
Abstract
Hyperactivity of voltage-gated sodium channels underlies, at least in part, a range of pathological states, including pain and epilepsy. Selective blockers of these channels may offer effective treatment of such disorders. Currently employed methods to screen for sodium channel blockers, however, are inadequate to rationally identify mechanistically diverse blockers, limiting the potential range of indications that may be treated by such agents. Here, we describe an improved patch clamp screening assay that increases the mechanistic diversity of sodium channel blockers being identified. Using QPatch HT, a medium-throughput, automated patch clamp system, we tested three common sodium channel blockers (phenytoin, lidocaine, and tetrodotoxin) with distinct mechanistic profiles at Nav1.2. The single-voltage protocol employed in this assay simultaneously measured the compound activity in multiple states, including the slow inactivated state, of the channel. A long compound incubation period (10 s) was introduced during channel inactivation to increase the probability of identifying “slow binders.” As such, phenytoin, which preferentially binds with slow kinetics to the fast inactivated state, exhibited significantly higher potency than that obtained from a brief exposure (100 ms) used in typical assays. This assay also successfully detected the use-dependent block of tetrodotoxin, a well-documented property of this molecule yet unobserved in typical patch clamp protocols. These results indicate that the assay described here can increase the likelihood of identification and mechanistic diversity of sodium channel blockers from a primary screen. It can also be used to efficiently guide the in vitro optimization of leads that retain the desired mechanistic properties.
Introduction
VGSCs can assume one of four distinct sets of conformational states: closed, open, fast inactivated, and slow inactivated. Most VGSC-blocking drugs in clinical use exhibit pronounced use and state dependence, preferentially binding to channels that are in the inactivated state(s). This state-dependent inhibition tends to discriminate against sodium channel activity in cells with sustained depolarization/ectopic firing, as is thought to occur in pathophysiological conditions such as ischemia and epilepsy, but leaves physiologically functioning channels little changed. Such functional selectivity is thought to improve the safety margin of therapeutic drugs.
VGSC-blocking drugs with higher affinity for the fast inactivated state than for the closed state have been widely investigated (for review, see ref. 1 ). Some of these drugs (e.g., lidocaine) bind to the fast inactivated state rapidly, 2 whereas others (e.g., phenytoin) do so with relatively slow kinetics. 2,3 More recent studies suggest that selective stabilization of the slow inactivated state may be clinically beneficial as well, as was shown for lacosamide, a novel anticonvulsant/analgesic. 4 –6 Other agents, such as tetrodotoxin (TTX), are conventionally classified as use-independent blockers. In the case of TTX, however, although use-/voltage-dependent block is not typically evident using relatively short use- or state-dependent protocols (e.g., a 100 ms preconditioning depolarization), it is observed in longer-duration protocols that appropriately account for the relatively slow kinetics of the interaction between TTX and the channel. 7
The diverse mechanisms of VGSC-blocking drugs documented to date, however, did not arise through rational design. Rather, they were determined empirically following the recognition of their clinical utility. Target-based efforts for the rational development of selective VGSC-blocking drugs would benefit greatly from “front loading” of mechanistic information, which can be obtained early in the process by conducting screens that appropriately identify blockers with diverse mechanisms for desired therapeutic effects. Unfortunately, existing screening methods are not optimal for achieving this goal.
In this study, we designed a patch clamp screening assay capable of identifying and distinguishing VGSC blockers that act via a range of mechanisms. We validated the assay by testing and recapitulating the mechanisms of three well-characterized, mechanistically distinct VGSC blockers at Nav1.2 (i.e., phenytoin, lidocaine, and TTX) using QPatch HT, an automated patch clamp system. 8 It is unlikely that many molecules possessing some of these characteristics/mechanisms would have been identified using existing screening approaches. Thus, the assay described here represents a significant improvement over existing methods in the range of mechanisms of sodium channel block that can be identified and distinguished in a screening campaign, providing for greater diversity and the potential for discovering novel VGSC-blocking drugs.
Materials and Methods
Chemicals
Phenytoin and lidocaine (Sigma, St. Louis, MO) were kept as 300 mM stocks in 100% dimethyl sulfoxide at −20°C. TTX (Sigma) was dissolved in a citrate buffer provided by the vendor and kept as a 1 mg/mL stock at −20°C. The highest working concentration of each compound was made from its respective stock, and serial dilutions to all lower working concentrations were in turn made from the highest working concentration.
Cell Preparations for QPatch HT
CHL1610 cells stably expressing rat Nav1.2 were cultured in Dulbecco's modified Eagle's medium that was supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 400 μg/mL G418 and maintained in 5% CO2 at 37°C.
Cells grown to ∼90% confluence were rinsed once with Ca2+- and Mg2+-free Dulbecco's phosphate-buffered saline and incubated in 5 mL Detachin (Genlantis, San Diego, CA) at 37°C for 10 min. Dissociated cells were then gently mixed with 10 mL of CHO SFM II serum-free media (Invitrogen, Carlsbad, CA) supplemented with 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), transferred into a 15-mL conical tube, and centrifuged at 650 rpm for 3 min at room temperature. After removing the supernatant, cells were resuspended by gentle trituration in 5 mL of CHO SFM II media (also containing 25 mM HEPES) and transferred to the QPatch stirring station (the QStirrer), where they were gently and continuously stirred to ensure that they remained individualized and in suspension. Cells were allowed to recover in the QStirrer for at least 15 min prior to the start of an experiment.
QPatch Electrophysiology
Experiments were conducted in the unattended mode of the QPatch HT. All test solutions (±compounds) were held in glass inserts placed in 96-well plates. The final density of cells delivered to the QPlate was ∼2–3×106 cells/mL. Approximately 47% of the cells completed the screening protocol (described below). For these cells, the average seal, whole-cell, and series resistance (80% compensated) values were 1.7, 0.8, and 9.7 MΩ, respectively.
For experiments studying the activity of compounds, cells were held at −100 mV except when the voltage protocol was executed. Solutions were delivered to the cells as applications of brief bursts, such that each liquid application consisted of a burst of three puffs (5–6 μL/puff ) separated by 2 s. Each experiment contained four liquid application periods, starting with the control solution followed by three applications of the same concentration of a test compound (or control solution in the case of control cells). The time interval between each liquid application was approximately 1 min. The voltage protocol was executed only once, 5 s after each liquid application. All compounds tested in the present study reached steady-state inhibition at the end of this liquid application protocol. Currents were sampled at 25 kHz and filtered at 3 kHz. No fewer than nine control cells were tested for each 96-well drug plate. Control cells for each drug plate were used to correct for current rundown in data generated from that drug plate only. For experiments studying recovery from and voltage dependence of inactivation, experimental conditions are given in the figure legends. For all experiments, series resistance was 80% compensated and solutions perfusing cells were at room temperature (i.e., ∼22°C).
The extracellular solution contained (in mM) the following: NaCl (132), KCl (5.4), CaCl2 (1.8), MgCl2 (0.8), HEPES (10), and glucose (10). The pH (adjusted with NaOH) and osmolarity were 7.4 and 305–315 mOsm, respectively. The intracellular solution contained (in mM) the following: CsCl (45), CsF (100), EGTA (5), HEPES (10), and glucose (5). The pH (adjusted with CsOH) and osmolarity were 7.4 and 290–300 mOsm, respectively.
Data Analysis
Data analysis was performed using Sophion's QPatch assay software in combination with Origin 7.0 (OriginLab, Northampton, MA) and Microsoft Excel (Redmond, WA). Currents from the first (i.e., control) and fourth (i.e., the last compound) applications were used to calculate % inhibition values. All concentration–response data were fitted to a logistic function as follows: R=100/(1+c/IC50) p , where R is the % inhibition, p is the Hill coefficient, c is the compound concentration, and IC50 is the concentration at which 50% inhibition occurred. The voltage dependence of inactivation was fitted to a Boltzmann function to determine V 1/2, the voltage at which 50% of the channels were inactivated. Data fitting was performed using Origin 7.0. Where applicable, two-way analysis of variance was performed to determine statistical significance. Data are expressed as mean±SEM.
Results
In an effort to design a single-voltage protocol capable of assessing channel availability in each of the conformational states of Nav1.2 (i.e., closed, open, fast inactivated, and slow inactivated) in a “one-stop” experiment, we first studied the kinetics of recovery from the fast and slow inactivated states. They were induced by a preconditioning voltage pulse to 0 mV, with pulse durations of 100 ms and 10 s, respectively. Recovery from either state was monitored with a double-pulse protocol as shown in Figure 1A and B. As summarized in Figure 1C, the time course of recovery from both fast and slow inactivation at −90 mV can be well approximated with a double-exponential function, with time constants of 5.9 and 495.1 ms (fast) and 698.2 and 5869.0 ms (slow), respectively. Although these protocols likely induced more than one fast inactivated state and more than one slow inactivated state, we will refer to the state(s) induced by the 100 ms depolarization (to 0 mV) as the fast inactivated state and those by the 10 s depolarization (also to 0 mV) as the slow inactivated state. 9
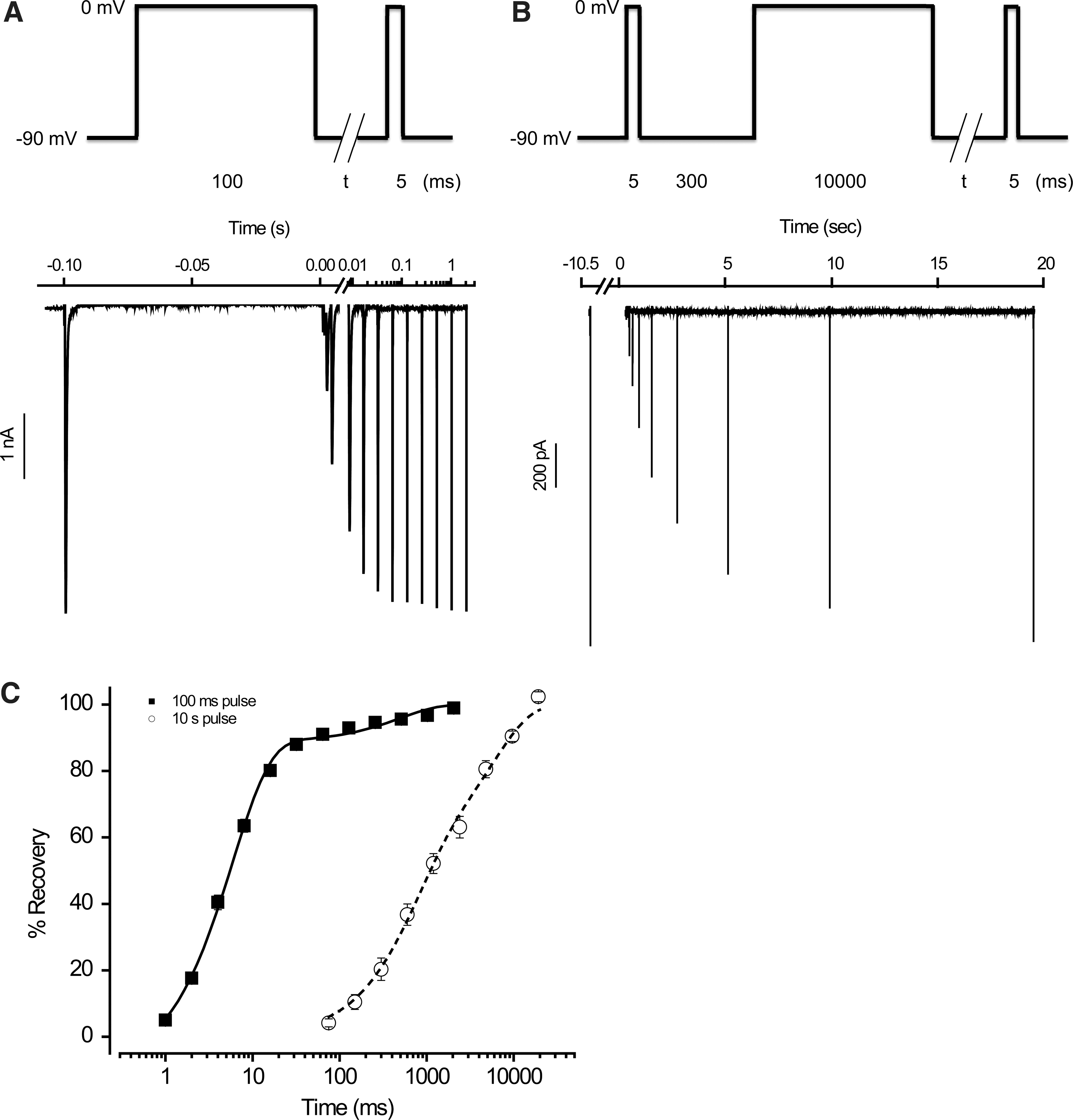
Recovery from inactivation of Nav1.2 channels.
We next determined the voltage dependence of steady-state inactivation using voltage protocols shown in Figure 2A and B. In the case of slow inactivation, a period of 300 ms at −90 mV was introduced to allow for full recovery from fast inactivation before assessing occupancy in the slow inactivated sate (i.e., t=300 ms in Fig. 2B). Under these conditions, V 1/2 was −48.4 mV for fast inactivation, −55.3 mV for slow inactivation, and −60.5 mV for total inactivation (i.e., t=0 in Fig. 2B), as shown in Figure 2C. These values are in general agreement with those from the literature. 10 –15
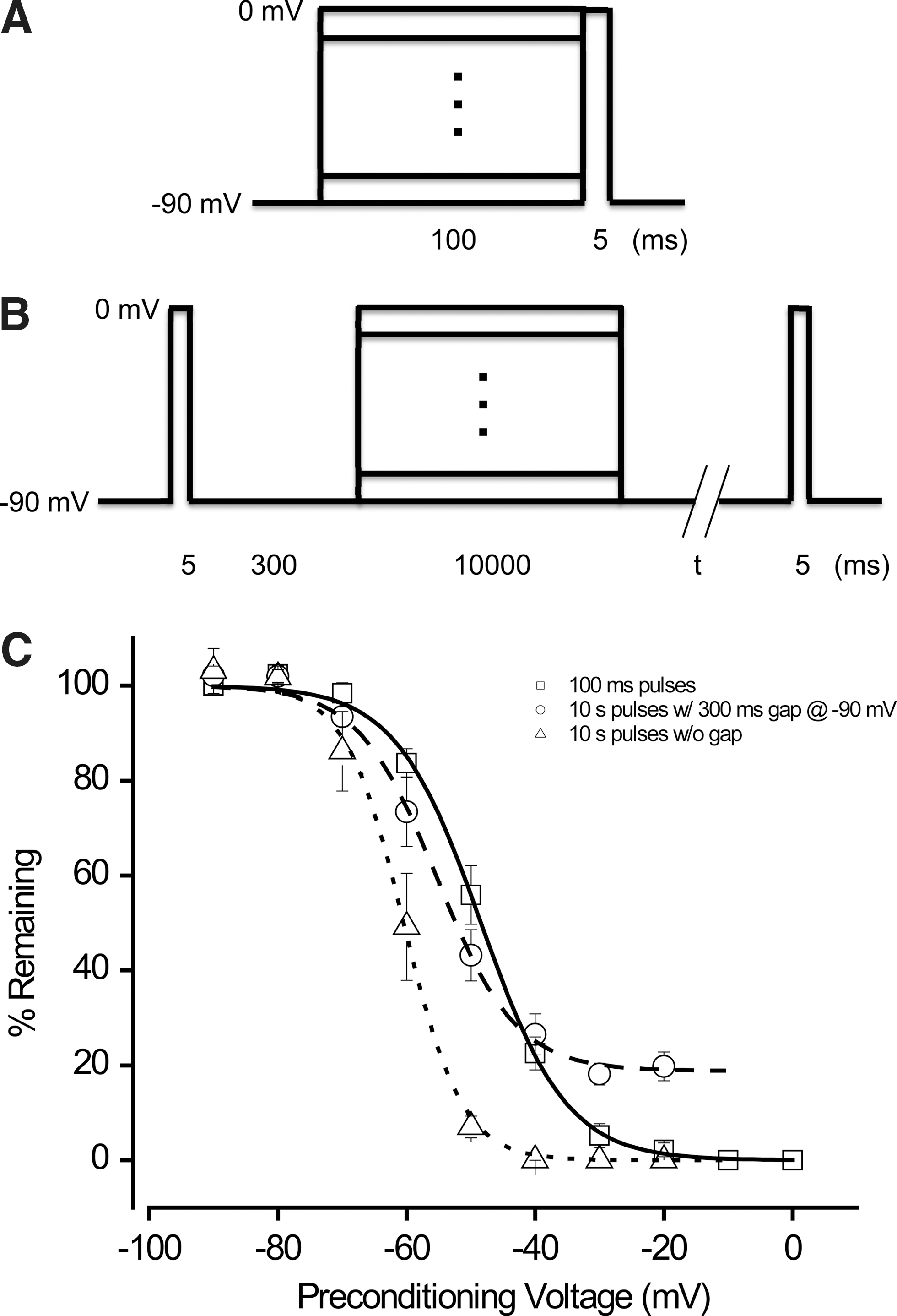
Voltage dependence of steady-state inactivation of Nav1.2 channels.
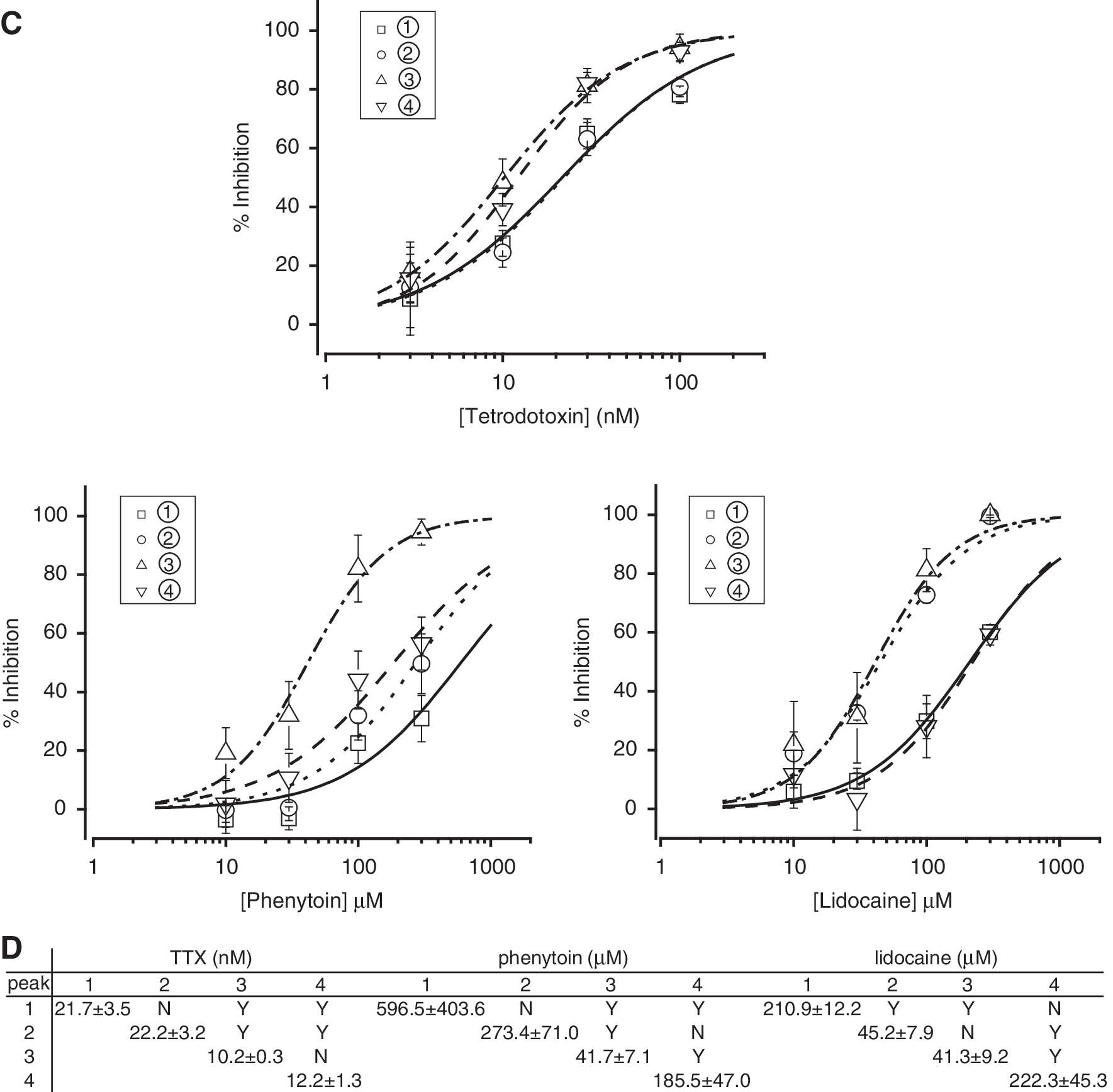
On the basis of these results, we designed a single patch clamp protocol to simultaneously assess the effect of compounds in various states of the channel. This protocol, shown in Figure 3A, included brief (i.e., 5-ms) periods of depolarization to 0 mV at four time points (corresponding to peaks 1–4) to assess channel availability in various states. Peak 1 reflected channel availability in the closed state. Peak 2 reflected channel availability after partial recovery from fast inactivation. Peak 3 reflected channel availability after partial recovery from the fast inactivated state and significant entrance into the slow inactivated state. Finally, peak 4 reflected channel availability after partial recovery from slow inactivation. Examples of these currents are illustrated in Figure 3B for control solution alone and in the presence of a blocking concentration of TTX, phenytoin, or lidocaine. As shown in Figure 3C, these three compounds inhibited Nav1.2 with distinct profiles when assessed by these peak currents. For example, lidocaine blocked Nav1.2 more potently in the fast inactivated state (peak 2) than in either the closed (peak 1) or slow inactivated (peak 4) state (Fig. 3C). At first glance, block by phenytoin in the fast inactivated state (as indicated by peak 2) was not significantly different from that in either the closed state (peak 1) or slow inactivated state (peak 4), as shown in Figure 3C. However, closer inspection revealed that most of the ∼6.5-fold increase in potency obtained for peak 3 over peak 2 resulted from binding of phenytoin to the fast inactivated state, which was only manifested after the channel remained in this state for a prolonged period of time (i.e., 10 s in this case vs. 100 ms for peak 2). Phenytoin may also have somewhat higher affinity for the slow inactivated state than for the closed state, as shown in Figure 3C (also, see Discussion). Interestingly, this protocol also revealed small (∼twofold) but significant differences between TTX potency obtained from peaks 1 and 2, on the one hand, and that obtained from peaks 3 and 4, on the other hand. TTX blocked the channel with higher affinity after prolonged depolarization (i.e., 10 s) than either after a short depolarization (i.e., 100 ms) or in the closed state (Fig. 3D).
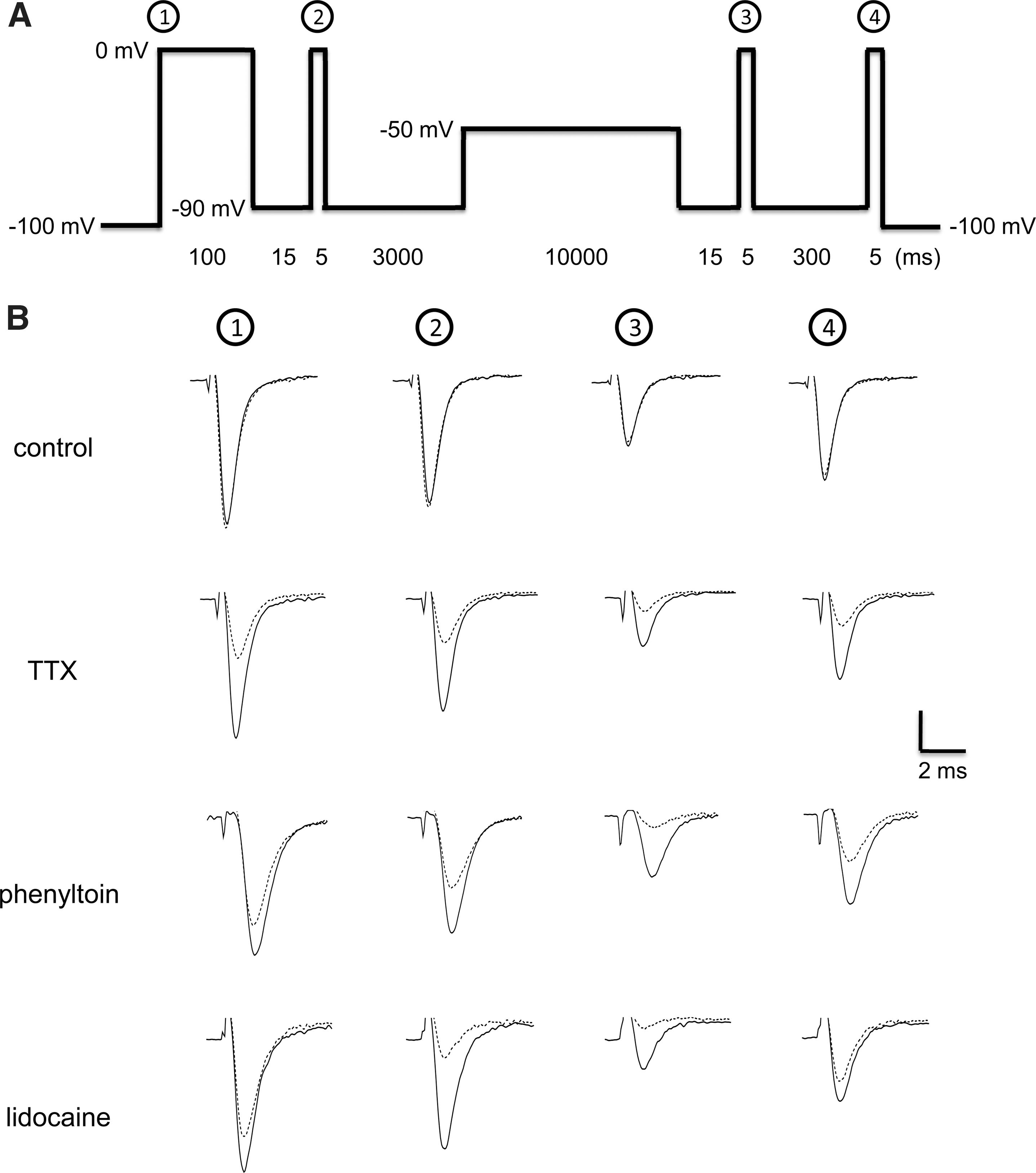
Simultaneous measurement of concentration-dependent responses of sodium channel blockers in multiple states of Nav1.2 channels.
Discussion
We developed a patch clamp assay in which a single-voltage protocol enabled the simultaneous determination of a compound's activity in all major sets of conformational (closed, open, fast inactivated, and slow inactivated) states of the VGSC Nav1.2. This assay protocol represents a significant improvement in several aspects over published protocols that are typically used for screening use/state-dependent sodium channel blockers. First, in addition to being able to detect compounds that bind to the open, closed, and fast inactivated states, this protocol also detects compounds that bind to the slow inactivated state, thereby broadening the mechanistic diversity of blockers that can be identified at one time. It can also guide the work of structure–activity relationships in a more directed manner, for instance, by developing blockers that bind selectively to the fast or slow inactivated state, depending on the mode of block desired for a particular therapeutic effect. This approach is superior to existing patch clamp paradigms that identify blockers without specific differentiation of their relative activities in the fast and slow inactivated states. 16,17 Hyperactive neurons in many pathological states tend to be chronically depolarized, causing sodium channels to enter the slow inactivated state. Blockers that specifically target this state may therefore offer a therapeutic advantage by selectively blocking the electrical activity in these neurons. The novel anticonvulsant lacosamide, for example, has been shown to bind selectively to the slow but not the fast inactivated state of sodium channels, 5,6 highlighting the clinical relevance of such blockers.
Second, by introducing a long (i.e., 10 s) depolarizing pulse, the present protocol increases the likelihood of identifying blockers that preferentially bind with slow association rates to the fast inactivated state. Phenytoin is a good example of a blocker that belongs to this category. 3 Such blockers have a higher probability of being dismissed as false negatives when using a typical use-dependent protocol or a two-pulse protocol with short (e.g., 100-ms), fast inactivation–inducing depolarizations. In the present assay, the affinity of phenytoin for the fast inactivated state after 100 ms of depolarization/channel inactivation was not significantly higher than that for the closed state. After 10 s of depolarization, however, the affinity of phenytoin for the fast inactivated state was greatly increased, consistent with the idea that the rate of phenytoin binding to the fast inactivated state is relatively slow. 3 This is in sharp contrast to lidocaine, a blocker with fast binding kinetics, whose effect on the fast inactivated state was complete within 100 ms of depolarization; increasing the duration of depolarization (to 10 s) did not change the affinity of lidocaine for this state. Phenytoin also appeared to prefer the slow inactivated state to the closed state, although it is possible that this apparent potency difference was the result of “contamination” due to slow drug dissociation from the fast inactivated state. Lidocaine, on the other hand, does not distinguish between the two states. These results are in general agreement with earlier studies. 3,18
Third, the protocol described in this study also detects the use dependence of TTX block, a particular feature of this molecule that typical use-dependent protocols or two-pulse protocols with short, fast inactivation–inducing depolarizations fail to detect. TTX binds with high affinity to the extracellular pore of TTX-sensitive sodium channels, including Nav1.2. Although TTX is generally considered to block these channels in a use-independent manner, 19 use-dependent block of sodium channels by this compound has been observed in many preparations, including cardiac and skeletal muscle cells. 20,21 It has been also demonstrated for neuronal Nav1.2 channels, in which the affinity of TTX is increased by membrane depolarization. 7,22 Ironically, high-frequency (e.g., 10-Hz) stimulation protocols designed to evaluate use dependence are often not long enough in duration to detect the type of use-dependent block produced by TTX, which occurs on a slower time scale (i.e., many seconds) and is thought to involve a trapped-ion mechanism. 7,23 Consistent with results obtained using two-pulse protocols with short depolarizations, peak 2 in the present protocol, which occurred only 115 ms after the start of the 100 ms depolarization, failed to show any difference in TTX potency than that in the closed sate. In contrast, the TTX potency obtained from peaks 3 and 4, which were elicited after >10 s of depolarization, both exhibited a ∼2-fold increase over that in the closed state, in good agreement with previous observations. 7,22,24,25
In summary, using three well-characterized VGSC blockers with distinct pharmacological profiles (i.e., TTX, phenytoin, and lidocaine), we demonstrated in this study the utility of a “one-stop” patch clamp assay for screening VGSC blockers. Compared with conventional patch clamp screening paradigms, this assay has the advantage of being able to identify and distinguish a wider range of state-dependent blocking mechanisms in a single protocol/run. The advent of high-throughput patch clamp platforms makes it possible and indeed, essential, for such assays to be implemented at the earliest stages of the drug discovery process. Of particular interest is the potential of this assay to identify compounds that bind to the slow inactivated state selectively or to the fast inactivated state with slow kinetics. Compounds with such properties may be of particular therapeutic potential and are often not rationally identified and optimized based on these molecular mechanisms. It should be noted that properties of certain VGSC blockers (e.g., slow rate of dissociation from fast inactivated state that significantly overlaps with the kinetics of slow inactivation) may preclude clear mechanistic differentiation using a single screening protocol or, for that matter, even more sophisticated protocols designed for detailed mechanistic studies. 18 Nonetheless, the higher mechanistic resolution (i.e., the ability to identify VGSC blockers with a wider range of mechanisms) afforded by the present assay over conventional patch clamp screening paradigms can greatly facilitate the identification and optimization of novel leads with desired mechanistic properties throughout the high-throughput screening and lead optimization processes. Finally, this assay can be readily configured for other subtypes of VGSCs using various automated patch clamp platforms.
Footnotes
Disclosure Statement
No competing financial interests exist.