Larry A. Sklar received his B.S. in Chemistry at the University of Chicago and Ph.D. in Chemistry at Stanford University. He was a Helen Whitney Postdoctoral Fellow at University of California-Santa Cruz and the Methodist Hospital at the Baylor College of Medicine. He was an Established Investigator of the American Heart Association and Associate Member at Scripps before taking a joint appointment as Director of the National Flow Cytometry Resource at Los Alamos National Laboratory and the University of New Mexico (UNM) where he is Regents Professor of Pathology and Distinguished University Professor. He also serves as Associate Director of Basic Research and Co-Director of the Program in Cancer Biology and Biotechnology in the National Cancer Institute (NCI) Designated University of New Mexico Cancer Center and the Director of Translational Technology in UNM's Clinical and Translational Science Center. He has more than 300 publications and patents in leukocyte biology, molecular assembly in signal transduction and cell adhesion, and high throughput flow cytometry for drug discovery. He is principal investigator and Director of the University of New Mexico Center for Molecular Discovery for the National Institutes of Health (NIH) Roadmap Molecular Libraries Initiative. He is a founder of IntelliCyt and was UNM's 53rd Annual Research Lecturer.
Bruce S. Edwards obtained his Ph.D. in Biology at the University of Colorado, Boulder. He was an NIH Postdoctoral Research Fellow and Cetus/Shell-Triton Postdoctoral Research Fellow at the University of Wisconsin Clinical Cancer Center in Madison. He came to New Mexico as an Associate Scientist and then Scientist at the Lovelace Medical Foundation (LMF) in Albuquerque. He founded and directed the flow cytometry core facility at the LMF. He subsequently moved to the University of New Mexico where he is currently Research Professor of Pathology, Director of the Cancer Research and Treatment Center Shared Flow Cytometry Resource, and co-leader of the High Throughput Screening Core of the NIH-designated New Mexico Center for Molecular Discovery. He is an inventor of novel technologies developed and patented at UNM that have led to the successful implementation of high throughput flow cytometry. He is a founder of IntelliCyt Corporation, a company whose mission is to commercialize these technologies and develop novel flow cytometry applications.
Dr. Sklar and Dr. Edwards, please describe how your careers developed and evolved. When did your collaboration begin and what do each of you bring to the collaboration?
Larry Sklar (LS): I have a Bachelor's degree and Ph.D. in physical chemistry, and I became interested in looking at biological problems using physical/chemical tools, which included instrumentation. After I completed a post-doc, I went to Scripps for my first professional job, and I worked with an immunology group that had early flow cytometers. I began using my physical chemistry tools to gather cellular measurements. One of the projects we were working on involved white cells and inflammation; I was interested in the binding of inflammatory ligands to receptors on white cells. I wanted to see if that could be studied with flow cytometry. In fact, we were able to measure ligand binding in real-time—as it was occurring—by looking at the flow cytometry data. We learned that we could acquire these measurements in a homogeneous way, without washing away the fluorescent ligand from the cells; we could watch the cells get brighter over time. That led me to pursue new applications of flow cytometry and instrumentation improvements that would allow us to use flow cytometry to look at ligand binding events—the initiating events—and the cellular responses that follow. That brought me to the University of New Mexico and to the position as director of the National Flow Cytometry Resource at Los Alamos National Laboratory.
Once Bruce gives you his background, we'll be able to tell you how we came to work together to do flow cytometry. The readers will be able to see how our backgrounds led us to think about drug discovery.
Bruce Edwards (BE): After I finished college, my wife and I moved to Boulder, Colorado, where my first job involved working in a support capacity with the National Oceanographic and Atmospheric Association's Space Environment Laboratories. Even though I was interested in biology, this is what was available at the time. I moved on from doing basic support services to learning to do computer programming. I learned a variety of programming techniques on mainframes; this was the 1970s.
I then completed my Ph.D. in biology at the University of Colorado, with a focus on microbiology, although I did a lot of work in molecular biology as well and was particularly interested in cell biology. Afterwards I did a post-doc in Madison, Wisconsin, and that is where I became heavily involved in cell biology, working in the Wisconsin Clinical Cancer Center for four years. I was working on natural killer cell activity, doing basic biology and immunology research.
I then moved to New Mexico, where I continued to combine my computer programming skills with my basic biological research interests, initially working at the Lovelace Medical Foundation, now the Lovelace Respiratory Research Institute. At that time, flow cytometry was just becoming commercially available, and I established the flow cytometry core lab at the Lovelace Foundation, where I worked for about 13 years.
LS: Both Bruce and I had an interest in instrumentation, and Bruce also had the computer skills. We were both doing work on leukocytes. A change in organization at Lovelace led Bruce to move to the University of New Mexico, and we began to look at ways in which we could interact. Whereas I had been doing measurements of real-time interactions of ligands and receptors, we realized toward the late 1990s that if we had the right delivery system for the flow cytometer, we could not only deliver samples faster, but we could deliver them repetitively. We also realized that the ability to measure fluorescence on or inside cells without a wash step offered the potential to measure not only cell responses but also molecular assemblies—binding interactions—on microspheres. If you could combine rapid, repetitive delivery of samples and the ability to make measurements on cellular or molecular targets, you were doing something akin to drug discovery.
We began to look at different ways to deliver samples, interacted with several companies, and sought and received grants to help us build the technology for what you could call pre-drug discovery. We had to bridge the gap between what was available in the flow cytometer market and where we wanted to go. We had some very complementary interests in biology and instrumentation, and we began to define our roles based on what needed to be done. I tell Bruce that he got to choose what he wanted to do and I got what was left over!
BE: I get to wear several hats, running things like assay development, but also building instruments and writing software. Maybe I do have more fun than Larry!
Please describe the mission of the Center for Molecular Discovery at UNM (UNMCMD). What is its relationship to the NIH's Molecular Libraries Initiative?
LS: UNMCMD is a center that focuses on our home-grown technologies. We are a specialty center for screening by flow cytometry for the Molecular Libraries Network. As a member of the network, we are charged with producing compounds that have interesting biological activities, which the NIH calls “probes,” and we work with other centers and with target providers around the world to develop and run these projects.
BE: This initiative represents the introduction of academia into the drug discovery enterprise, and I believe it will expand the scope of diseases that will be studied and targeted for drug development.
LS: Bruce brings up a good point. We are a production center, but we are firmly entrenched in a university. Our colleagues have academic aspirations; they want to have projects they can move forward, and they want to be in a position to publish and write grants as individuals. We try to encourage and promote this in our particular environment and to balance that with the production requirements of the network.
What are some of the disease areas that have been the focus of programs at UNMCMD?
BE: We have a focus on cancer because we are associated with a cancer center. We have performed screening studies on transporters expressed on cancer cells. We have also done screens involving Bcl-2 family proteins, which are involved in cell apoptosis, with John Reed, M.D., at Sanford-Burnham Institute for Medical Research. Cancer cells are particularly good at producing Bcl-2 family proteins that can inhibit the apoptotic process. We have done screens to detect molecules that can interfere with that ability.
We also have an infectious disease focus, and one of our assays, developed by Hattie Gresham, Ph.D., at our institution, involves the study of drug-resistant strains of Staphylococcus aureus and the development of compounds to inhibit quorum sensing, an approach that differs from the use of antibiotics. Rather than killing the cells, it prevents their production of toxic factors responsible for their pathology, a process that is normally triggered when the cells achieve a critical threshold concentration or quorum.
We have also been involved in developing compounds that are ligands for a variety of targets. In fact, Eric Prossnitz, Ph.D., at our center identified a new class of estrogen receptors, GPR30. We also do a lot of protein–protein interaction studies that may not be specifically disease related.
LS: There is a complementary question that has to do with our mission. While we are in an environment where there is interest and expertise in certain diseases, the technology requires that we focus on suspension cells. When you think about cells in suspension, what naturally comes to mind are white blood cells, blood cancers, and the role of white blood cells in infection, allergy, and inflammation. We do screens that would be of interest to the institutes that are part of the NIH, such as the NCI, National Heart, Lung, and Blood Institute, and National Institute of Allergy and Infectious Diseases.
The other types of cells researchers work with in suspension are bacteria and fungi, and we have studied yeast as model systems for discovery. We get requests from time to time from people who want to develop screens using pathogenic organisms or to look at interactions of viruses with cells under nonreplicating conditions. So we do not think only in terms of diseases; rather we focus on the unique opportunities offered by the instrument platform, including performing cell-based and bead-based assays in which we can study protein–protein interactions or binding interactions in a homogeneous way.
Additionally, the technology underlying flow cytometry—the multiple color detectors, multiple lasers, and complex cell or bead samples—can be used to look at multiple targets simultaneously, and we routinely do multiplexing. We have looked at as many as six targets at a time, and we think it is possible to do high throughput screening with up to 20 targets. For example, you could take a complex blood sample with multiple subsets of white cells and look at what each of them is doing, or you could engineer cell lines so you would be able to look at multiple targets simultaneously, such as multiple transporters. In this way, flow cytometry is analogous to high content screening.
What are the basic principles underlying flow cytometry and its traditional applications in science and medicine?
BE: Flow cytometry was developed in the 1970s for the purpose of studying the DNA cell cycle in cancer cells and identifying aberrant DNA expression. The core technology provides the ability to look at single particles or cells in a moving stream as they flow across the path of a laser beam, where they are interrogated to detect fluorescent probes attached to the cells or particles. It is very much like microscopic imaging in that the secret lies in the probes.
Flow cytometry is particularly good at analyzing many cells very rapidly. For example, we can analyze 50,000–75,000 cells per second, and in the 5–10 microseconds that a cell is in the beam of the laser, we can detect the fluorescence of up to 16 different probes, giving you 16 pieces of information about a cell or a particle in a small fraction of a second. A particularly useful feature of flow cytometry that we at UNMCMD do not yet take advantage of in screening is the ability not only to do complex, multiparameter analyses very quickly, but also to isolate individual cells that have a particular expression profile via a process called cell sorting. This technology was developed based on ink-jet technology, which allows you to break up the particle stream, trapping a cell of interest in a droplet, and then put an electrostatic charge on the droplet, causing it to be sorted into a tube or into the well of a microtiter plate. This is how chromosomes were collected for the Human Genome Project in the 1990s.
A sticking point with flow cytometry has been the ability to measure multiple collections of cells rapidly. For example, if I had two bioassays in two different tubes, historically I would have had to put those tubes into the flow cytometer manually to analyze each of them. We have been able to speed up the process of analyzing multiple collections of cells, and we can now routinely analyze 40 collections of cells in about one minute. I am now working on technology that will quadruple this. In each of the samples loaded into the flow cytometer we can look at thousands of cells, and we can collect information on as many as 20 color-coded cell subsets.
What has surprised you the most about how the technology has evolved?
LS: When we started, back in the late 1990s, we had a very limited picture of the flow cytometer. It was a machine that you had to pressurize and then pump samples into from a multiwell plate. It became apparent that flow cytometers did not have to function by using pumps to push samples in; we could instead use pumps to pull things into the flow cytometer. There are now all sorts of different delivery systems: instruments that sip; others that started out using hydrodynamic focusing and now rely on acoustic focusing; handheld flow cytometers, etc. These advances have given us an opportunity to continue to upgrade the technology and make it more flexible.
BE: On the biological side, I am particularly surprised by the new types of probes being developed. The two trends we see are the increasing flexibility and versatility of the fluorescent probes and the way the instruments are becoming more miniaturized and more available to the masses.
LS: The price of the instrument has dropped to the point that every person in a laboratory could have his or her own flow cytometer. They are now in the $40,000 range. This means that flow cytometers do not have to live in the realm of shared resources that are provided to an institution. Although cell sorting is not yet available at a personal level, cell analysis is presently available as an individualized laboratory tool. From the discovery perspective, just like with plate readers, people are now in a position to pilot their own discovery pipeline and to set targets up at their own discretion. I think that will have a major impact on how discovery is done.
Flow cytometry principles compatible with homogeneous mix and measure assays. (A) A small volume of sample stream surrounds a cell as it passes through the excitation laser beam so that the fluorescence signal from unbound fluorophore in the stream may be small relative to that of fluorophore bound to the cell. (B) The voltage pulse signal elicited by the cell represents the difference between cell and sample stream fluorescence. Although pulse height may be reduced in the presence of background fluorescence (black vs. red pulse), it is a quantitatively accurate measure of bound fluorophore relative to other cells measured under the same conditions. This allows fluorescent probes with affinities up to 100 nM to be detected without requiring a wash step.
Following on Bruce's comment about probe development, one of the probes in particular was a real surprise to me. It has always been a challenge in the flow cytometry field to get information about topography—about where molecules are in a cell. With Yang Wu, Ph.D. at UNMCMD, we have been working with a group at Carnegie Mellon University's Technology Center for Networks and Pathways, called the Molecular Biology and Imaging Center, run by Alan Waggoner. Alan's group has developed probes that you can track. You get different signals back depending on whether the probes are inside or outside the cell. We have a joint project focusing on G-protein–coupled receptors (GPCRs), which when activated are transported from the outside to the inside of a cell.
This technology is especially useful to look for ligands for orphan receptors. Another strategy is to take receptors with known ligands and to look for molecules that cause them to be transported without interacting with their ligand binding site. In high throughput screening for drug discovery, one of the things we would like to be able to do is to change the location of a molecule in a cell with or without eliciting its normal physiological response, and this technology may help us accomplish that.
Can you please describe some specific case studies that illustrate the types of programs being pursued at UNMCMD?
LS: We think of projects that can leverage the potential of the technology platform in different ways. A good example is the use of flow cytometry to study low molecular weight GTPases. Kinases, which hydrolyze ATP, have been prime targets for drug discovery. In the GTPase world, a fundamental molecule is Ras, which is associated with many cancers. The Ras protein now represents a family of more than 100 proteins that are involved in many different aspects of cell physiology, topography, differentiation, and trafficking. We were interested in identifying small molecules that could regulate the ability of GTPases to interact with GTP. This assay was developed in collaboration with Angela Wandinger-Ness, Ph.D., in the pathology department here at UNM, and Zurab Surviladze, Ph.D., who led the development of that target at UNMCMD.
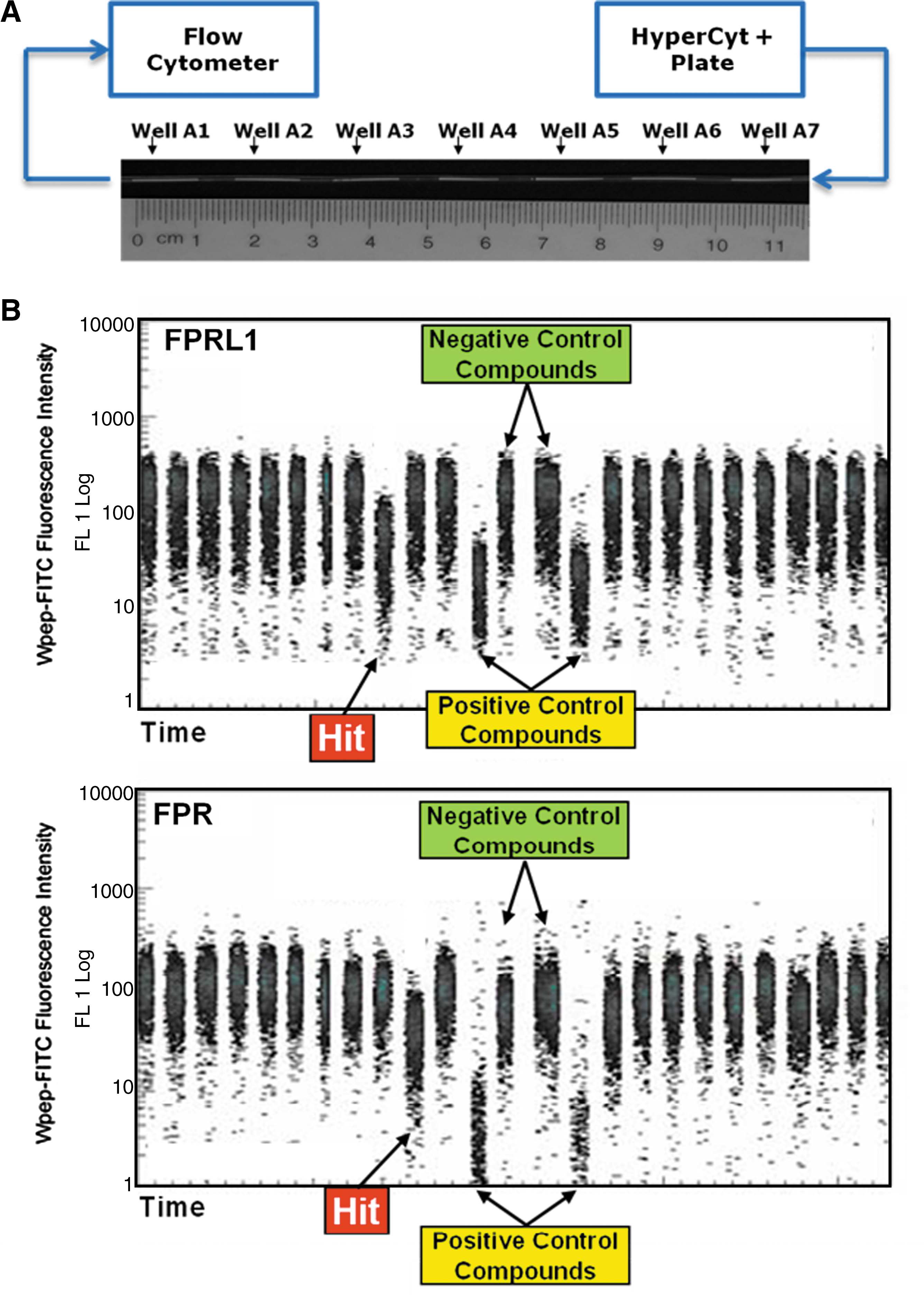
HyperCyt method for flow cytometry high throughput screening. (A) Samples are aspirated from microplate wells and delivered to a flow cytometer as a series of fluid volumes (∼2 μL each) separated by air bubbles. Illustrated are fluid samples (white) as they appear in the tubing used for sample transport. (B) The samples are detected in the flow cytometer as discrete clusters of events that appear at uniform time intervals. Each cluster represents ∼2000 cells sampled from an individual well. In this assay a fluorescent ligand binds to its receptor on intact cells and the objective is to screen for small molecules capable of displacing the ligand from its receptor. Cells expressing the formylpeptide receptor (FPR) were color-coded with a red fluorescent dye so that they could be distinguished from cells expressing the closely related receptor FPRL1 that were analyzed in parallel in the same wells. A “hit” was detected as a decrease in green cell fluorescence intensity associated with displacement of bound ligand. In this example a hit compound selective for FPRL1 (top panel) was detected in a well adjacent to a well that contained a hit compound selective for FPR (bottom panel). Wells containing negative and positive control compounds were used to define the range of cell fluorescence intensity for each receptor.
We were able to take a set of color-coded microspheres—having different intensities of red fluorescence—and put a different low molecular weight GTPase on each microsphere, creating bead sets with glutathione displayed that could bind glutathione-S-transferase (GST) fusion proteins of these GTPases. We used a green fluorescent GTP as our signal and looked for molecules that could regulate the binding of GTP to this family. From a pharmacological perspective, you would expect to see all possible combinations of molecules: some would hit one GTPase at a time; some would hit several GTPases in subfamilies; and others would hit them all. Some could increase GTP binding and others could decrease GTP binding. We saw many of these different types of pharmacological combinations, and some of the molecules we uncovered are being pursued further.
As an example, low molecular weight GTPases regulate protein trafficking in cells. If you can regulate GTPases you have the potential to regulate the molecules they traffic, such as growth factor receptors. In one case, project leaders Angela Wandinger-Ness and Laurie Hudson, Ph.D., are starting to move molecules that came out of these studies into cancer models. This is very interesting for us, because it is a real opportunity for the platform and demonstrates the potential to move these compounds forward into preclinical translation.
Some of the molecules that have come out of our studies are already approved drugs, and we have a strong interest in the area of repositioning approved drugs. We think that for a center like ours, repositioning gives our academic and clinical colleagues the potential to take existing molecules into clinical trials. This is a major goal at NIH and is complementary to the Molecular Libraries Initiatives' efforts to identify new chemical entities.
One aspect of your research focuses on drug resistant transporters. How does a cell or pathogenic organism acquire resistance by this mechanism?
BE: Targeting transporters is another good case study to talk about. Transporters are present in virtually every type of cell in the body and their main job is to protect the cells from toxic molecules. This is another one of those fields in which the harder you look, the more diversity you find. There are multiple transporters, and several different structural families have been identified. The ones of particular clinical interest are those over-expressed by cancer cells. They protect the cancer cells from chemotherapeutic drugs, which are intended to kill the cancer cells. The transporters allow the cancer cells to reject the chemotherapeutic agents. There is a lot of interest in identifying ways to disable this mechanism in cancer cells without affecting healthy cells.
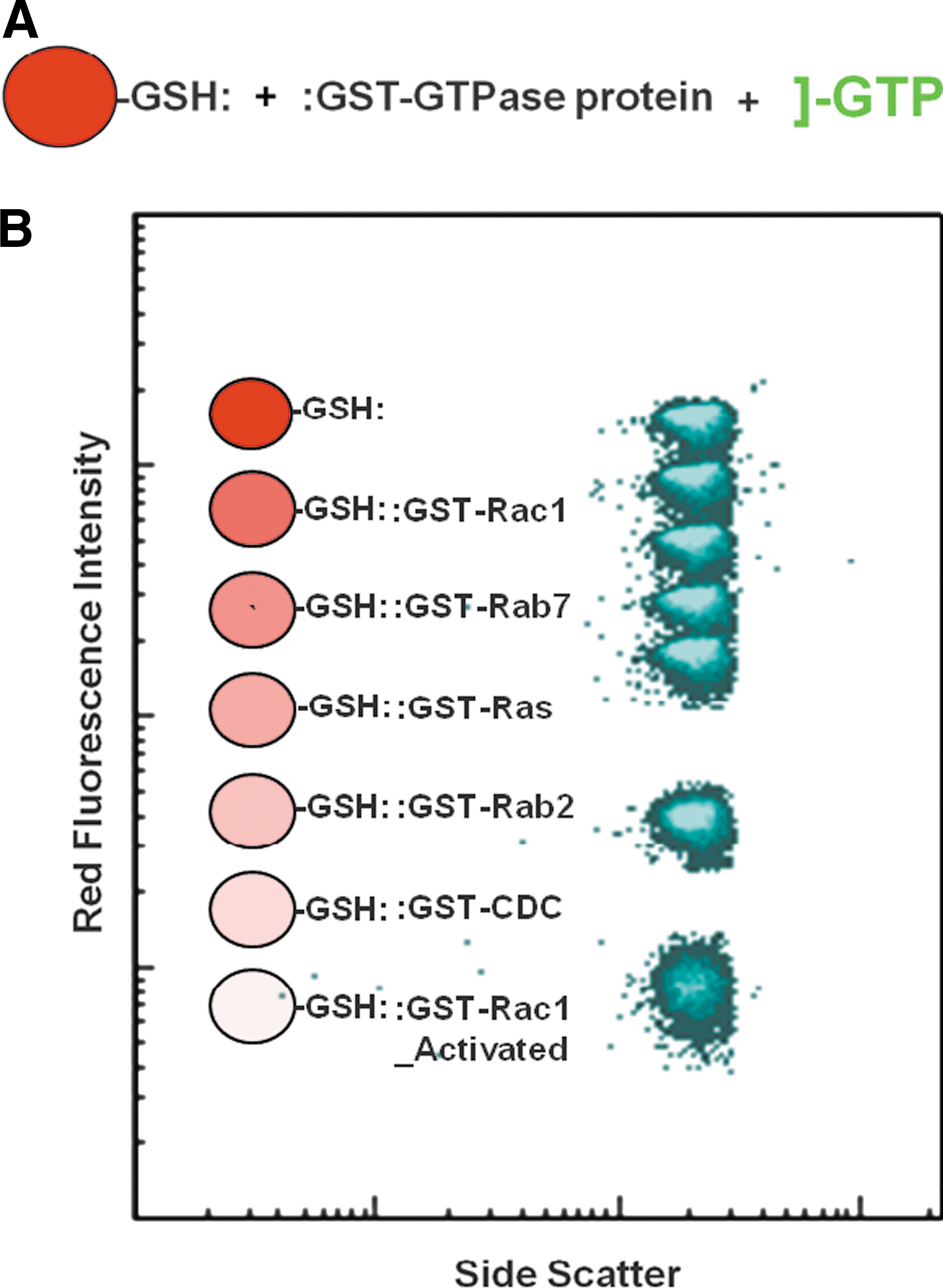
Multiplexed screen to detect potential regulators of signaling by Ras and related low molecular weight GTPases. (A) General assay method. Microspheres with covalently linked glutathione were coated with GTPase fusion proteins and incubated with test compounds in the presence of green fluorescent GTP. Hit compounds were detected by a decrease in microsphere green fluorescence intensity resulting from competitive displacement of fluorescent GTP. (B) Six sets of microspheres with distinct levels of red fluorescence intensity were coated with six different GTPases. All sets were incubated with test compound in the same well and subsequently analyzed in parallel. Changes in green fluorescence intensity of each set were monitored independently by electronically gating the analysis of individual color-coded microsphere populations. A seventh microsphere set with glutathione alone was used to detect nonspecific changes in fluorescence intensity resulting, for example, from innate fluorescence of a test compound.
We want to be able to study multiple transporters at one time, so that if we identify a compound that can affect one transporter, we want to be able to determine if this is a general effect involving multiple transporters or if we can identify compounds selective for different transporters. This is where the ability to look at multiple targets in one assay comes in very handy. With Richard Larson, M.D., Ph.D., and Irena Ivnistki-Steele, Ph.D. at UNM, we approached this problem by producing cells that selectively express different sets of transporters. We would then color-code the cells so we could easily identify which transporter was being expressed by which cells. We then developed a transporter assay that showed whether a fluorescent substance known to be a substrate of the transporter was being extruded from the cell. In this way we could determine which transporter was being affected. We were able to use this assay to identify multiple compounds that were either selective or cross-reactive for a variety of transporters.
LS: Cancer cells typically develop multiple mechanisms of resistance, but it is almost always the case that surviving cancer cells will have among their mechanisms of resistance a pump that is over-expressed. For example, blood cancer cells tend to over-express molecules called ABCB1 and ABCC1, solid tumors such as breast cancer tend to over-express ABCG2, and it is now believed that melanoma cells may over-express ABCB5. There are well over 50 drug transporters in this family alone and each is associated with removal of toxins from cells.
It is also interesting that in bacterial and fungal resistance there are often transporters present that reduce the susceptibility of the pathogenic organisms to therapy. Surviving pathogens tend to over-express one or more transporters. This is of interest to us because we are studying yeast as a model system and are also looking at transporters; now we can look at these two projects together. In fact, the NIH has awarded us support to look at transporters in general across fungal and mammalian species, where we can take advantage of multiplexing to look at large numbers of targets and look for similarities and differences in transport properties for various species.
For example, a fungal infection might develop in a cancer patient because of his or her weakened immune system. We would like to be able to discriminate between molecules that act on fungal transporters versus those that work on mammalian transporters, and to discriminate between transporters that function in healthy cells to maintain the blood–brain barrier, for example, from those over-expressed in cancer cells. The problem becomes very interesting in terms of chemical biology and being able to use tools that can do multiplexing in suspension cells. You can imagine the advantages of being able to combine a chemotherapeutic agent with a drug that prevents cancer cells from pumping it out.
BE: I would add that historically one of the problems with developing drugs that target transporters is that they tend to have serious toxic side effects. A legitimate hypothesis is that if you had more selective transporter inhibitors you might be able to inhibit only the cancer cell transporters and cause less toxic side effects. We think multiplexing will allow us to identify more selective drugs more quickly.
LS: One of the other enabling features of flow cytometry is that with the many fluorescent colors we can use to develop assays we can look at the different substrate interaction sites on transporters that allow them to pump multiple substrates. One of the areas we are working on now is to probe multiple sites on a pump that allows for the inhibition of multiple substrates. We have the capability to use multiple substrates that emit different fluorescent colors to determine the inhibitor profile of an individual transporter.
Does flow cytometry offer any special advantages for enabling personalized medicine?
LS: These same types of applications described above make it possible to develop personalized medicine strategies, particularly in the case of blood cancers. Blood cancer cells are typically identified based on specific combinations of antibodies or other properties that can be measured in a flow cytometer. We envision being able to take small samples of blood—for example 10 million white cells in about 10 mL of blood—put about 10,000 cells in each of 1000 wells of a multiwell plate, and determine the effects of treating a patient with blood cancer that is resistant to chemotherapy with another molecule that would either enhance the effect of the chemotherapeutic agent or have a desirable impact on the behavior of the cells on its own. We are just starting to do those types of studies, showing that we can impact outcomes based on identifying molecules that can improve the survival of cells in vitro. If these compounds are already approved drugs, then they could be taken directly into patients who are failing chemotherapy. This concept is more difficult to translate to solid tumors as you have the problem of delivery.
What is of interest as well in blood cancers is that blood cancer cells tend to survive in niches, such as bone marrow. Their survival may be impacted by their interactions with other cells. One of the things we will have to figure out is how to combine personalized medicine approaches tested on suspension cells with their survival in niches by mimicking physiological survival. Obviously there is still a lot of research to be done.
BE: I would also add that flow cytometry is an excellent way to look at biomarkers, and those types of applications are being developed. For example, you can generate sets of color-coded beads, with each bead expressing a different protein that you might expect to find in blood or some other body fluid. You can then run an assay in which you look for a particular analyte in the fluid and detect patterns linked to clinical disease, prognosis, outcomes, etc. Luminex pioneered this technology, even though they do not call their instruments flow cytometers.
LS: There is a cell-based analogue of this biomarker work as well, in which Pharma is using this technology to look at toxicity of molecules during the discovery process. Toxicity assays are being adapted for use on flow cytometers to perform compound profiling.
High throughput flow cytometric analysis of ABC transporter activity. JC1 duplex assay in which IgMXP3 cells (ABCG2, dim red/FL8 fluorescent) and Jurkat DNR cells (ABCB1, bright red/FL8 fluorescent) are enclosed in separate electronic gates (A, circles) to allow separate analysis of the green JC1 fluorescence intensity response (FL1) of cells sampled from a 384-well plate (B and C, respectively). JC1 is pumped out of cells by each of the ABC transporters resulting in dim green cell fluorescence intensity. Blockade of transporter activity prevents JC1 efflux and results in a concomitant increase in green fluorescence intensity. In the plots of Time vs. FL1 on the right, each discrete cluster of dots represents cells sampled from a single well. Arrows indicate cells from negative vehicle control wells (N), wells containing the positive control compound nicardipine (P), and the well containing lasalocid (Las), a compound that resulted in a significant assay response for both transporters. The illustrated time-resolved dot plots (B and C) represent only a 30- to 40-second segment of the full 384-well data acquisition sequence (∼11 minutes total). Ivnitski-Steele et al, Assay Drug Dev Technol 2008;6:263–276.
How do you envision your research moving forward in light of the six-year $15.5 million grant awarded to you by the NIH three years ago?
LS: There are multiple elements associated with this grant: a user-driven component, which defines how we work with collaborators around the world and their targets; and a center-driven component, which is the way we do research at UNMCMD. The development of yeast model systems during the first two years of funding has evolved into our work with transporters in pathogenic organisms and cancer as well as the project involving GPCRs that I just spoke about. Other elements and activities associated with this grant include funding from the NIH stimulus budget that is allowing us to renovate a building here at the university and move into contiguous space with integrated modular equipment. Bruce also has a grant focused on bringing those elements to reality.
BE: We are somewhat unique among the screening centers in that we are evolving the platform technology as we are performing our screening activities. When we started we were working in 96-well plates and screening libraries of about 10,000 molecules. Within the first year we moved up to 384-well plates and the compound libraries kept increasing in size. The library is now up to about 330,000 compounds, and we have had to evolve the technology to increase throughput and keep up with this growth. I have been focusing on this effort over the last few years, as well as developing technology capable of doing more sophisticated types of data analysis. A recently funded RO1 grant is funding my work on increasing the throughput of flow cytometry screening four-fold, allowing us to move from 384-well to 1536-well screens. We are validating that technology now.
LS: We will be integrating that effort with the new instrumentation, which is coming through a mechanism called a G20 supplement grant for shared resource consolidation. I would also like to point out that the figure of 10,000 data points that Bruce mentioned is now up to 8 million data points five years later, with a technology that people often told us was not possible to use for high throughput screening.
What led you to found IntelliCyt Corporation? What is the main focus of the company and what role do you play in guiding its research and business activities?
LS: One of the ways Bruce and I have differentiated is that my role in the company is rather restricted, whereas Bruce has a very different role. My “day job” has to do with keeping the center, UNMCMD going. The company was envisioned as a mechanism for getting the technology out to the world. The university has a science and technology corporation—a technology transfer group—and we sign over the technology to them. The individual who was interested in this technology, Terry Dunlay, was one of the founders of Cellomics, the business model of which was to put a sample handling front-end on a commercial microscope.
Terry was looking for a business opportunity. Alan Waggoner, who is the director of the center I mentioned at Carnegie Mellon, was one of the other founders of Cellomics, and we have been colleagues for many years. Alan was also very interested in exploring the potential for flow cytometry as a discovery tool. He directed Terry toward us, and Terry made a home here in Albuquerque and set up the company. At that point I made the decision to get out of the way and to be more like an uncle, cheering everyone on. Bruce has taken a different perspective, but together our goal is to bring to the company various opportunities based on biology.
BE: A lot of people were interested in the technology we were developing, and part of the reason we formed the company was to free ourselves from what was becoming an increasingly time- and energy-consuming burden of transferring the technology to the public domain. People were requesting access to the technology and the software, and setting up a commercial enterprise was an opportunity to do that efficiently while retaining the ability to pursue academic research. During the first couple of years I was writing much of the software, and I still work with the company as a consultant.
LS: At UNMCMD, we are charged with being a vehicle for outreach for identifying groups that want to be target providers for the network. Some of these people may also want to have the technology in their own laboratories. So we have tried to make our role for the network and what the company does as synergistic as possible. We talk about our work and the fact that the screening is done on a platform that is now available to the community. They have access to both the screening capabilities of the network and to the technology to do the screening in their laboratories if they so choose.
Do you have any advice for others interested in starting up a technology company?
LS: For me, I have more than a full day's work to do at the university. I think people have to decide what they want out of a company: to make a lot of money, to get their technology out to the public, or, as Bruce said, to use it as a defensive mechanism. We needed to be able to show that certain things were possible with the technology, but there was also the hope that the improvements that could happen in a corporate setting would continue to give us a competitive edge. All of those things played into the way I envisioned this company. The idea that the company would be an additional source of major revenue was not a big factor for me.
BE: I agree. Both Larry and I were very interested in finding someone who was both capable and interested in establishing and building the company. We did not envision ourselves in that role.
What new technologies are you developing that are centered around flow cytometry?
LS: Ligand binding and protein–protein interactions that can be studied in homogeneous assays without the need for expensive detection reagents. For high throughput screening our target is currently 200 wells per minute, which is somewhat slow relative to other technologies, but it has the advantages of homogeneous resolution of free and bound ligand, as well as multiplexing and the capability to measure multiple targets or multiple responses of a single target. For example, you can look for phosphoproteins in cell populations, at epitope expression on the outside of a cell, or at intracellular markers. All of those techniques have been developed for flow cytometry, and you can mix and match them for complex sample handling. In terms of our own center, we are moving in that direction—to be able to exploit all the intrinsic capabilities of the detection system, which are currently limited by sample handling. That is how we see our mission evolving—to be able to do the types of practical experiments that cannot be done readily with other technologies.
BE: Mostly just doing it better and faster. We are more application-oriented now.
Do you see a change in how new medicines will be developed in the next decade?
BE: On the one hand, I see that giving academia the ability to do sophisticated screens is likely to result in the generation of novel probes. However, whether those will turn into drugs will have a lot to do with legal protection and the cost of drug development. I think academics will be more involved in developing diagnostic and prognostic biomarkers in the future. But in terms of developing new drugs, I am not sure the legal and financial environment makes it possible for new drugs to come out of academia.
LS: There is a notion that discovery is not a science, it is simply a mechanical process and that having been industrialized there is nothing left to learn. I doubt that is true. Drug discovery is not just a matter of throwing lots of small molecules at targets. There are many different kinds of targets and there are different kinds of compound and fragment libraries. Discovery science is not nearly as advanced as one might imagine. It is not all about hooking plate readers or detectors to robots.
Having this capability in an academic environment means that people will be trying things that are very risky but that have big potential rewards. I believe the federal government is willing to support that, and this is leading to new kinds of partnerships. I recently attended a Kauffman Foundation meeting in Kansas City. This is a foundation interested in promoting entrepreneurship through academia–industry relationships and public–private partnerships.
These collaborations are no longer simply two-way partnerships. The federal government is not only giving money to these partnerships, it is also making decisions about what areas are worth promoting. You see this with the Clinical and Translational Science Center program and the NCI's Experimental Therapeutics program. These programs are becoming very much results and translation oriented.
Another type of partnership, called Venture Philanthropy, was an eye-opener to me. In this setting, foundations with their own agendas are involved in partnerships intended to move things along and into people. It has become clear that these partnerships do not only involve universities handing things to Pharma, but that Pharma has things they do not know what to do with that they are asking academic scientists to look at. Reagents sitting on the shelf in Pharma companies that failed in their original purpose may have the potential to be repurposed even before they have been purposed, because they failed in their original purpose.
I think there will be new types of partnerships, and for us, with a niche technology, within about a year away from being in our new space with new equipment, and with the funding we have from the government and from our institution, we feel that we are going to participate in these partnerships and be part of a larger network of interactions.
Footnotes
Suggested Reading
1
Principal Investigator and Director;
2
Co-Leader, Assay Development and Optimization Core;
3
Co-Leader, High Throughput Screening and Implementation Core;
Center for Molecular Discovery, University of New Mexico, Albuquerque, New Mexico.