Geva-Zatorsky N, Dekel E, Cohen AA, Danon T, Cohen L, Alon U. Protein dynamics in drug combinations: a linear superposition of individual-drug responses. Cell 2010;140:643–651.
Abstract: Drugs and drug combinations have complex biological effects on cells and organisms. Little is known about how drugs affect protein dynamics that determine these effects. Here, we use a dynamic proteomics approach to accurately follow 15 protein levels in human cells in response to 13 different drugs. We find that protein dynamics in response to combinations of drugs are described accurately by a linear superposition (weighted sum) of their response to individual drugs. The weights in this superposition describe the relative impact of each drug on each protein. Using these weights, we show that one can predict the dynamics in a 3-drug or 4-drug combination on the basis of the dynamics in drug pairs. Our approach might eliminate the need to increase the number of experiments exponentially with the number of drugs and suggests that it might be possible to rationally control protein dynamics with specific drug combinations.
Commentary: Drug combination therapies are of great interest but finding optimal drug combinations presents a huge combinatorial problem. If one assumes nonlinear behavior for drug combination effects, then each drug/concentration combination has to be examined experimentally and the number of experiments rises exponentially as KN (K = number of concentrations tested; N = number of drugs). Therefore, exploring even a few drug combinations becomes essentially intractable. This article presents data suggesting that the solution to this problem may be far less cumbersome. The authors use a dynamic proteomics approach where protein expression in response to drug treatment was followed at different time points in a H1299 lung cancer cell line library of tagged proteins. To follow expression over time, the proteins were tagged using YFP in a manner where the fusion protein is expressed from its endogenous chromosomal locus under native regulatory elements thus enabling live cell imaging. Approximately 200–400 cells were imaged at each time point and a total of 15 proteins were followed with 13 drugs. Each drug alone often caused opposite responses in fluorescent protein levels but surprisingly when 2-drug combinations were tested the expression dynamics were found to be midway between the dynamics observed for single-drug doses. In fact the 2-drug effects could be fit to a linear superposition of the single-drug effects (see figure). One exception was the PI3K inhibitor Wortmanin where combinations could not be explained by linear superposition but for the other 12 drugs the dynamics could be described by a weighted sum of the effects for each drug. Even more promising, it was found that 3 and 4 drug combinations could also be fit using the weights determined in the 2-drug combination experiments. This study suggests that a predictive weighted sum function can be produced by examining only single and double combinations. This reduces the complexity of the combinatorial problem greatly as the number of measurements is now manageable. For example, exhaustively testing 100 drug combinations at just one concentration (with vehicle controls) would require 1030 experiments, but this article suggests that one can cover these combinations with just 5,000 experiments, which cover all drug pairs. While it remains to be seen if such linear superposition models can be used to make precise predictions on drug combinations in other assays with different proteins, the findings described here offer some hope in screening for optimal drug combination mixtures. Contributed by Doug Auld.
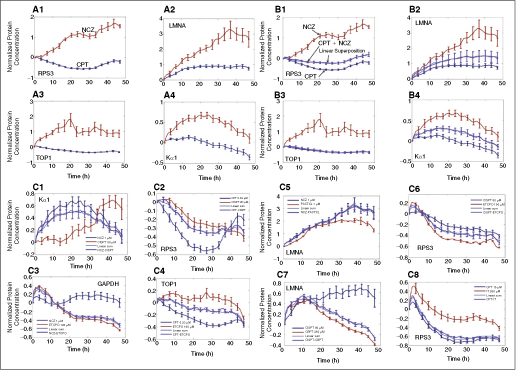
Protein dynamics in drug combinations can be described as a linear superposition of the dynamics under single drugs applied separately. (A) Proteins respond differently to each drug. Shown are protein fluorescence levels as a function of time after the addition of drugs at time t = 0. Protein fluorescence levels P(t) are normalized to initial level at time of drug addition P(0), as follows (P(t) − P(0))/P(0). Ribosomal protein RPS3 (1), nuclear lamina protein LMNA (2), TOP1 (3), and Kα1 (α-tubulin) with 0.33 µM CPT (blue) and 1 µM NCZ (red) (4) are shown. Error bars represent the standard errors of 3 or more independent experimental repeats. (B) Dynamics in drug pair is a linear superposition of the dynamics in each drug alone. Shown are same proteins and drugs as in A. Dynamics in the presence of both drugs is in purple. The linear superposition is in light blue. Error bars represent the standard errors of 3 or more independent experimental repeats. (C) Dynamics in drug pair is a linear superposition of the dynamics in each drug alone. Proteins and drug combinations are indicated. Dynamics in the presence of both drugs is in purple, linear superposition is in light blue. Error bars represent the standard errors of 3 or more independent experimental repeats.
ENLIGHTENING PROTEIN METHYLTRANSFERASE ASSAYS
Ibáñez JL, McBean YM, Astudillo M, Luo A. An Enzyme-coupled ultrasensitive luminescence assay for protein methyltransferases. Anal Biochem 2010; doi: 10.1016/j.ab.2010.03.010.
Abstract: Epigenetic regulation through protein posttranslational modifications is essential in development and disease. Among the key chemical modifications is protein methylation carried out by protein methyltransferases (PMTs). Quantitative and sensitive PMT-activity assays can provide valuable tools to investigate PMT functions. Here, we developed an enzyme-coupled luminescence assay for S-adenosyl-l-methionine (AdoMet/SAM)-based PMTs. In this assay, S-adenosyl-l-homocysteine (AdoHcy/SAH), the by-product of PMT-involved methylation, is sequentially converted to adenine, adenosine monophosphate, and then adenosine triphosphate (ATP) by 5′-methylthioadenosine/AdoHcy nucleosidase (MTAN), adenine phosphoribosyl transferases (APRT), and pyruvate orthophosphate dikinase (PPDK), respectively. The resultant ATP can be readily quantified with a luciferin–luciferase kit. This assay is featured for its quantitative linear response to AdoHcy and the ultrasensitivity to 0.3 pmol AdoHcy. With this assay, the kinetic parameters of SET7/9 methylation were characterized and unambiguously support an ordered mechanism with AdoMet binding as the initial step, followed by the substrate binding and the rate-limiting methylation. The luminescence assay is also expected to be generally applicable to many other AdoMet-dependent enzymes. Additionally, the mix-and-measure 96-/384-well format of our assay makes it suitable for automation and high throughput. Our enzyme-coupled luminescence assay therefore represents a convenient and ultrasensitive approach to examine methyltransferase activities and identify methyltransferase inhibitors.
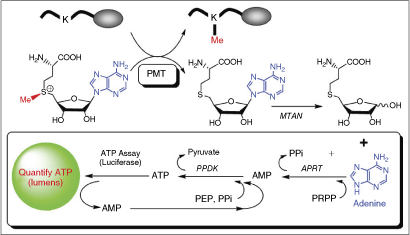
Commentary: Bioluminescence can provide for highly sensitive and robust assays for high-throughput screening. The commonly used bioluminescent enzyme firefly luciferase (FLuc) requires both ATP and D-luciferin to produce a luminescent signal and both of these substrates have been used in the design of assays for many enzyme classes. The present article describes an assay for protein methyltransferases (PMT) where the products of the reaction are converted through a series of coupling enzymes to ultimately produce ATP, which is readily coupled to the bioluminescent reaction of firefly luciferase (see figure). The assay was optimized for PMT sensitivity using excess coupling enzymes in a common buffer system, which did not hinder the activity of any of the enzyme. The assay was configured such that the PMT reaction was allowed to proceed in the presence of 5′-methyladenosine/AdoHcy nucleosidase (MTAN) that relieved product inhibition by S-adenosyl-l-homocysteine (AdoHcy) and produces adenine for subsequent conversion to ATP. The luminescent signal was then generated by adding a standard luciferase detection reagent formulated to measure ATP concentrations. The assay was validated using the PMTs G9a and SET7/9. The luciferase-coupled format enabled the collection of initial rate data where the substrates for the PMT were varied and the measured kinetic constants (eg, kcat, KM) were found to be within 3-fold of reported values. As well, the assay was used to measure the mechanism of the PMT where the data were consistent with an ordered mechanism, similar to reports using more conventional assay systems. The luciferase-coupled PMT assay showed sensitivity to AdoHcy of 0.3 pmol, which allowed for lower concentrations of enzyme and cofactors to be used compared with other assay formats. The coupled assay system described here could be applied to other enzymes that use S-adenosyl-L-methionine as a cofactor. Although fluorescent, radiometric, and AlphaScreen assays exist for some PMTs, the luciferase assay could be used as a primary or orthogonal assay to complement these assays. Contributed by Doug Auld.
Mechanism of enzyme-coupled luciferase assay. The methylation by-product S-adenosyl-l-homocysteine (AdoHcy) is sequentially converted to adenine, AMP, and then ATP with Salmonella enterica 5′-methylthioadenosine/AdoHcy nucleosidase (seMTAN), Saccharomyces cerevisiae adenine phosphoribosyl transferase (scAPRT), and Clostridium symbiosum pyruvate orthophosphate dikinase (csPPDK). The ATP is quantified by commercially available luciferease assay kit (ATPlite from PerkinElmer).
THE PAINS OF SCREENING
Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2010;53:2719–2740.
Abstract: This report describes a number of substructural features that can help to identify compounds that appear as frequent hitters (promiscuous compounds) in many biochemical high-throughput screens. The compounds identified by such substructural features are not recognized by filters commonly used to identify reactive compounds. Even though these substructural features were identified using only one assay detection technology, such compounds have been reported to be active from many different assays. In fact, these compounds are increasingly prevalent in the literature as potential starting points for further exploration, whereas they may not be.
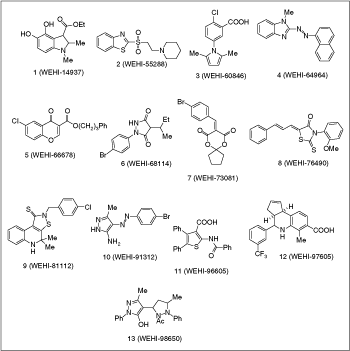
Commentary: Compounds that have proven to be “frequent hitters” in high-throughput screening (HTS) can lead to many false starts if researchers are unaware of these. This article comes from a group where >40 HTS campaigns have been performed on a collection of ∼93K compounds. In these efforts, certain scaffolds have been proven to be associated with “pan assay interference compounds” (or as the authors term it, PAINS). The authors initially examine 6 assays based on AlphaScreen™ to define the compounds associated with PAINS. In this effort, using a screening concentration of between 10 and 30 µM, it was found that 73,164 compounds were inactive, 12,077 showed activity in only one of the AlphaScreen assays, and 7,972 compounds hit in 2 or more with 2,062 showing activity in 4 or more of the AlphaScreen assays. These data were then used to determine if any of the compounds that hit in at lease 4 of the assays also showed significant inhibition (>85%) in at least 1 of 30 additional HTS campaigns, not necessarily based on AlphaScreen technology. Compounds hitting in 4 or more AlphaScreen assays that also showed >80% inhibition in at least 2 other HTS campaigns were also flagged as PAINS. Next the set of PAINS were subjected to substructural analysis to define common scaffolds associated with these compounds. Many of the structures contained potential reactive sites such as alkenes that could interfere with assay components (see figure). Other groups included rhodanine-based compounds, catechols, and quinones, which have also been flagged as showing reactive or redox-dependent behavior by other groups. Many other chemotypes were also found as PAINS and their structures are given in the supplemental tables of this article. In this regards, this group should be commended for providing a database that should help prevent the proliferation of false leads from HTS campaigns. Of course, there are exceptions to every rule and throwing out frequent hitters without consideration of the mechanism of action could ignore potentially useful leads. For example, the authors note that 5% of marketed drugs are flagged as PAINS. However, if one determines that the mechanism of action underlying the activity observed in the HTS was due to a trivial interaction in the assay (eg, absorbance of light, interaction with the reporter, or reaction with detection reagents), then the compound should be discarded. This work also underscores another emerging theme among frequent hitters, which is similar to the aggregation phenomena, the reactivity associated with PAINS is subtle so these compound may selectively react in some assay conditions but not others, making it difficult to determine if the activity is genuine. One approach to this problem is to construct orthogonal assay systems where the exact assay is re-run but with one component switched (such as the enzyme reporter or fluorophore used to detect activity). Carefully constructed orthogonal assays could readily flag PAINS at an early stage following the HTS. In this regards, the authors emphasize that more rigorous reporting of HTS data in journals is required where proof of the mechanism of action underlying the activity identified in the HTS is required along with careful reporting of the experimental methods. Also, research into the nature of assay interference with publications of the results as was done in this article would seem to be a wise use of time and resources, particularly given the current proliferation of public chemical biology databases. HTS assays reduce complicated biology to easily measurable signals, but this reductionist approach can introduce new complications that need to be understood to prevent painful lessons. Contributed by Doug Auld.
Problematic cul de sac compounds that have incurred wasted resources through being followed up to varying degrees at our institute. We have found chromones such as 5 to be highly susceptible to nucleophilic attack at the 2-position, while β-amino sulfones (and ketones) such as 2 readily form reactive retro Michael alkenes. Compounds 6–9 are also susceptible to attack by biologically relevant nucleophiles. The other compounds are problematic for reasons that are either discussed in the text or remain unknown.
ASSAYS IN A DROP
Granieri L, Baret J-C, Griffiths AD, Merten CA. High-throughput screening of enzymes by retroviral display using droplet-based microfluidics. Chem Biol 2010;17:229–235.
Abstract: During the last 25 years, display techniques such as phage display have become very powerful tools for protein engineering, especially for the selection of monoclonal antibodies. However, while this method is extremely efficient for affinity-based selections, its use for the selection and directed evolution of enzymes is still very restricted. Furthermore, phage display is not suited for the engineering of mammalian proteins that require posttranslational modifications such as glycosylation or membrane anchoring. To circumvent these limitations, we have developed a system in which structurally complex mammalian enzymes are displayed on the surface of retroviruses and encapsulated into droplets of a water-in-oil emulsion. These droplets are made and manipulated using microfluidic devices and each droplet serves as an independent reaction vessel. Compartmentalization of single retroviral particles in droplets allows efficient coupling of genotype and phenotype. Using tissue plasminogen activator (tPA) as a model enzyme, we show that, by monitoring the enzymatic reaction in each droplet (by fluorescence), quantitative measurement of tPA activity in the presence of different concentrations of the endogenous inhibitor PAI-1 can be made on-chip. On-chip fluorescence-activated droplet sorting allowed the processing of 500 samples per second and the specific collection of retroviruses displaying active wild-type tPA from a model library with a 1,000-fold excess of retroviruses displaying a non-active control enzyme. During a single selection cycle, a >1,300-fold enrichment of the active wild-type enzyme was demonstrated.
Commentary: Demonstrated in this work is the coupling of a molecular biology approach with microfluidic devices to generate enzyme assays on the pL scale. The technique of phage display can generate large numbers of protein variants (∼1012), allowing for efficient selection and identification of specific variants. Here the authors adapt this approach to display enzymes on the surface of murine leukemia virus (MLV). For the assay, the virion MLV particles are encapsulated into a 12 pL droplet using a microfluidic device (see figure). The droplets were generated by flow-focusing in an oil containing a fluorogenic substrate for the enzyme so that each droplet represents the equivalent of a microtiter plate well, serving as a separate assay or microreactor. To test the system, the authors used human tissue plasminogen activator (tPA) that was fused through its N-terminus to the transmembrane domain of platelet-derived growth factor receptor. This allowed expression of active enzyme on the surface of the MLV particle. Drops containing the virions were produced and incubated with a fluorescent substrate for plasmin. After incubation, the droplets were injected to a reading module where laser excitation of the drop-containing microfluidic channels was used to record the epifluorescence of each drop that provided a throughput of 500 samples/s. The endogenous inhibitor PAI-1 was also mixed at different concentrations and negative controls for each emulsion were included to normalize the data and measure the inhibition. This confirmed that the microvolume assay could accurately measure inhibition potency (IC50 measured 1.1 µg/mL vs. 2.7 µg/mL obtained in a large volume assay). Next mixtures of active and inactive tPA enzymes (at 1:100 or 1:1,000 ratios) were used to test the sorting efficiency. Positive droplets were scored at percentages close to the expected values (1.3% and 0.3% for the respective ratio above), which were consistent with previous sorting experiments that showed a false positive rate of <1 in 104. The pL volumes in this assay system provide for an ultra-HTS system with a million-fold reduction in cost based on the assay volume employed. This study demonstrates a nice integration of molecular biology approaches with microfluidic engineering technology. Contributed by Doug Auld.
Microfluidic devices. (A) Twin drop maker for the generation of emulsions with 2 different droplet species (± inhibitors). (B) Re-injection device for fluorescence measurements. (C) Drop maker for the encapsulation of particle mixtures. (D) Sorting device. The geometry of the sorting module was calculated as described previously (Baret et al., Lab Chip 2009;9:1850–1858). Green spots indicate the focus of the laser beam for the fluorescence measurements. Colored rectangles indicate the sections shown in the microscopic images.
WHOLE BLOOD AND MORE
Stern E, Vacic A, Rajan NK, Criscione JM, Park J, Ilic BR, et al. Label-free biomarker detection from whole blood. Nat Nanotechnol 2010;5:138–142.
Abstract: Label-free nanosensors can detect disease markers to provide point-of-care diagnosis that is low-cost, rapid, specific, and sensitive. However, detecting these biomarkers in physiological fluid samples is difficult because of problems such as biofouling and nonspecific binding, and the resulting need to use purified buffers greatly reduces the clinical relevance of these sensors. Here, we overcome this limitation by using distinct components within the sensor to perform purification and detection. A microfluidic purification chip simultaneously captures multiple biomarkers from blood samples and releases them, after washing, into purified buffer for sensing by a silicon nanoribbon detector. This 2-stage approach isolates the detector from the complex environment of whole blood, and reduces its minimum required sensitivity by effectively pre-concentrating the biomarkers. We show specific and quantitative detection of 2 model cancer antigens from a 10 µL sample of whole blood in <20 min. This study marks the first use of label-free nanosensors with physiological solutions, positioning this technology for rapid translation to clinical settings.
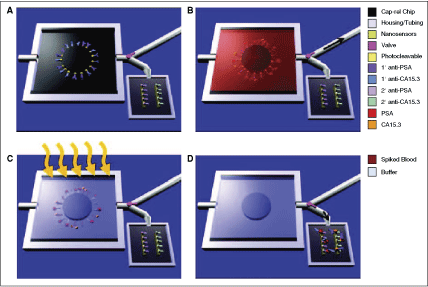
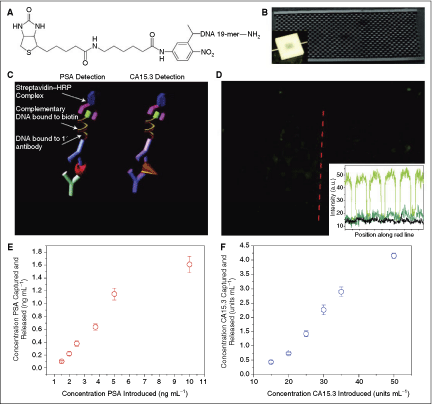
Schematic of MPC operation. (A) Primary antibodies to multiple biomarkers, here PSA and carbohydrate antigen 15.3 (CA15.3), are bound with a photocleavable cross-linker to the MPC. The chip is placed in a plastic housing and a valve (pink) directs fluid flow exiting the chip to either a waste receptacle or the nanosensor chip. (B) Whole blood is injected into the chip with the valve set to the waste compartment (black arrow shows the direction of fluid flow) and, if present in the sample, biomarkers bind their cognate antibodies. (C) Washing steps follow blood flow, and the chip volume (5 µL) is filled with sensing buffer before UV irradiation (orange arrows). During UV exposure, the photolabile cross-linker cleaves, releasing the antibody–antigen complexes into solution. (D) The valve is set to the nanosensor reservoir (black arrow shows the direction of fluid flow) and the 5 µL volume is transferred, enabling label-free sensing to be performed to determine the presence of specific biomarkers.
Commentary: Reports describing microfluidic devices of high complexity have increased lately and so have the descriptions of systems capable of processing real-world samples such as patient blood or serum. Yet, the majority of these publications mostly describe individual components and rarely a fully integrated device. In the work by Stern and colleagues, a sophisticated microfluidic system capable of processing whole blood is described, which operates by the capture–release principle that has traditionally been difficult to realize in microfluidic settings. The new chip first captures protein analytes, such as low-abundance cancer protein biomarkers, by passing the blood through a section coated with anti-analyte antibodies. A wash step follows the capture where fresh buffer is flowed through the chip. The release and detection steps incorporate several unique features that allow the authors to attain exquisite assay sensitivity. First, a second antibody is applied in a manner similar to sandwich ELISA, thereby greatly increasing the specificity of antigen detection. Second, the authors incorporated a photocleavable tag onto the sandwich antibody that allowed detection signal to be generated only upon light-driven cleavage of the tag from the sandwich complex (first figure). To achieve this, that antibody was derivatized with a 19-mer DNA that in turn was labeled with a distal biotin tag via a photocleavable nitroaromatic linker (second figure). The biotin tag served as an affinity attachment point for a standard streptavidin-HRP reagent. Thus, after antigen capture and wash, the second antibody tagged with the photocleavable biotin tag was added allowing the formation of highly antigen-specific sandwich complex. After removal of excess antibody, the application of UV light enabled the biotin tag carrying the HRP signal-generating component to be released and high-sensitivity detection to be achieved downstream. Multiple validation experiments were performed, including the loading and detection of fluorescently labeled chicken ovalbumin and subsequently the detection of 2 key biomarkers, prostate-specific antigen (PSA) and carbohydrate antigen (CA15.3) (second figure). For PSA, detection of the antigen in the range of single-digit nanogram per milliliter was achieved using whole blood samples. While the proof-of-principle study appears to be very successful, considerable amount of future development work remains to be done in order to turn this into a fully operational point-of-care product, including the complete integration of the microfluidic device into an instrument with the requisite user-friendly sample handling and data analysis features, and determining the fabrication and stability parameters for the chip. Contributed by Anton Simeonov.
MPC operation. (A) Molecular structure of the photocleavable cross-linker. Primary antibody conjugation was performed with the amino group (right) and binding to chip-bound avidin occurred through the biotin group (left). (B) Scanning electron micrograph of a representative (w = 4 mm) × (l = 7 mm) × (h = 100 µm) MPC capture–release chip. The inset is an optical image of MPC operation during washing. (C) Schematic representation of PSA and CA15.3 detection using a modified ELISA technique. (D) Fluorescence optical micrograph of an anti-OVA functionalized MPC following OVA–FITC-spiked whole blood flow and washing. The inset plots the pixel intensity (gray value, determined by ImageJ) vs. position for the red cut line (green data plot) and similar cut lines from images of post-UV irradiation and transfer (blue) and of an anti-PSA functionalized MPC following OVA–FITC-spiked blood flow and washing. The same exposure times were used for all images. (E) and (F) Scatter plots showing the concentration of PSA (E) and CA15.3 (F) released from the MPC vs. the concentration of PSA and CA15.3 introduced in whole blood, respectively. Each data point represents the average of 3 separate MPC runs, and error bars represent 1 standard deviation.
MICROFLUIDIC CLINICAL DIAGNOSTICS SYSTEM, FOR REAL
Kagebayashi C, Yamaguchi I, Akinaga A, Kitano H, Yokoyama K, Satomura M, et al. Automated immunoassay system for AFP-L3% using on-chip electrokinetic reaction and separation by affinity electrophoresis. Anal Biochem 2009;388:306–311.
Abstract: Implementation of the on-chip immunoassay for α-fetoprotein (AFP)-L3% was achieved using a fully automated microfluidic instrument platform that will prepare the chip and run the assay with a total assay time of <10 min. Reagent/sample mixing, concentration, and reaction in microfluidic channels occur by the electrokinetic analyte transport assay (EATA) technique, enabling the integration of all assay steps on-chip. The determination of AFP-L3%, a biomarker for hepatocellular carcinoma, was achieved by the presence of Lens culinaris agglutinin in the separation channel, causing separation of the fucosylated isoform, AFP-L3, from the nonfucosylated AFP-L1 by lectin affinity electrophoresis. Laser-induced fluorescence (LIF) detection was used to quantitate the labeled immunocomplexes. The limit of detection (LOD) was 0.1 ng/mL AFP, and assay precision of <2% coefficient of variation (CV) was obtained for quantitation from 24 to 922 ng/mL total AFP in spiked serum samples. Assay precision of <3% CV was obtained for AFP-L3% measurements from 8.5% to 81%. Furthermore, good correlation of test results for 68 patient serum samples with a commercially available reference method (LiBASys assay for AFP-L3%) was obtained, with r2 = 0.981 and slope = 1.03.
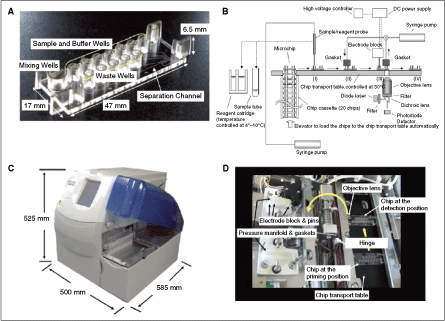
(A) Molded plastic chip with top wells for reagents and precision microchannels formed on the bottom surface. Detection of the fluorescence signal is done through the thin film closing the channels on the bottom of the chip. (B) Schematic diagram of the fully automated microchip electrophoresis immunoassay analyzer. Plastic microchips are supplied from the chip cassette, which accommodates 20 chips. The microchip is fed to the chip transport table, which is temperature controlled at 30°C. The chip is transferred to position I, and the reagents and sample are applied to the designated wells by the sample/reagent probe. The chip is then transferred to position II, and positive pressure (20 psi) is applied so that all reagents and sample are loaded onto the chip channel. The chip is then transferred to position III, and the electrodes are inserted into the anode, cathode, and handoff wells. After negative pressure (∼5 psi) is applied briefly to equilibrate each zone, electric field is applied between the anode and cathode wells to initiate ITP. When TB boundary reaches to the handoff junction, electric field is switched to the handoff and anode wells to start the capillary zone electrophoresis separation mode. The immunocomplex is excited by a laser, and emission fluorescence is detected. After electrophoresis, the used chip is transferred to position IV and then disposed in the waste bin. (C) External view of the immunoassay analyzer. The dimensions of the tabletop instrument are 500 mm (width) × 585 mm (depth) × 525 (height) mm. (D) Internal view of the immunoassay analyzer. Chips are visible at the priming position II and the detection position III on the temperature-controlled chip transport table. The locations of the LIF detector objective lens, electrode block pins, and pressure manifold gaskets are indicated, with the electrode block and pressure manifold rotated away from the chip to view the components. The yellow arrows indicate the direction of chip movement on the chip transport table and the rotation of the electrode block and pressure manifold.
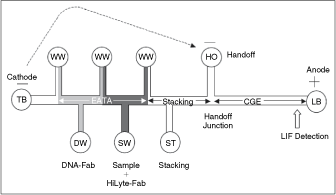
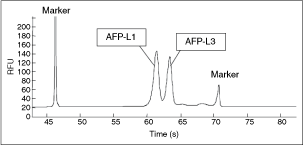
Commentary: The above report can be compared with a somewhat older publication where the team from Wako Corporation presents an actual fully productized instrument that incorporates a microfluidic device and is capable of a fully automated processing of patient samples to measure the levels of total α-fetoprotein (AFP) and the concentration and percent of the L3 glycoform (AFP-L3), the latter being a key biomarker for the progression of noncancerous liver diseases to hepatocellular carcinoma. In addition to the typical challenges associated with processing highly variable and complex human serum samples (in extreme cases, human serum can contain floating lipid droplets and/or precipitating proteins), the analysis for AFP brings an additional hurdle: AFP-L3 only differs from AFP-L1 (the baseline glyco-isoform) by the degree of fucosylation of the AFP protein and typically AFP-L3 is a minor fraction of the total AFP. A detection platform for AFP-L3 has to be capable of detecting low-nanogram levels of AFP and AFP-L3 as low as 10% of total. Because of the small molecular differences between the major and minor AFP biomarkers, there are no fucosylation-specific monoclonal antibodies and the only way to distinguish the 2 species is to exploit their minor differences in binding affinity toward Lens culinaris agglutinin (LCA), a carbohydrate-binding protein that is sensitive to the degree and type of glycosylation on AFP. The team has met all these challenges by producing a chip where the binding, separation, and detection of the AFP analytes are all performed in a homogeneous liquid format by using a combination of lectin affinity electrophoresis and sample concentration by isotachophoresis (first figure). Serum samples were mixed with 2 anti-AFP antibodies recognizing distinct epitopes: the first antibody was fluorescently labeled with HiLyte Fluor 647 dye, allowing a laser-induced fluorescence detection using the commonly available red diode laser as excitation source, while the second antibody was derivatized with a 250-bp DNA fragment that conferred a high and uniform negative charge to the molecule. Thus, in an electrophoretic field, only the sandwich complex comprising AFP and both antibody reagents will be capable of migrating as a sharp peak (due to the incorporated DNA) and be detectable by fluorescence (due to the HiLyte dye). To achieve separation of the complex from the unbound antibodies, poly(dimethylacrylamide) sieving matrix was added to the microfluidic channel, while the resolution of the AFP-L3 form was accomplished by the inclusion of LCA in the buffer: during electrophoresis, the binding of LCA only to the AFP-L3 sandwich complex, but not to AFP-L1, resulted in a small but sufficient slowing down of that species, ultimately allowing its detection as a later peak on the electropherogram (second and third figures). Separation of the sandwich complex from the excess proteins in the serum, as well as the concentration of the complex into a narrow plug within the channel, was achieved by the application of isotachophoresis with sample stacking (second figure), also described in an earlier publication (Kawabata et al. Electrophoresis 2008;29:1399–1406). Using the final instrument, the team performed calibration and reproducibility runs followed by tests of serum samples containing variable AFP-L1 and AFP-L3. Sensitivity down to the 0.1 ng/mL level for both AFP-L1 and AFP-L3 was observed, representing a dramatic improvement over existing platforms. Further, the new tabletop instrument was demonstrated to consume very small amounts of reagents and to deliver results within 10 min of test initiation, making it especially suitable for point-of-care use in doctors’ offices or small clinics. It also appears to be setting the bar fairly high for future reports of microfluidics systems. Contributed by Anton Simeonov.
Schematic chip diagram showing EATA method. Waste wells (WWs), trailing buffer (TB) well, leading buffer (LB) well, handoff (HO) well, DNA-Fab well (DW), sample well (SW), and stacking buffer (ST) well are shown. Vacuum applied to the WWs loads the reagents and sample to chip channel segments, and voltage applied between cathode and anode mixes the sample and reagents for the binding reaction. Switching the voltage from the TB well to the HO well switches from ITP stacking mode to CGE mode, and the separation of AFP-L3 from AFP-L1 occurs in the LCA-filled CGE separation channel prior to LIF detection.
Typical electropherogram of AFP-L1 and AFP-L3 peak separation. Fluorescent markers were co-electrophoresed to identify the AFP peak positions. The first marker at 46.3 s is HiLyte dye conjugated to 2,000 bp DNA, and the second marker at 70.8 s is a slow-migrating dye conjugate. RFU, relative fluorescence units.
ENZYMELESS SNP GENOTYPING
Bowler FR, Diaz-Mochon JJ, Swift ND, Bradley M. DNA Analysis by dynamic chemistry. Angew Chem Int Ed 2010;49:1809–1812.
Abstract: No abstract.
Dynamic chemistry applied to single nucleotide polymorphism (SNP) analysis.
Commentary: Of the multitude of methods currently available for conducting single nucleotide polymorphism (SNP) genotyping, a large majority rely on the recognition action of an enzyme: in the Taqman technology the exonuclease proofreading ability of the PCR enzyme is used to generate signal by the release of allele-specific fluorophore in conjunction with the target amplification, while in most allele-specific primer extension assays the polymerase recognizes the different alleles by the degree of strand complementarity between primer and SNP-containing template. Nonenzymatic alternatives exist, exemplified by molecular beacon and similar-format assays, where no enzyme is involved in the SNP detection, but these have suffered from limited utility largely due to difficulties in designing probes for random SNPs. The work by Bowler and colleagues provides a first-in-kind example of a genotyping method driven by dynamic chemistry, where the act of molecular recognition (of a perfect-complement nucleic acid sequence, in the present case) brings 2 reactants into spatial proximity and thus enables a chemical coupling reaction to conjoin the units. Conversely, the lack of a recognition event keeps the 2 reactants well separated in solution where practically no reaction takes place. To achieve dynamic chemistry here, the authors probe the SNP site by hybridizing the target DNA strand (prepared via PCR amplification of the initial DNA sample) to a peptide nucleic acid (PNA) probe that instead of a regular base contains a reactive amino group in the SNP position. The “typing” consists of adding the 4 bases derivatized with an aldehyde group: only the base that engages in Watson–Crick pairing with the SNP base on the DNA strand is capable of molecular recognition that brings it in close proximity to the PNA probe and allows the chemical coupling reaction between the amino group of the PNA and the aldehyde of the base to take place, thereby covalently tagging the PNA probe with the correct base (figure and scheme). Detection of the outcome of this typing reaction is achieved by mass spectrometry analysis of the PNA. When a PNA probe (sequence within Table 1 of the article) was hybridized to 4 different DNA templates and typed with the 4 aldehyde bases, highly selective incorporations of the correct bases were observed in all 4 typed base contexts (Table 3 of the article). The method was also demonstrated by the successful genotyping of templates corresponding to the SNP involved in cystic fibrosis (Figure 3 of the article), including successful calls of heterozygous samples. While this new method seems to work, a major hurdle that has yet to be overcome is the general difficulty associated with selection of PNA length and sequence to address each SNP. In addition, the unpredictable solubility and aggregation properties of PNAs, and the relatively high costs of PNA production need to be factored in. Contributed by Anton Simeonov.
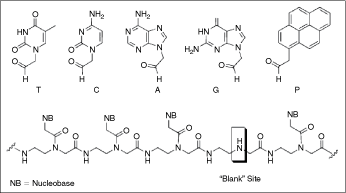
Top: The 4 aldehyde-modified nucleobases (T, C, A, and G) and 1-pyrene acetaldehyde (P). Bottom: General structure of a modified “blank” PNA strand.
NANOFLUIDIC EPIGENETICS: THE BEGINNING
Cipriany BR, Zhao R, Murphy PJ, Levy SL, Tan CP, Craighead HG, et al. Single molecule epigenetic analysis in a nanofluidic channel. Anal Chem 2010;82:2480–2487.
Abstract: Epigenetic states are governed by DNA methylation and a host of modifications to histones bound with DNA. These states are essential for proper developmentally regulated gene expression and are perturbed in many diseases. There is great interest in identifying epigenetic mark placement genome wide and understanding how these marks vary among cell types, with changes in environment or according to health and disease status. Current epigenomic analyses employ bisulfite sequencing and chromatin immunoprecipitation, but query only one type of epigenetic mark at a time, DNA methylation, or histone modifications and often require substantial input material. To overcome these limitations, we established a method using nanofluidics and multicolor fluorescence microscopy to detect DNA and histones in individual chromatin fragments at about 10 Mbp/min. We demonstrated its utility for epigenetic analysis by identifying DNA methylation on individual molecules. This technique will provide the unprecedented opportunity for genome wide, simultaneous analysis of multiple epigenetic states on single molecules.
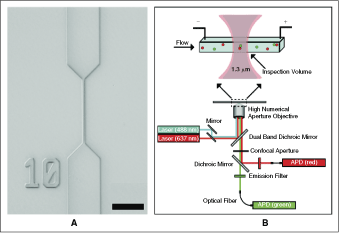
Experimental platform. (A) A differential interference contrast optical micrograph of a typical nanofluidic channel used in SCAN. The narrow region, with a 500 nm wide and 250 nm deep cross section, was used during fluorescence detection. We formed 432 of these channels on a single 100 mm diameter fused silica wafer. The scale bar is 10 µm. (B) Schematic diagram of a wafer mounted on a confocal fluorescence microscope. Two overlapped lasers illuminated a 1.3 µm length of the nanofluidic channel and formed an inspection volume of 0.16 fL. Collection of the dim, fluorescent signature for each molecule was achieved using a confocal aperture, which spatially restricted the optical collection to the inspection volume, and avalanche photodiodes (APDs), which provided single photon detection.
Commentary: Nowadays, not a single issue of Science or Nature gets published without an epigenetics-related paper. While there have been multiple studies reporting on novel histone modifications, on epigenetic mechanisms of disease progression, or on cross-talk between epigenetic signaling events, there has been a general paucity of new methodologies to profile histone modifications in a comprehensive manner or to monitor the enzymatic activity of histone-modifying enzymes. The major methods to analyze epigenetic marks include bisulfite sequencing for DNA methylation and chromatin immunoprecipitation for histone methylation and acetylation patterns, but the DNA and histone modifications could not be detected together and chromatin immunoprecipitation has remained an overall difficult method to scale up. A step in the right direction is the method described in this Analytical Chemistry report where the combined detection of DNA methylation and histone protein is achieved within a nanofluidic chip, possibly allowing future profiling studies of increased sophistication. The device contained channels with dimensions of <1 mm and was manufactured via photolithography techniques. Analytes were transported via electrokinetically driven flow (first figure) while fluorescence analysis was achieved by laser illumination and single photon detection. Differential labeling of the DNA and histone components of the nucleosome combined with 2-color coincidence analysis allowed the platform to detect species containing either DNA (methylated or nonmethylated) or histone protein (carrying a certain mark or not). Initial device performance was tested with simple staining of the DNA contained within a nucleosome with red-fluorescent TOTO-3 alone or in combination with a nucleosome containing a GFP-tagged histone H2B (green-channel fluorescence); 2-color coincidence analysis was used to detect a species bearing both the DNA and histone features, that is the nucleosome, and to distinguish it from histone-free linker DNA. The authors then demonstrated the detection of methylated vs. unmethylated DNA by using a methyl-DNA-binding protein (MBD1): to achieve 2-color detection, MBD1 was labeled with AlexaFluor 488 and the DNA was stained with TOTO-3. By registering the 2-color coincident events, the authors were able to determine the fraction of methylated DNA relative to total DNA (second figure). While the present report does not show the simultaneous detection of histones and methylated DNA or the detection of a specific histone modification (such as methylation or acetylation, which could be achieved by the application of an appropriate fluorescently labeled antibody), the authors indicate that the new device could also be used to pre-fractionate chromatin species based on methylation level and other features, akin to flow cytometry, thereby potentially offering an alternative to chromatin immunoprecipitation. Contributed by Anton Simeonov.
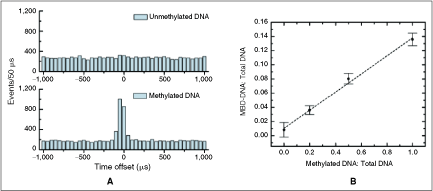
Detection of DNA methylation. (A) Unmethylated (top) and methylated (bottom) DNA samples labeled with TOTO-3 were both incubated with a molar excess of MBD1 probes labeled with Alexa Fluor 488 and then analyzed for 15 min. The emergent peak in the bottom panel demonstrates SMD of methylated DNA. The molar excess of labeled MBD1 contributed to the background of uncorrelated events within each experiment. (B) We analyzed mixtures of methylated and unmethylated DNA. The proportion of dual-color labeled MBD-DNA was shown to increase with methylated DNA, as described by a linear fit with R2 = 0.99. Error bars represent the propagated error from SMD of both the bound and unbound molecules.
FRAGMENT SCREENING RESONATES
Navratilova I, Hopkins AL. Fragment screening by surface plasmon resonance. ACS Med Chem Lett 2010; ASAP Online.
Abstract: Fragment-based drug discovery is a validated approach for the discovery of drug candidates. However, the weak affinity of fragment compounds requires highly sensitive biophysical techniques, such as nuclear magnetic resonance (NMR) or X-ray crystallography, to identify hits. Thus the advantages of screening small fragment libraries are partly offset by the high cost of biophysical analyses. Here we present a method for biosensor-based fragment screening using surface plasmon resonance (SPR). In order to reduce the false positive detection rate, we present a novel method of data analysis that incorporates multiple referencing with ligand efficiency. By implementing all necessary steps for assay design, data analysis, and interpretation, SPR-based fragment screening has potential to eliminate all nonspecific (false positive) binders. Therefore, given the advantages of low protein consumption, rapid assay development, and kinetic and thermodynamic validation of hits, SPR can be considered as a primary screening technology for fragment-based drug discovery.
Commentary: Fragment-based screening and surface plasmon resonance have been around for some time and there are already reports that combine these technologies. This contribution from Navratilova and Hopkins, however, provides both a convincing rationale for expansion of this technique and straightforward methods for data analysis that should reduce the number of false positives that can plague fragment screening efforts. Fragment screening is, historically, most effective in the domains of crystallographic and NMR-based screening formats. More traditional HTS platforms are often not compatible with the high concentrations needed in screening small molecule fragments and false positives can dominate the results of such efforts. Successes from fragment-based screening continue to be reported but the authors argue that the extended timelines and costs associated with fragment approaches hinder the field. The use of surface plasmon resonance would certainly combat these issues. To underscore this point, the author’s proof-of-principle screening of 656 fragments for binding of carbonic anhydrase II was accomplished in 4 weeks using only 27 µg of protein. The authors further detail important controls (in terms of reference proteins, blank channels, and control compounds) but the primary advancement involves their data analysis (see first figure). After a standard adjustment for nonspecific binders based upon binding to a reference protein, the authors implicate ligand efficiency as a means to hone in upon fragments that bind with high affinity. The ligand efficiency was calculated as the binding energy of the fragment per non-hydrogen atom of the fragment. Filtering the results with a ligand efficiency cutoff of 0.333 (−0.333 kcal/mol) reduced the number of false positives to a manageable number (4 characterized false positives as judged by nonstoichiometric sensorgram responses). Overall, the screen was well performed (Z′ = 0.63) and yielded a hit rate of 1.8%, which the authors point out is comparable with similar efforts that use crystallographic and NMR-based screening formats. This study greatly validates the use of SPR methods in fragment-based screening efforts by applying a practical data analysis method that should assure that the results of a relative high-throughput form of fragment screening yields true binders. Contribution by Craig Thomas.
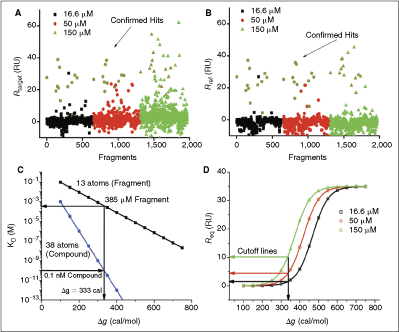
Fragment data analysis. (A) Fragment data overlay for binding referenced for blanks and blank reference flow cell. Fragments were injected at 3 concentrations: black ▪, 16.6 µM; red •, 50 µM; and green ▴, 150 µM. Dark yellow points represent confirmed hits. (B) Fragment data overlay referenced for blanks, blank surface, and reference surface with immobilized SAPk2 protein. (C) Affinity vs. ligand efficiency dependence for compound (blue •, 38 atoms) and fragment (black ▪, 13 atoms); arrows represent calculation of ligand efficiency necessary for a fragment to lead to a compound binding to target with affinity 0.1 nM. (D) Cutoff curves calculated from Equation 4 based on fragment (13 atoms). Arrows represent Req cutoff values for each concentration necessary for fragment efficiency 333 cal/M per heavy atom.
PAINLESS DIOXYGENASES
Hagel JM, Facchini PJ. Dioxygenases catalyze the O-demethylation steps of morphine biosynthesis in opium poppy. Nat Chem Biol 2010;6:273–275.
Abstract: Two previously undetected enzymes involved in morphine biosynthesis and unique among plants to opium poppy have been identified as non-heme dioxygenases, in contrast to the functionally analogous cytochrome P450s found in mammals. We used functional genomics to isolate thebaine 6-O-demethylase (T6ODM) and codeine O-demethylase (CODM), the only known 2-oxoglutarate/Fe(II)-dependent dioxygenases that catalyze O-demethylation. Virus-induced gene silencing of T6ODM and CODM in opium poppy efficiently blocked metabolism at thebaine and codeine, respectively.
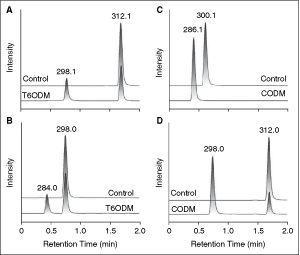
Commentary: The opioids are one of the best-studied classes of medicinal compounds. It is somewhat of a surprise, therefore, to realize that a major component of their biosynthetic pathway (the key demethylation of thebaine) remained uncharacterized. Uncharacterized, that is, prior to this communication from Hagel and Facchini. A major key to their success was a lack of a presupposition that the enzyme responsible for thebaine’s O-demethylation was not necessarily a member of the cytochrome family. These authors and others had previously established poppy varieties with a metabolic block of the O-demethylase step that are enriched in the precursor molecule (the aforementioned compound thebaine). Microarray profiling revealed several candidate genes of which one gene was a putative deoxygenase. The gene (DIOX1) was expressed with a tag that allowed the authors to identify 2 protein products which, when purified, were shown to have O-demethylase activity on multiple substrates including thebaine (see figure). The newly characterized enzymes were named thebaine 6-O-demethylase and codeine O-demethylase. The manuscript further characterizes the substrate specificity, catalytic efficiency, and mechanism of these enzymes. One may ask why this communication warrants a high degree of attention. From a practical standpoint, the production of codeine and other semisynthetic opioids are governed by poppy varieties that are enriched in their production of thebaine. The identification of T6ODM will certainly allow poppy varieties engineered to further thebaine production altering the cost of such botanical medicines. The authors also suggest that such varieties of poppy could alter the illicit market of heroin production if coordinated correctly. Beyond these social and economic interests, Hagel and Facchini’s work prompts a purely scientific appreciation of a well-conducted study of a historically important molecule that interconnects biology, chemistry, and medicine. Contribution by Craig Thomas.
Extracted ion chromatograms showing the substrates and products of T6ODM and CODM enzyme assays. In each panel, the upper (control) extracted ion chromatogram corresponds to an assay performed with boiled enzyme, whereas the lower (T6ODM or CODM) extracted ion chromatogram shows an assay performed with native enzyme. Reaction products were unambiguously identified using collision-induced dissociation analysis. (A) T6ODM assay with thebaine as the substrate (m/z 312.1) and codeinone as the product (m/z 298.1). Neopinone, which is unstable and spontaneously rearranges to codeinone in aqueous solutions, was not detected. (B) T6ODM assay with oripavine as the substrate (m/z 298.0) and morphinone as the product (m/z 284.0). (C) CODM assay with codeine as the substrate (m/z 300.1) and morphine as the product (m/z 286.1). (D) CODM assay with thebaine as the substrate (m/z 312.1) and oripavine as the product (m/z 298.0). T6ODM assays were analyzed after 1 h to minimize the spontaneous formation of codeinone or morphinone adducts. CODM assays were stopped after 4 h.