
Abstract
To identify a new protective or therapeutic intervention for hepatitis C virus (HCV) infection, we performed efficient structure-based virtual screening to identify novel inhibitory agents for HCV. To this end, we selected NS5B, an RNA-dependent RNA polymerase (RdRp), as the target for the treatment of HCV infection. To decipher the dockable nature of various RdRp X-ray crystals, we docked the crystal ligand (inhibitor) to the crystal receptor (enzyme). The accuracy of regeneration of the crystal pose indicates the amenability of the RdRp binding pocket for structure-based virtual screening. We also utilized a consensus scoring scheme to reduce false positives, thereby ensuring efficient virtual screening. In this study, each molecule that ranked in the top 1% among all screening molecules gained 1 consensus point in a scoring function. Thus, after virtual screening of 57,177 chemicals from the Maybridge Screening collection, 14 molecules gained 8 points across 11 scoring functions. One of them, an isoxazole, showed significant dose-dependent inhibition of HCV RdRp activity and replication. In this study, we have developed a structure-based virtual screening method using HCV RdRp for efficient identification of novel inhibitors.
Introduction
The HCV is a positive-sense single-strand RNA virus and a member of the Hepacivirus genus within the family Flavivirida. 9 The 9.6-kb genome of HCV translates to a single viral polyprotein composed of about 3,000 amino acids including four structural proteins (C, E1, E2, and p7) and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B). 10,11 Among the nonstructural proteins, NS5B, an RNA-dependent RNA polymerase (RdRp), 12 is essential for viral RNA replication and has been considered as an attractive target for the chemotherapeutic inhibition of HCV infection. 13,14 The structure of RdRp has been extensively characterized by using X-ray crystallography. 15 –31 This structural information provides a strong base for the development of an efficient structure-based virtual screening method for new anti-HCV agents.
In this research, we attempted to develop an efficient, structure-based virtual screening procedure for identifying novel anti-HCV agents. Most of the resolutions of the X-ray protein crystals in the Protein Data Bank (PDB) are lower than 2 Å, and this error could interfere with the high hit rate of structure-based virtual screening. If a 2-Å error occurs at the binding site, time and money would be wasted. To overcome this intrinsic flaw of crystallography, we docked the crystal inhibitor back into the crystal enzyme to identify the suitability of crystal proteins for virtual screening. At the same time, the optimal parameters of docking were optimized. We also used the consensus scoring scheme to reduce the false positives. To test the candidates for virtual screening, we used a replicon system 32 for HCV replication and an RdRp activity assay 33 for testing the inhibitory activity of RdRp's function. Results showed that 14 potential drug candidates, downsized from a total of 57,177 chemicals, earned the highest consensus scoring points, 8, among 11 scoring functions. The screening molecules were then purchased from the Maybridge Screening Collection (Maybridge Chemical Library, Thermo Fisher Scientific). 34 One of them appeared to inhibit HCV virus replication. Further dose-dependent tests of the RdRp activity assay showed inhibitory activity specific to HCV RdRp.
Experimental
Best Crystal Structure and Binding Pocket Selection
For enhancing the hit rate of the virtual screening for HCV NS5B, we examined the 26 three-dimensional complex crystals of HCV RdRp obtained from the PDB. According to the position of inhibitor binding sites, the complex crystals were divided to central or “palm” domain 35,36 (PDB code: 1YVF, 23 2AWZ, 25 2AX0, 25 2AX1, 25 2FVC, 26 2GC8, 18 2GIQ, 20 2IJN, 28 2QE2, 37 2QE5 37 ) and “thumb” domain 35,36 (PDB code: 1NHU, 27 1NHV, 27 1OS5, 22 2BRK, 16 2BRL, 16 2DXS, 19 2GIR, 20 2HAI, 21 2HWH, 38 2HWI, 38 2I1R, 30 2O5D, 29 3CIZ, 15 3CJ0, 15 3CJ2, 15 3CJ3, 15 3CJ4, 15 3CJ5, 15 3BSA, 31 3BSC, 31 and 3BR9 31 ). In each crystal complex, the crystal inhibitor was removed, leaving the binding site empty. Each complex inhibitor structure was randomized and then subjected to the molecular mechanics optimization, MMFF94 39,40 in ChemBioOffice 2008 package (CambridgeSoft Corporation), and docking it back to the original binding sites. We used LigandFit in Accelrys Discovery Studio 2.0 package (Accelrys) as our virtual screening and docking tools. The spatial root-mean-square deviation (RMSD) was calculated between the crystal pose and the after-docking pose. The lowest RMSD of 0.203 Å for the crystal inhibitor of 3CJ2 indicates both the correctness of the quality of the 3CJ2 crystallization and our HCV RDRP virtual screening procedure.
Docking and Consensus Scoring
LigandFit (Accelrys Software) is a docking strategy designed for high-throughput screening and for accurately docking ligands into protein binding sites. Some significant designs are as follows: a cavity detection algorithm is adopted for candidate active site regions. A Monte Carlo shape-based method is used for matching the ligand conformation into the active site shape. A grid-based method is to optimize ligand binding conformation for lowering the protein–ligand interaction energies. To reduce errors from grid interpolation, a nonlinear interpolation scheme, that is, a soft van der Waals potential and a soft electrostatic potential, was designed by developers. 41 In this study, the docking cavity is the occupied space of crystal 3CJ2 ligand by setting the sphere radius of hydrogen to 2.0 Å and the radius of heavy atom (other atoms) to 2.5 Å. Crystal protein residues around binding sites are not allowed to move. Note that various scoring functions exist and differ in the descriptions of protein–ligand interactions. The interactions include a variety of sophisticated forms of noncovalent interactions, such as polar interactions, nonpolar interactions, solvent effect, and entropy effect. Different scoring functions perform differently in different types of protein complexes. No single scoring function can handle all equally excellent. In this study, the performances of 11 scoring functions were considered, which are LigScore1, 41 LigScore2, 41 DockScore, 41 PMF, 42 PMF04, 43 Ludi1, 44,45 Ludi2, 44,45 Ludi3, 46 PLP1, 47,48 PLP2, 47,48 and Jain. 49 If the compound's score of a particular scoring function rank within top 1%, the compound gains 1 point for consensus scoring. Maybridge compounds after virtual screening gaining 8 points among 11 scoring functions were purchased for further anti-HCV activity tests.
In Vitro HCV RdRp Activity Assay
Renilla luciferase reporter activity developed by us 32 represents the HCV RdRp activity. BHK-NS5B-FRLuc reporter cells were seeded in a 24-well plate at a density of 4 × 103 cells per well and treated with the compound in various concentrations. After incubation at 37°C for 4 days, cells lysates were harvested to measure the reporter gene expression inside the cell by using the Dual-GloTM Luciferase Assay System (Promega Corporation) following the manufacturer's instructions. The relative activity of NS5B polymerase was determined by normalizing the level of Renilla luciferase against the level of Firefly luciferase.
In Vitro HCV Cellular Replication Assay
The activity of HCV cellular replication is represented by the levels of NS5B protein in the Huh7-harboring HCV subgenomic replicon cells, designed Ava5. The Ava5 cells were kindly provided by Dr. C. Rice (Rockefeller University, NY) 50,51 and were maintained in Dulbecco's modified Eagle's medium with 10% heat-inactivated fetal bovine serum, 5% antibiotic–antimycotic, 5% nonessential amino acids, and 1 mg/mL G418 and incubated at 37°C with a 5% CO2 supplement. The Ava5 cells were seeded in a six-well plate at a density of 2 × 105 cells per well and treated with the compound at various concentrations. After incubation at 37°C for 4 days, total cellular RNA was extracted using Trizol reagent (Invitrogen). The levels of HCV subgenomic RNA were detected by RT-quantitative polymerase chain reaction (RT-qPCR) with primers corresponding to NS5B gene; forward primer 5′-GGA AAC CAA GCT GCC CAT CA-3′, and reverse primer 5′ -CCT CCA CGG ATA GAA GTT TA-3′. The copy number of HCV RNAs in each sample was normalized to cellular glyceraldehyde 3-phosphate dehydrogenase (forward primer: 5′-GTC TTC ACC ACC ATG GAG AA-3′; reverse primer: 5′-ATG GCA TGG ACT GTG GTC AT-3′) levels from three independent experiments with the ABI Step One Real-Time PCR-System (ABI Warrington). Briefly, real-time PCR was carried out in 10 μL reaction volume containing 200 ng cDNA, 5 μL Power SYBR Green PCR Master Mix, and 0.4 μM primer pair. The PCR was conducted under the following conditions: denaturation at 95°C for 10 min, 40 cycles of amplification at 95°C for 15 s, 60°C for 1 min, and finally, 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s.
Cytotoxicity Assay
Cell viability was determined by the colorimetric 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenil)-2H-tetrazolium (MTS) assay as described previously. 32 The Ava5 and BHK-NS5B-FRLuc-1 cells were seeded in a 96-well plate at densities of 5 × 103 and 8 × 102 cells per well, respectively, and treated with the compound at various concentrations. The CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) was used to determine the cell viability after 4 days of incubation. The absorbance was detected at 490 nm.
Statistical Analysis
GraphPad Prism (version 5.0; SmartDrawNet) was used for all statistical analyses and graphical illustrations. Data were presented as means ± standards deviation for at least three independent experiments. The statistical significance was analyzed by using Student's t-test. The significant difference was considered as P < 0.05 or P < 0.01.
Results
Binding Pocket Selection
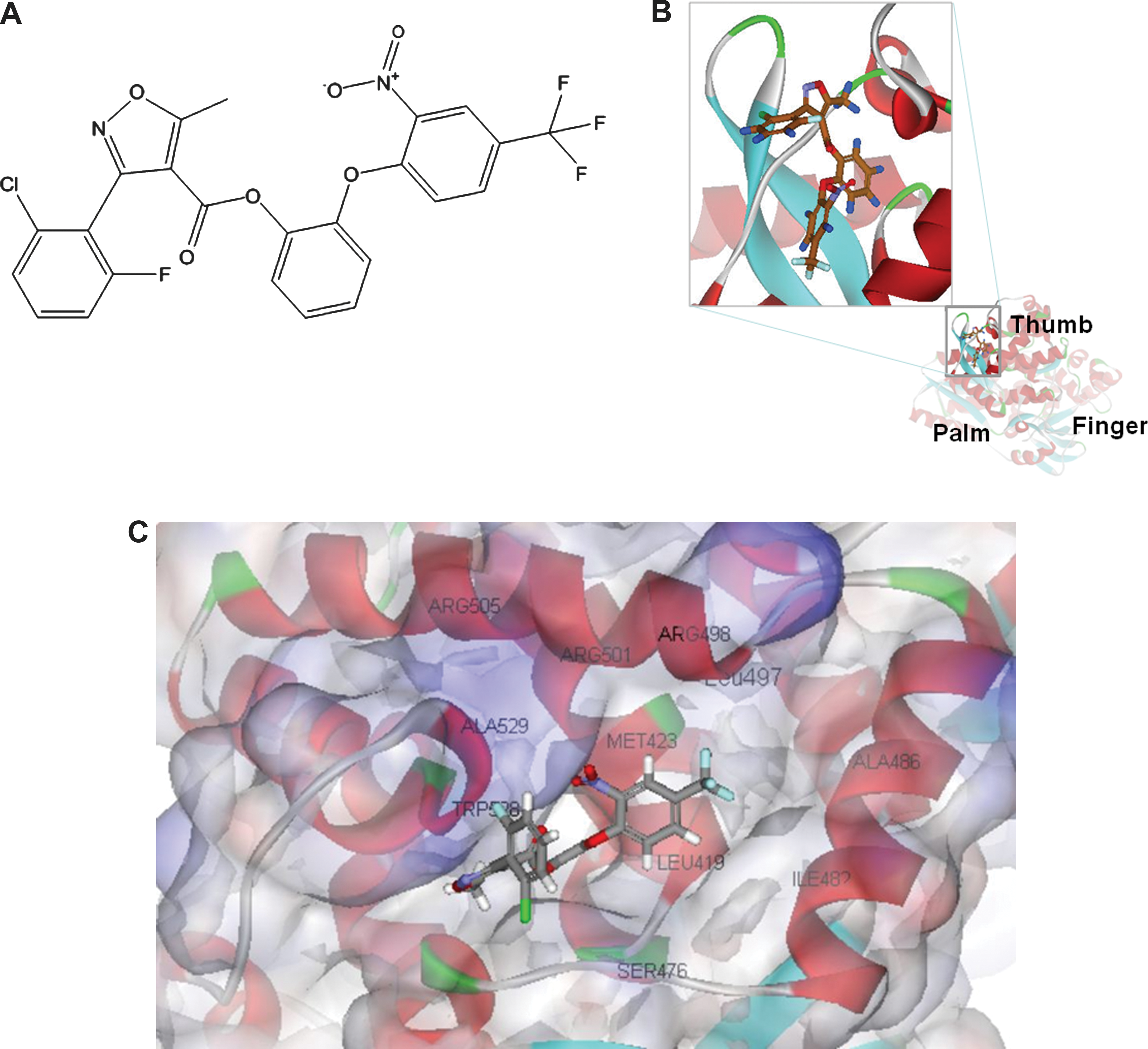
We attempted to conduct a highly accurate (high hit rate) structure-based virtual screening for HCV with limited number of chemicals. X-ray crystallography can resolve structure at the atomic level and provide comprehensive structural insights. However, the average resolution as mentioned above is still not good enough for efficient structure-based virtual screening of HCV. The average resolution of the available RdRp crystals in the PDB was calculated to be 2.16 Å. This resolution factor as aforementioned could decrease the efficiency and accuracy of virtual screening, and this could lead to extra cost and time or even result in failure. Thus, the quality of the “dockable nature” of the binding pockets from crystal complexes under equal screening methodologies must be examined. To check the amenability of the crystal complexes for docking, we adopted a re-docking procedure in which a structure-randomized crystal ligand was docked back into the original crystal binding pocket. The spatial overlap between the crystal's pose (the X-ray bound pose) and the after-docking pose of the crystal inhibitor was calculated by RMSD. The regain of ligand crystal pose is usually one of central criteria for the feasibility of docking method, 52 but here we attempted to emphasize the suitability of crystal binding sites. Under an “equal base” of docking procedure, by checking the RMSDs (Table 1), we assessed the quality of the binding site of each crystal complex. The results showed that the docking pose of the 3CJ2 inhibitor was almost similar to the original crystal structure, with an RMSD of 0.203 Å. Figure 1A shows an almost ideal structural overlap of the crystal pose and the after-docking pose of the 3CJ2 inhibitor, labeled with the calculated RMSD. Thus, the binding site of 3CJ2 was used for the structure-based virtual screening of RdRp, as it did not require any modifications in surrounding crystal residues or other ligand-based information. Figure 1B shows the enlarged portion of the cocrystal inhibitor and its relative position on 3CJ2, located in between the thumb and palm domain. Figure 1C also represents the surrounding residues LEU418, MET423, THR427, SER476, ILE482, ALA486, LEU497, ARG498, ARG501, ARG505, ALA529, and TRP528. Further, while finding the lowest RMSD between the crystal pose and the docking pose, we simultaneously optimized the docking procedure.

A List of the Protein Data Bank IDs of Available RNA-Dependent RNA Polymerase Crystals and the Root-Mean-Square Deviations Between the Crystal Inhibitor Pose and the After-Docking Pose
3CJ2, first listed, has the lowest RMSD and is considered to be amenable for structure-based docking.
PDB, Protein Data Bank; RMSD, root-mean-square deviation.
Virtual Screening and Consensus Scoring
To lower the RMSD between the crystal pose and the docking pose, we extensively adjusted the docking parameters. At the same time, we obtained the best virtual screening procedure with optimal docking parameters. The docking details are described in the Experimental section. We used 57,177 molecules from the Maybridge Screening Collection 34 as the molecular screening library. To eliminate the false positives, we used a total of 11 scoring functions implemented in the LigandFit wizard to perform consensus scoring. After virtual docking, each molecule was given 11 scores. In this research, we adopted stringent criteria for limiting compounds. If the compound's score in a scoring function was within the top 1% of total library docking poses, by definition that compound gained 1 consensus point. The 14 molecules that gained more than 8 consensus points in the 11 scoring functions selected are listed in Table 2. These compounds were put into the HCV replication assay and the RdRp activity assay. The isoxazole CS01 (Fig. 2A) shows a significant inhibitory effect on HCV replication and RdRp activity. The IUPAC name of CS01 is 2-(2-nitro-4-(trifluoromethyl)phenoxy)phenyl 3-(2-chloro-6-fluorophenyl)-5-methylisoxazole-4-carboxylate. The inhibitor binding model (Fig. 2B) shows the docking pose of CS01 on HCV RdRp. Note that this binding site is thought to differ from the structural effect of nucleoside class (palm domain) and to possibly interfere with the breathing of RdRp, like the allosteric binding of 3CJ2 inhibitor (thumb).
Structures of 14 Molecules Gaining 8 Consensus Points in 11 Scoring Functions
The criterion is that if the compound's score belonging to a scoring function is within top 1% of total library docking poses, it gains 1 consensus point by definition.
Inhibition of RdRp Activity and HCV Replication
A cell-based RdRp reporter assay was recently described in our previous manuscript. 33 The RdRp activity was measured by a Renilla luciferase assay. To identify the RdRp inhibitors, 14 compounds from virtual screening with the concentration of 5 and 10 μM were put into the RdRp reporter screening, that is, testing for HCV replication. Of the chemicals used, CS01, shown in Table 2, exhibited an inhibitory effect on reporter activity compared with a well-characterized RdRp inhibitor 2′-C-methylcytidine (NM107), 53 –56 which served as a positive control (Fig. 3A). Upon an increase in the concentration of CS01, a significant reduction of reporter activity was observed, with the effective concentration of 50% inhibition (EC50) value being 3.9 ± 0.2 μM (Fig. 3B). Notably, about 88% of the reporter activity was inhibited at a CS01 concentration of 15 μM, which showed no detectable cytotoxic effects by MTS analysis (data not shown). To further verify whether this compound can inhibit viral replication, HCV subgenomic replicon-harboring cells, Ava5, were incubated with increasing concentrations of CS01 for 4 days. RT-qPCR was then performed to quantify intracellular HCV RNA. As shown in Figure 3C, CS01 reduced viral RNA levels significantly in a dose-dependent manner. The EC50 value, normalized for endogenous glyceraldehyde 3-phosphate dehydrogenase mRNA, was 6.1 ± 0.3 μM. Similarly, MTS analysis failed to show cytotoxicity at levels up to 15 μM of CS01 (data not shown). Taken together, our approach based on binding site selection, virtual screening, and consensus scoring successfully identified a novel compound that inhibits HCV RdRp activity and also blocks viral RNA replication.
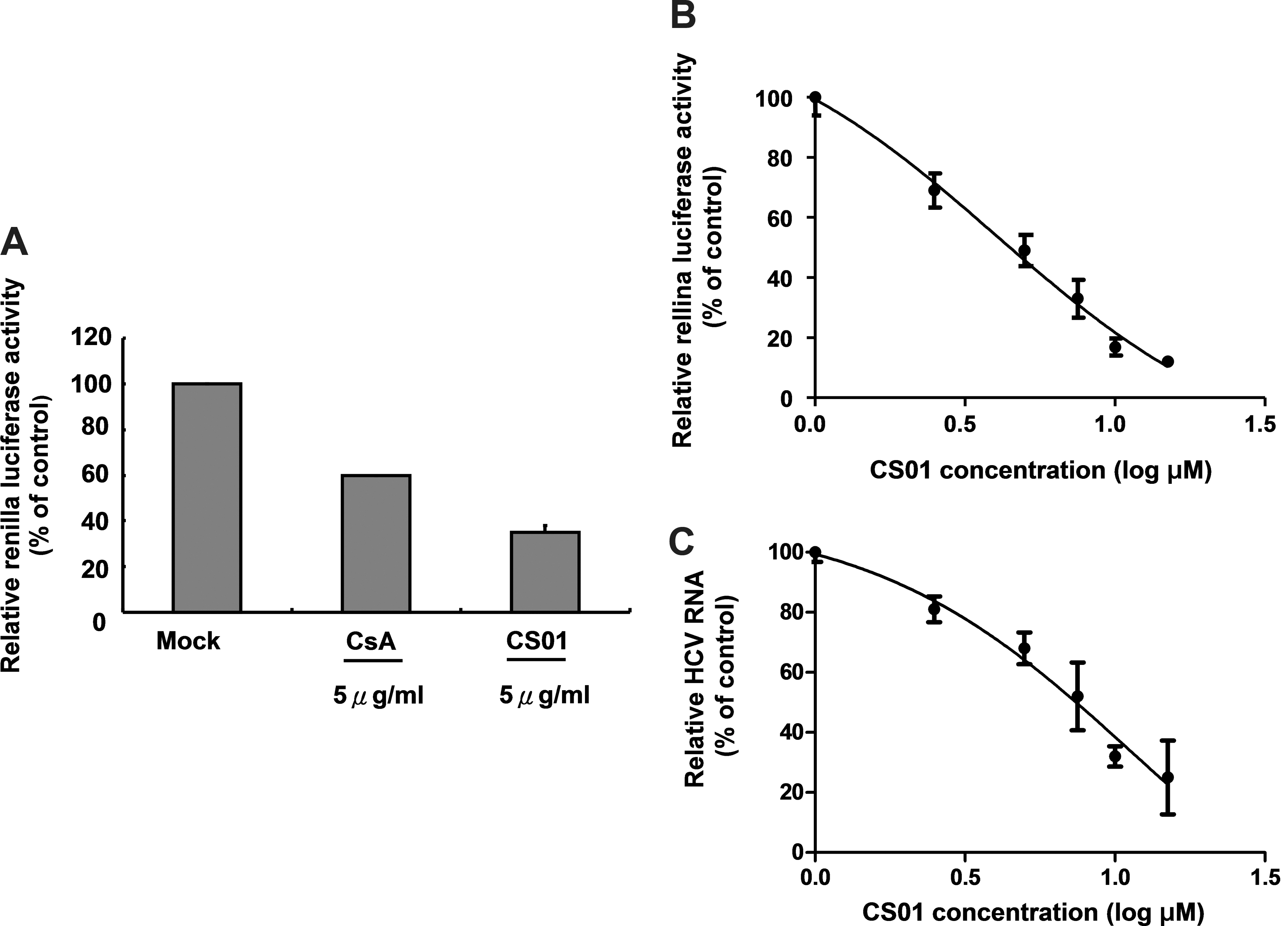
The inhibitory effect of CS01 on HCV RdRp activity and replication.
Discussion
In the present study, we identified a novel inhibitor of HCV RdRp through structure-based virtual screening with limited number of chemicals and in vitro RdRp assay. About 3–4 million individuals worldwide are expected to be infected with HCV each year. At present, no vaccine is available for HCV prevention, and the current therapy for chronic HCV infection combines IFN-α and the nucleotide analog RBV. Unfortunately, anemia and depression are common side effects of this treatment. The adverse side effects of current drugs necessitate an alternative therapy. 7 To successfully develop new anti-HCV drugs, it is essential to identify as many potential candidate drugs as possible. One of the promising target proteins of anti-HCV, NS5B, an RdRp 12 essential for the viral RNA replication, was chosen as the study target. In our experience, many structure-based virtual screenings fail or are not as efficient as expected. The factors responsible for this could be many: the strategy of virtual screening, the accuracy of scoring functions, or whether including ligand-based pharmacophore, and so on. Besides these, the resolution of the X-ray crystal itself could be a crucial factor, as we know that a 2-Å deviation can lead to failure of structure-based virtual screening. Thus, for all available RdRp crystal complexes, it is essential to check whether the protein crystal is amenable for docking and virtual screening. One straight-forward way to do this is to adopt RMSD to examine the regenerated pose after a run of docking and to measure the spatial differences between the docking pose and the crystal pose. Under optimal docking settings, the 3CJ2 crystal inhibitor had the lowest RMSD (0.203 Å), which was far below the average RMSD of the available RdRp crystal inhibitors (2.16 Å). It must be noted that the resolution of the 3CJ2 X-ray crystal (1.75 Å) is higher than the average value (2.18 Å). In Table 1, the docking results show that better the X-ray resolution, higher will be the chances of regaining the crystal pose. The binding pockets around the palm part generally showed RMSDs above 3 Å, which are unsuitable for efficient structure-based virtual screening. The docking settings were simultaneously optimized when searching the lowest RMSD, like sampling of ligand conformation, grid energy, grid extension, interactions of rigid body minimization, and so on. In addition to the resolution issue of X-ray crystals, the hydrogen atoms on crystal waters were totally missing because of the intrinsic characteristic of crystallography. This means that if we added hydrogen atoms to crystal oxygen, it would cause additional uncertainty, effecting the real orientation of hydrogen and impeding efficient docking. Thus, for the time being, we discounted and removed all crystal water oxygen atoms for virtual screening. Of 57,177 chemicals from the Maybridge Screening Collection, 14 chemicals gained 8 points among 11 scoring functions. One of them, analyzed under the newly developed RdRp activity assay, showed inhibitory activity similar to that of the well-characterized RdRp inhibitor, NM107. The hit rate was thus up to 7.1% when compared with a previous study 32 in which the hit rate was 2.3%. The two screening methods were only slightly different in the ways of handling consensus scoring, whereas the docking procedure was the same. On reviewing the literature, we found that virtual screening of drugs for HCV RdRp is limited, and the hit rate is generally below 1%. In the present study, however, the hit rate as we expected would be much higher if we used cell-free assay. The reason for this is that the cell-free RdRp assay could eliminate the effects of drug metabolism, transportation, and toxicity. Thus, an in-house in vitro cell-free assay for anti-HCV RdRp activity is currently under development. In future, we plan to further refine the structure-based virtual screening procedure, both to improve the accuracy of the scoring function and to consider the effects of cell toxicity and drug transportation. Currently, virtual screening methods are being considered as promising approaches to accelerate drug discovery. 57 –59 In the present study, we successfully identified a potent inhibitor of HCV NS5B by using a structure-based virtual screening of the Maybridge Screening Collection and the in vitro cell-based reporter assay. If more diverse potential drug candidates can be identified, then greater is the possibility of developing new therapies for inhibiting HCV. From the extensive interplay between in silico virtual screening and in vitro evaluation shown here, a more diverse structural core of potential inhibitors may be discovered. The new core can be further modified to improve its anti-HCV activity. In future, we hope that more potential drug candidates will be available, from which the most effective therapy for HCV, with minimum or no side effects, can be developed.
Footnotes
Acknowledgments
The authors thank the Taiwan National Science Council (Grant No. NSC95-2113-M-037-016-MY2, NSC97-2113-M-037-004, and NSC97-2311-B-037-001-MY3) and the Kaohsiung Medical University (Grant No. KMU-EM-99-2-4) for their financial supports. The authors are also grateful to the Taiwan National Center for High-Performance Computing for computer time and facilities.
Disclosure Statement
No competing financial interests exist.