and OSA.