Heijtz RD, Wang S, Anuar F, Qian Y, Björkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci U S A 2011;347:3047–3052.
Abstract: Microbial colonization of mammals is an evolution-driven process that modulate host physiology, many of which are associated with immunity and nutrient intake. Here, we report that colonization by gut microbiota impacts mammalian brain development and subsequent adult behavior. Using measures of motor activity and anxiety-like behavior, we demonstrate that germ free (GF) mice display increased motor activity and reduced anxiety, compared with specific pathogen free (SPF) mice with a normal gut microbiota. This behavioral phenotype is associated with altered expression of genes known to be involved in second messenger pathways and synaptic long-term potentiation in brain regions implicated in motor control and anxiety-like behavior. GF mice exposed to gut microbiota early in life display similar characteristics as SPF mice, including reduced expression of PSD-95 and synaptophysin in the striatum. Hence, our results suggest that the microbial colonization process initiates signaling mechanisms that affect neuronal circuits involved in motor control and anxiety behavior.
Commentary:Professor Jeffrey Gordon, who studies microbial symbiotic relationships at the Washington University School of Medicine in St. Louis, has suggested that there are 10 times as many microbial cells than human cells in the body—meaning that we are really 90% microbial and only 10% human. Microbes found in our gut play a role in both normal physiology and pathology. This article suggests a role of gut microbiota in mammalian behavior. In this work, either germ-free (GF) mice (which do not contain microorganisms, including aerobic and anaerobic bacteria and fungi) or mice that have a normal gut microbiota but are free of specific pathogens (SPF) were used to evaluate anxiety-like behavior. GF mice differed from SPF mice when tested in two common behavioral tests of anxiety. When GF mice were introduced to gut microbiota derived from SPF mice at day 30 (so-called conventionalized [CON] mice), it was found that the CON mice normalized to the SPF mouse behavior. Therefore, introduction of gut microbiota into developing GF mice resulted in modification of anxiety-like behavior. Examination of monoamine neurotransmitters in the brains of GF mice showed higher turnover of neurotransmitters such as dopamine, adrenaline, and serotonin. The expression patterns of genes previously linked to behaviors related to anxiety and fear in the brains of these mice also showed lower expression levels in GF mice within several key regions of the brain (see figure). The exact mechanism by which gut microbiota may control anxiety-like behavior in these mice remains to be found, but these results lend further insight into the how microbiota influence development. Recently, some epidemiological studies have shown an association between pathogen exposure during early human development and behavioral disorders such as autism and schizophrenia. Gaining an improved understanding on how residing microorganisms influence both our health and behavior should continue to be an important goal of biomedical research. Contributed by Doug Auld.
GF mice show altered expression of anxiety and synaptic plasticity-related genes. (A) Representative autoradiograms showing NGFI-A mRNA expression at the level of the frontal cortex of SPF and GF mice (OFC, orbital frontal cortex; AO, anterior olfactory region). (A′) Bars show expression of NGFI-A mRNA (nCi/g) in the OFC and AO of SPF and GF mice. (B) Representative autoradiograms showing BDNF mRNA expression at the level of amygdala and dorsal hippocampus of SPF and GF mice (BLA, basolateral amygdala; CA1, CA1 region of the dorsal hippocampus). (B′) Bars show expression of BDNF mRNA (nCi/g) in the BLA and CA1 region of SPF and GF mice. (C) Representative autoradiograms showing dopamine D1 receptor (Drd1a) mRNA expression at the level of the striatum and nucleus accumbens of SPF and GF mice (STR, striatum; Accb, nucleus accumbens, shell region). (C′) Bars show expression of Drd1a mRNA (nCi/g) in the STR and Accb of SPF and GF mice. (D) Representative autoradiograms showing Drd1a mRNA expression at the level of the dorsal hippocampus of SPF and GF mice (DG, dentate gyrus; PtCx, parietal cortex, somatosensory area). (D′) Bars show expression of Drd1a mRNA (nCi/g) in the DG and PtCx of SPF and GF mice. All data (A′–D′) are expressed as means ± SEM, n = 8 per group. Filled bars represent GF mice. Open bars represent SPF mice. *P < 0.05, **P < 0.001 compared with SPF mice.
Assays: Easy as TEV?
Djannatian MS, Galinski S, Fischer M, Rossner MJ. Studying G protein-coupled receptor activation using split-TEV assays. Anal Biochem 2011; DOI:10.1016/j.ab.2011.01.042.
Abstract: G protein-coupled receptors (GPCRs) constitute the largest receptor family in mammals and represent important drug targets. Signaling through GPCRs mediates physiological effects which are strongly dependent on the cellular context. Therefore, the availability of assays monitoring GPCR activation applicable in different cell types could help to better understand GPCR functions and to realize the potential of known as well as novel substances. Here, we introduce a split-TEV assay to monitor GPCR activation through the stimulation-dependent recruitment of β-arrestin 2. Inactive N- and C-terminal fragments of the TEV protease are coupled to a GPCR and β-arrestin 2, respectively. Ligand-dependent interaction of the two fusion proteins leads to functional complementation of the TEV protease, followed by the cleavage of an artificial transcription factor and successive reporter gene activation. The presented split-TEV assay system is highly sensitive and was successfully applied in heterologous cell lines as well as in primary cultured neuronal and glial cells. We show that assay performance strongly depends on the endogenous properties of different cell types. The sensitivity and flexibility makes split-TEV assays a valuable tool to analyze GPCR activation in different cell types in a rapid and cost-effective way.
Commentary:Several assay formats have been developed to measure the interaction of GPCRs with β-arrestins. In one format, a 27-kDa protease from the tobacco etch virus (TEV) is used to release a transcription factor by proteolysis following the interaction of two protein partners (Tango® format). The transcription factor is then free to activate a reporter gene that expresses an enzyme such as firefly luciferase. The enzymatic mechanism of the TEV protease is similar to serine proteases, but the nucleophile in the canonical Ser-Asp-His triad is cysteine instead of serine, making the protease resistant to many serine protease inhibitors. The cleavage specificity of TEV is highly specific as well (recognition sequence ENLYFQG/S; cleavage between the Q and G/S), so the protease is not generally toxic to cells. This article describes a different version of this format in which a split TEV protease is used to measure the interaction of β-arrestin 2 with GPCRs (see figure). The transcription factor was attached to the GPCR C-termini via a TEV cleavage site and consisted of a fusion between the Gal4 DNA-binding domain and the herpes simplex VP16 activation domain as well as the C-terminal fragment of TEV. The N-terminal of TEV was then fused to β-arrestin. Upon interaction of the GPCR with β-arrestin, the transcription factor was released by the reconstructed TEV protease, resulting in expression of firefly luciferase through a Gal4 responsive reporter gene. The tagged GPCR receptors used in the split-TEV assay demonstrated normal responses when typical cAMP or calcium assays were used. In this work, the split TEV assay was compared to the full length TEV assay (e.g., Tango, so-called “full-TEV” in the article) by using three different GPCRs with either transient transfection or stable cell line protocols. In this study the split-TEV system showed improved signal to background compared to the full-TEV format especially in transient transfections measuring GPCR activation. The split-TEV system used high-affinity TEV cleavage sites, which could explain the improved performance. One common aspect of the TEV assay demonstrated in this article is that this assay is cell-type dependent, with certain cell lines showing high background due to TEV-independent cleavage of the transcription factor. This is likely due to the presence of endogenous agonists in certain cell lines; for example, secretion of dopamine by PC12 cells. Therefore, assay performance of such systems is highly dependent on such endogenous cellular components (agonist ligands, receptors, GRKs, etc.). Overall, this article provides a good discussion of the factors to consider when employing a TEV-based reporter system to measure GPCR activation. Contributed by Doug Auld.
Design of split-TEV and full-TEV assays to monitor GPCR activation. (A and B). Illustration of split-TEV (A) and full-TEV (B) assays with functionally relevant domains depicted schematically. Both split- and full-TEV assays rely on the interaction of modified GPCRs with βArr2 on GPCR activation. Split-TEV assays are based on the functional reconstitution of inactive N- and C-terminal TEV fragments (N-TEV and C-TEV). N-TEV is fused to the C terminus of the GPCR, followed by a high-affinity TEV cleavage site (tevS) and the artificial transcription factor GV, whereas C-TEV is fused to βArr2 (A). Interaction of the two fusion proteins leads to the reconstitution of the TEV protease activity-releasing GV. Full-TEV assays use a “proximity approach” in which the full-length TEV protease (TEV) is fused to βArr2, whereas the GPCR is coupled to GV via a low affinity TEV protease cleavage site (tevS*) (B). Proximity of the two fusion proteins leads to TEV-mediated cleavage and release of the transcription factor. Full-TEV receptor constructs were used with an additional N-TEV domain (similar to split-TEV constructs [left]) or without N-TEV (similar to Tango constructs [right]). Common for both split and full-TEV assays is the TEV-dependent release of GV that finally activates firefly luciferase as a reporter gene (not shown). (C) Domain structure of constructs used for split-TEV (left) and full-TEV assays (right). For GPCR-N-TEV-tevS-GV and βArr2-C-TEV fusion constructs, the following modified constructs were also tested: GPCR-VC-N-TEV-tevSGV, which harbors a C-terminal tail of the human vasopressin AVPR2 receptor to provide a stronger interaction with βArr2; and βArr2Δ-C-TEV, the C-terminally truncated form of human βArr2. N-TEV, N-terminal fragment of TEV protease (aa 1–118); C-TEV, C-terminal fragment of TEV protease (aa 119–221); TEV, full-length TEV protease; VC, C terminus of the AVPR2 receptor (aa 343–371); tevS, natural TEV cleavage site ENLYFQ'G; tevS*, low-affinity TEV protease cleavage site ENLYFQ'L; GV, Gal4-VP16 transcription factor; bArr2, b-arrestin 2; βArr2Δ, truncated βArr2 (aa 1–382).
Chemofluor Assay
Mahendrarajah K, Dalby PA, Wilkinson B, Jackson SE, Main ERG. A high-throughput fluorescence chemical denaturation assay as a general screen for protein–ligand binding. Anal Biochem 2010; DOI:10.1016/j.ab.2010.12.001.
Abstract: Chemical denaturation of ligand–protein complexes can provide the basis of a label-free binding assay. Here, we show how the technique can be used as a sensitive/affordable screen of potential ligands from a pool of lead drug variants. To demonstrate, we characterized the binding of polyketide ligands based on the mTOR inhibitor rapamycin to the cellular immunophilin FKBP12. This used the intrinsic fluorescence of the protein to monitor the chemical denaturation of each FKBP12–ligand complex. The assay was then successfully modified to a 96-well plate-based screen. Both formats were able to differentiate binding affinities across a wide dynamic range.
Commentary:Methods that measure ligand-binding to proteins can be a critical step in the validation of compounds derived from high-throughput screening efforts. Evidence for direct binding of compounds to the protein target of interest provides the basis for mechanism of action studies and structure–activity optimization efforts. Several methods to measure ligand binding have been described and are well utilized. These include the use of surface plasmon resonance instruments, fluorescence-based measurements that monitor shifts in thermal denaturation profiles and Tm by using commercial PCR machines, and label-free methods in which the aggregation of unfolded proteins upon thermal-denaturation is monitored by light scattering to provide Tagg values (StarGazer®; Harbinger Biotechnology and Engineering Corporation). Proteins can also be stabilized to chemical denaturants by ligand binding. Chemical denaturation experiments provide thermodynamic values such as the ΔG of unfolding (ΔGD) and the Cm (the concentration of denaturant at the mid-point of the unfolding curve). Monitoring the perturbation of chemical denaturation folding equilibriums by ligand binding can be used to determine the Kd of the ligand. This article employs guanidine-hydrochloride to denature FKBP12 in the presence or absence of rapamycin and other analogs (see figure). A label-free approach is enabled by monitoring the intrinsic fluorescence of tryptophan in FKBP12 because this protein shows a strong change in fluorescence upon denaturation. The difference in ΔGD values for free or ligand bound FKPB12 can be used to calculate the Kd values of the ligands, and the necessary equations and methods are given in the supplementary material of the article. Comparison of these results to binding data derived from isothermal titration-based calorimetry showed that both methods yielded the same rank order ligand potency. The assay volumes were reduced to allow a 96-well format in which a concentration of 2 μM protein with a 10-fold excess of ligand was required for the measurement. UV-transparent F-type microtiter plates were used to allow excitation of tryptophan fluorescence at 280 nm with a fluorescent plate reader. The 96-well results agreed well with those obtained using a standard cuvette-based protocol. The 96-well approach using standard equipment and could be used to profile ligands at the lead optimization stage. Although based on traditional protein-chemistry methodology, this article provides a reminder of how this approach could be used to enhance biophysical characterization of ligand–protein binding when the protein possesses sufficient intrinsic fluorescence. Contributed by Doug Auld.
(A) Structure of FKBP12 bound to rapamycin (PDB:1FKL), prepared using PYMOL (www.pymol.org). (B–E) Chemical structures of rapamycin (B), ligand A (C), ligand B (D), and ligand T (E). (F and G) Equilibrium unfolding of FKBP12 with various rapalogs. Experiments were monitored using either a fluorimeter (F) or a fluorescence plate reader (G).
BTK Inhibitor Launches a Two-Pronged Attack on Arthritis
Di Paolo JA, Huang T, Balazs M, Barbosa J, Barck KH, Bravo BJ, Carano RAD, Darrow J, Davies DR, DeForge LE, Diehl L, Ferrando R, Gallion SL, Giannetti AM, Gribling P, Hurez V, Hymowitz SG, Jones R, Kropf JE, Lee WP, Maciejewski PM, Mitchell SA, Rong H, Staker BL, Whitney JA, Yeh S, Young WB, Yu C, Zhang J, Reif K, Currie KS. Specific Btk inhibition suppresses B cell– and myeloid cell–mediated arthritis. Nat Chem Biol 2011;7:41–50.
Abstract: Bruton's tyrosine kinase (Btk) is a therapeutic target for rheumatoid arthritis, but the cellular and molecular mechanisms by which Btk mediates inflammation are poorly understood. Here we describe the discovery of CGI1746, a small-molecule Btk inhibitor chemotype with a new binding mode that stabilizes an inactive nonphosphorylated enzyme conformation. CGI1746 has exquisite selectivity for Btk and inhibits both auto- and transphosphorylation steps necessary for enzyme activation. Using CGI1746, we demonstrate that Btk regulates inflammatory arthritis by two distinct mechanisms. CGI1746 blocks B cell receptor–dependent B cell proliferation and in prophylactic regimens reduces autoantibody levels in collagen-induced arthritis. In macrophages, Btk inhibition abolishes FcγRIII-induced TNFα, IL-1β and IL-6 production. Accordingly, in myeloid- and FcγR-dependent autoantibody-induced arthritis, CGI1746 decreases cytokine levels within joints and ameliorates disease. These results provide new understanding of the function of Btk in both B cell– or myeloid cell–driven disease processes and provide a compelling rationale for targeting Btk in rheumatoid arthritis.
Commentary:Bruton's tyrosine kinase (Btk) is a target for the treatment of a variety of diseases including autoimmune diseases, inflammatory diseases, leukemias, and lymphomas. Indeed, an irreversible inhibitor of Btk, PCI-32765, is currently in clinical trials for the treatment of leukemia and lymphoma. Btk is implicated in multiple signaling pathways with its role in the B-cell receptor (BCR) pathways being the most well-studied. Its biological role in inflammation, however, has been poorly defined. Mutations in human Btk cause X-linked agammaglobulinemia, a disease characterized by a lack of immunoglobulin-producing peripheral B cells. In mouse models with Btk mutations or deletions, there is a reduction in both B cells and immunoglobulin (Ig). Recently, several X-ray crystal structures have been reported that provide insight into the diverse conformations accessible to Btk, including several distinct inactive conformations (PDB ID: 3PIX, 3PIY, 3PIZ, 3PJ1, 3PJ2, 3PJ3, 3P08, 3GEN, 3K54). Type II inhibitors that target the inactive conformation of kinases became of interest because they were presumed to offer the possibility of more kinase selectivity over type I compounds that are indifferent to the activation state. There are selective type I inhibitors and less selective type II inhibitors; however, the allure of type II inhibitors stems from the success of the highly selective type II inhibitor Gleevec® for the treatment of chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors. This article presents data for CGI1746, a small molecule ATP-competitive inhibitor that stabilizes an inactive non-phosphorylated enzyme conformation of Btk (see figure). CGI1746 (IC50 = 1.9 nM and Kd = 1.5 nM) was found from hit-to-lead chemistry efforts following an initial 400 nM hit from a screen of a library built around an imidazo(1,2-a)pyrazzine scaffold. This compound has exceptional selectivity, and when screened against Ambit's large panel of 385 nonmutant kinases at 1 μM it only showed significant binding to Btk. In the inactive conformation, Tyr551 moves significantly and creates a large pocket called H3 into which the t-butylphenyl moiety of CGI1746 binds. This pocket is distinct from the allosteric pocket that most type II inhibitors utilize, and the conformation is also unique in that it is a DFG-in–inactive conformation, where DFG-in refers to the active conformation of the Asp-Phe-Gly residues of the activation loop. SAR analysis has indicated that this interaction with the H3 pocket contributes significantly to the overall selectivity and potency. This selectivity should help to eliminate unwanted effects due to off-target kinase activity, which is particularly important in the treatment of chronic diseases such as rheumatoid arthritis, the target of this article. CGI1746 is not only a potential drug compound, but it is also a useful probe compound due to its selectivity for Btk, and it was used to determine that Btk regulates inflammatory arthritis by two distinct mechanisms. B cells are antigen-presenting cells and they produce the auto-antibodies associated with rheumatoid arthritis. These auto-antibodies form immune complexes, which have pathogenic effects through activating Fcγ receptors (FcγR). Activating FcγRs are found on myeloid and dendritic cells. In particular, macrophage numbers in the joint are correlated with severe cartilage destruction. CGI1746 impacted B-cell activation and prevented arthritis in prophylactic B cell–dependent collagen-induced arthritis models (CIA). In inflammatory arthritis models driven by myeloid cell or FcγR activation, CGI1746 ameliorated disease and was shown to be important in the both the initiation and effector phases. CGI1746 was shown to be essential for FcγRIIa- and FcγRIII-induced cytokine (TNFα, IL6, and IL-1β) production in monocytes. It also blocked all Btk-dependent signaling events in B cells and B-cell proliferation in IgM+ and isotype-switched IgG+ B cells. Having a highly selective Btk inhibitor has allowed the biological role of Btk in disease to be further elucidated. Rheumatoid arthritis is a multifactorial disease and the ability to simultaneously disrupt several of the pathogenic pathways with inhibition of one target is an attractive strategy. Contributed by Mindy I. Davis.
Structural basis of CGI1746 selectivity. (a) Btk undergoes a conformational change upon binding CGI1746 resulting in sequestration of Tyr551. Ribbon diagram of the crystal structure of human Btk bound to CGI1746 (green and yellow) superimposed on human apo Btk (gray). The Tyr551 residues from each structure are shown as spheres. (b) Closeup view of the occupation of the H3 pocket by the t-butylphenyl moiety of CGI1746. Btk is shown as a transparent surface with residues of interest including Tyr551 rendered as sticks. (c) CGI1746 binds to Btk in an extended conformation and interacts with both the Btk ‘hinge’ and the H3 pocket. Side chains within 4.2 Å of CGI1746 are shown as sticks. The DGF motif is shown as sticks and colored pink. Select hydrogen bonds are shown as dashed lines. All residues mentioned in the text are labeled with the exception of Thr474, Glu475, His519 and Ser543, which are obscured by CGI1746 in this orientation. (d) Dasatinib (magenta) binds to Btk (blue) in a typical type I inhibitor orientation. Btk is shown in the same orientation as in (a). The activation loop including Tyr551 is not ordered in this structure, and the H3 pocket is not formed. (e) Comparison of the dasatinib (magenta) and CGI1746 (yellow) binding modes. Btk and CGI1746 are shown as in (c).
Global proteomic activity profiling by SMM
Wu H, Ge J, Yang PY, Wang J, Uttamchandani M, Yao SQ. A peptide aldehyde microarray for high-throughput profiling of cellular events. J Am Chem Soc 2011; DOI: 10.1021/ja109597v.
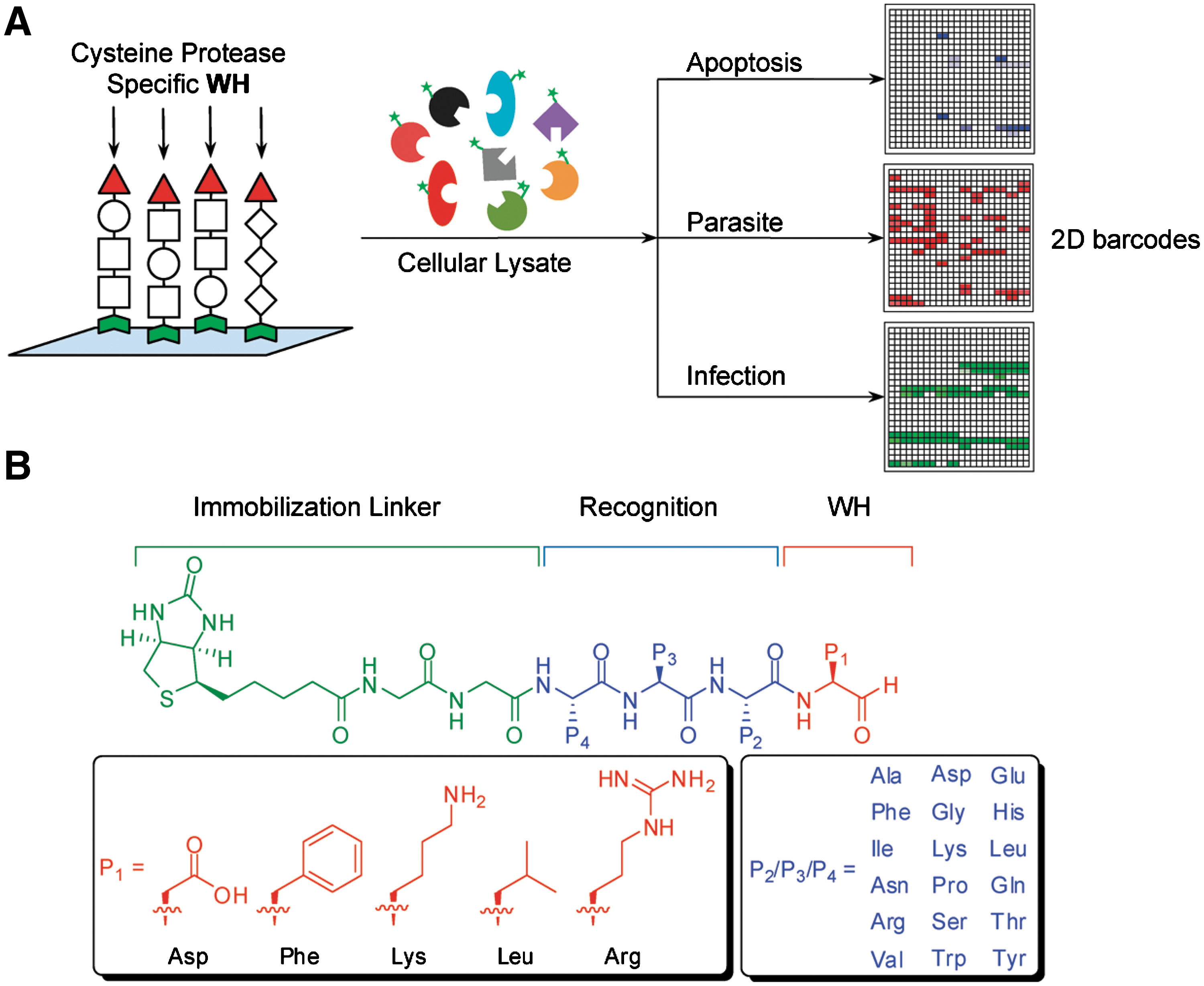
Abstract: Microarrays provide exciting opportunities in the field of large-scale proteomics. With the aim to elucidate enzymatic activity and profiles within native biological samples, we developed a microarray comprising a focused positional-scanning library of enzyme inhibitors. The library was diversified across P1–P4 positions, creating 270 different inhibitor sublibraries which were immobilized onto avidin slides. The peptide aldehyde-based small-molecule microarray (SMM) specifically targeted cysteine proteases, thereby enabling large-scale functional assessment of this subgroup of proteases, within fluorescently labeled samples, including pure proteins, cellular lysates, and infected samples. The arrays were shown to elicit binding fingerprints consistent with those of model proteins, specifically caspases and purified cysteine proteases from parasites (rhodesein and cruzain). When tested against lysates from apoptotic Hela and red blood cells infected with Plasmodium falciparum, clear signatures were obtained that were readily attributable to the activity of constituent proteases within these samples. Characteristic binding profiles were further able to distinguish various stages of the parasite infection in erythrocyte lysates. By converting one of our brightest microarray hits into a probe, putative protein markers were identified and pulled down from within apoptotic Hela lysates, demonstrating the potential of target validation and discovery. Taken together, these results demonstrate the utility of targeted SMMs in dissecting cellular biology in complex proteomic samples.
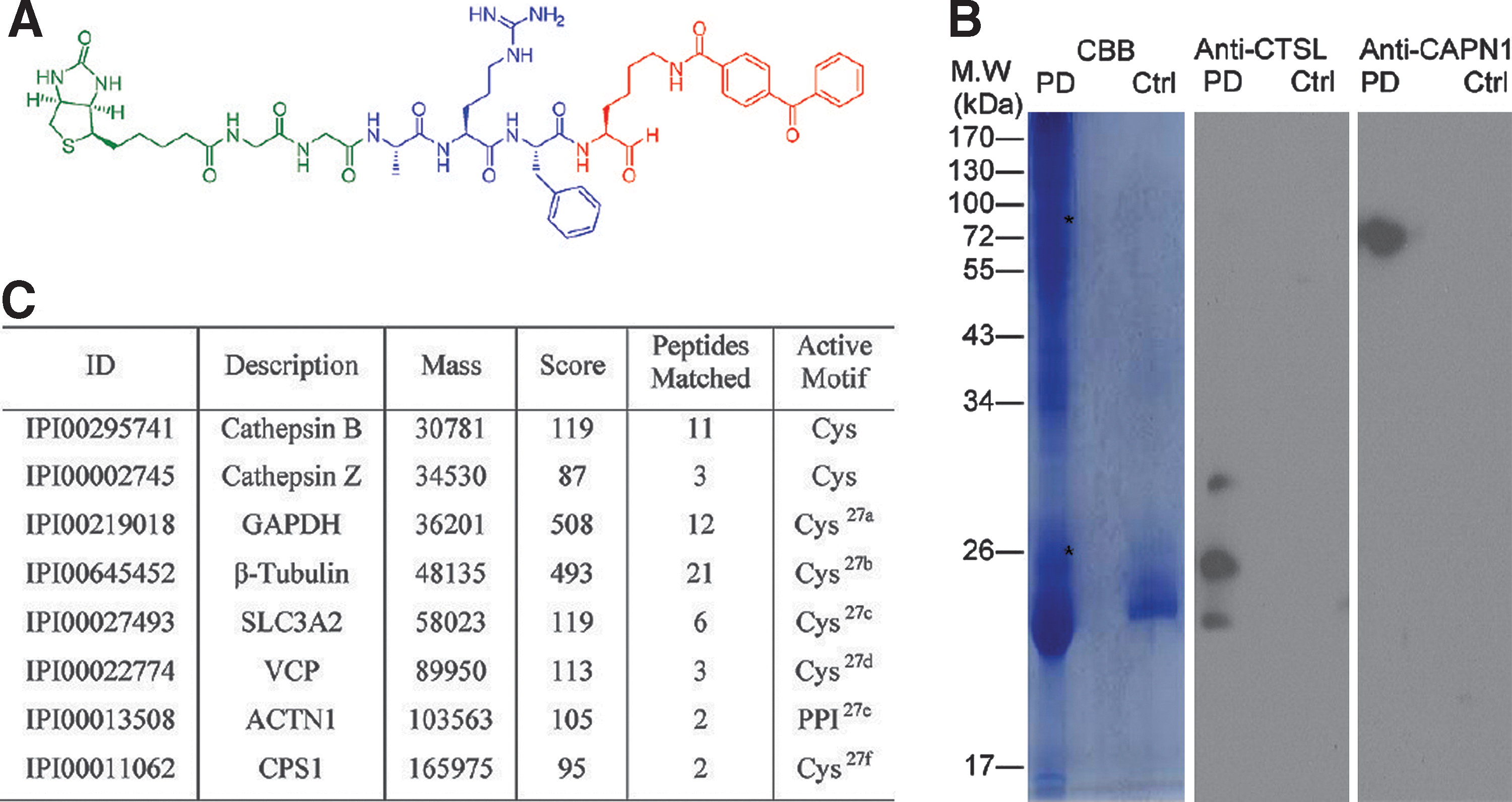
Commentary:Small molecule microarrays (SMMs) have been increasingly applied in ligand identification for isolated targets of interest (Uttamchandani et al., Current Pharmaceutical Design 2008;14:2428–2438). However, there is a paucity of reports demonstrating the use of SMMs for functional assessment of complex biological samples. The article highlighted here illustrates the feasibility of microarrays to obtain binding fingerprints in both normal and infected cell lysates. In addition, the authors also illustrated the method's potential to aid chemical probe development and protein target identification and validation. In this study, Wu et al. designed a peptide aldehyde-oriented SMM using known cysteine protease inhibitor properties and performed a large-scale functional fingerprinting assessment against a number of model cysteine proteases. A positional scanning library was strategically constructed across P1 to P4 positions (see first figure) to incorporate synthetically tractable and tight-binding inhibitors. For example, five different aldehyde warheads were installed at the P1 position to provide sufficient substrate specificity. This, together with the utilization of 18 different amino acids permuting P2 to P4 positions, led to the generation of almost 30,000 compound diversities. This customized SMM platform, containing known inhibitor controls, was first applied to recombinant forms of four cysteine proteases (Cy5-labeled), including rhodesein, cruzain, and caspase-3/-7. Binding and specificity profiles obtained from the SMM platform correlated well with those obtained from microplate-based enzyme assays, thus validating the design of the current microarray configuration. The SMM platform was subsequently evaluated for its ability to distinguish between cell states. By using the apoptosis pathway as a model, the distinction between apoptotic and nonapoptotic HeLa cell lines was enabled due to their characteristic binding profiles, with the former exhibiting similar binding patterns as those observed for purified caspases. The SMM platform was also used to examine lysates from T. brucei and P. falciparum, the causative parasites for African sleeping sickness and malaria, respectively. Distinct binding profiles on the microarrays were observed and were further attributed to the expression of rhodesein and calpain (see second figure, panels A and B), the key constituent proteases in these parasites, suggesting future applications for parasite diagnosis. Furthermore, the authors also attempted to obtain calpain-binding profiles of red blood cells (RBS) (Ca2+ activated) infected by different stages of P. falciparum (see second figure, panel C). Interestingly, the only calpain-binding profile was observed at the trophozoite stage (see second figure, panel D), which was suggested to correlate with the lack of secretion of falstatin (a potent cysteine protease inhibitor) by the parasite only at that stage. Additionally, the platform demonstrated its utility for chemical probe development and target identification. A photo-cross-linking probe was designed based on a ubiquitous hit identified from the above SMM experiments (see third figure, panel A). By applying the probe in activity-based protein profiling studies, several clan CA proteases were successfully pulled down, detected by immunoblotting or mass spectrometry (see third figure, panel B and C). The overall work is a welcome addition to the field of microassay-based activity profiling; the technology's value is further enhanced by its ability to obtain functional signatures at the proteome level in complex biological samples. Contributed by Wendy Lea.
(A) Overall strategy of the SMM platform for comparative profiling of biological events. Unique enzyme/inhibitor interaction profiles were generated upon screening prelabeled cellular lysates with the SMM. (B) Design of the peptide aldehyde SMM. The PSL was designed using five individual P1 aldehyde warheads (in red), targeting different subclasses of cysteine proteases, and diversity elements from P2 to P4 positions (in blue). P2, P3, and P4 were either individual amino acids for mapped positions or an isokinetic mixture of 18 amino acids (which excluded cysteine and methionine).
Profiling parasites and infected red blood cell lysates using the peptide aldehyde SMM. (A) Microarray profiles with T. brucei (BSF) and P. falciparum (trophozoite) lysates. (B) Venn diagram distribution of the top binders (>500 RFUs) in (A). (C) The typical life cycle of P. falciparum within the human blood stages. The black arc represents stages where falstatin was secreted. (D) Microarray profiles with P. falciparuminfected RBC lysates. The lysates were first activated with Ca2+.
Identification of protein targets by pull-down experiments with a selected probe modified with a photo-cross-linking moiety. (A) The chemical structure of the benzophenone (Bp)-containing probe used for pull-down experiments. Aldehyde warhead coupled with benzophenone is highlight in red, the peptide recognition element is indicated in blue, and the biotin tag is shown in green. (B) Western blot analysis of protein targets pulled down from Hela cell lysates using the probe shown in (A). Proteins were identified by the respective antibodies. Asterisks show the expected locations of cathepsin L and calpain-1. (C) Pull-down and LCMS/MS results, identifying potential protein targets that bound to the probe. CBB, Coomassie Brilliant Blue; PD, positive pull-down assay; Ctrl, avidin beads without probe; CTSL, cathepsin L; CAPN1, calpain-1; PPI, protein-protein interaction domain. For the complete table, see Table S1 in the Supporting Information of the original article.
Stabilized mRNA
Kormann MSD, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, Huppmann M, Mays LE, Illenyi M, Schams A, Griese M, Bittmann I, Handgretinger R, Hartl D, Rosenecker J, Rudolph C. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol 2011;29:154–157.
Abstract: Current viral vectors for gene therapy are associated with serious safety concerns, including leukemogenesis, and nonviral vectors are limited by low gene transfer efficiency. Here we investigate the therapeutic utility of chemically modified mRNA as an alternative to DNA-based gene therapy. A combination of nucleotide modifications abrogates mRNA interaction with Toll-like receptor (TLR)3, TLR7, TLR8 and retinoid-inducible gene I (RIG-I), resulting in low immunogenicity and higher stability in mice. A single intramuscular injection of modified murine erythropoietin mRNA raises the average hematocrit in mice from 51.5% to 64.2% after 28 days. In a mouse model of a lethal congenital lung disease caused by a lack of surfactant protein B (SP-B), twice weekly local application of an aerosol of modified SP-B mRNA to the lung restored 71% of the wild-type SP-B expression, and treated mice survived until the predetermined end of the study after 28 days.
Commentary:Nucleic acid therapeutics (represented by antisense oligos, siRNA, mRNA), as agents to control gene transcription or to enable production of functional proteins, have received a lot of attention during the past two decades. Despite multiple successes to date, major issues surrounding these classes of agents include lack of robust delivery methods, unpredictable safety profiles, and relatively rapid degradation in the body. In the work by Kormann and colleagues, mRNA was chemically modified by incorporation of 2-thiouridine and 5-methyl-cytidine by replacement of approximately a quarter of the uridine and cytidine bases during the mRNA synthesis via in vitro transcription. The modified mRNA, coding for the model red fluorescent protein (RFP), was shown to have an increased half-life and to be associated with a higher level of protein production in human and mouse epithelial cell cultures (see first figure). To test for improved delivery characteristics of the modified mRNA, the authors generated a dual-modified construct incorporating enhanced green fluorescent protein and luciferase reporter and administered the mRNA via direct spraying technique into the lungs of test mice: the expression level achieved with the modified mRNA was over four orders of magnitude higher than that achieved with the corresponding nonmodified mRNA. Having validated the new platform with model proteins, the authors moved on to testing more physiologically relevant therapeutic proteins. Mouse erythropoietin mRNA was delivered via intramuscular injection and both the protein production and the mouse hematocrit levels were monitored (figure 2 in the article). Both parameters were significantly improved for the animals treated with the modified mRNA relative to those receiving unmodified control treatment or untreated group. Lastly, the modified mRNA was tested in a genetic disease model of congenital surfactant protein B deficiency, a rare genetic disorder for which the only currently available treatment is lung transplantation. The administration of the modified mRNA coding for the surfactant protein was done as either a two-dose treatment or as a continuous treatment for 28 days. In both cases, increased survival of the treated animals was noted; this was accompanied by the expression of surfactant protein, as determined by Western blot and immunostaining (second figure). These studies provide strong support for the future testing of modified mRNAs as treatment agents for genetic diseases associated with protein deficiency. As noted by the authors, future studies will have to be carried out in order to fully map the protein size range achievable with the use of modified mRNA and the limitations associated with the use of this new therapeutic agent. Contributed by Anton Simeonov.
Modification of mRNA enhances transgene expression and decreases immune responses by reducing immunoreceptor binding. (a) RNA immunoprecipitation (RIP). PBMCs were transfected with 5 μg red fluorescent protein (RFP) mRNA, and the recovery ratios were determined by RIP using TLR3, TLR7, TLR8 and RIG-I specific antibodies. Boxes represent medians ±IQR (interquartile range). Whiskers represent the minimum and maximum observations. *P < 0.05; **P < 0.01 versus the unmodified mEpo mRNA group. (b) Modifications of RFP mRNA inhibit immune responses in vivo after intravenous administration. Data represent the mean ± s.e.m. after 24 h (n = 4 each). s2U(0.5), modified mRNA with 50% 2-thiouridine (s2U) incorporation; s2U(0.25)m5C(0.25), modified mRNA with 25% s2U and 25% 5-methylcytidine (m5C) incorporation. (c,d) A549 cells (c) and MLE12 cells (d) were transfected with 200 ng RFP mRNA. Transfection efficiency (blue and orange bars) and RFP mean fluorescence intensity (MFI) (small blue bars) were determined by flow cytometry (medians ± IQR are shown). *P < 0.05 versus the unmodified RFP group. Total RFP expression (small red bars) was calculated as proportion of RFP-positive cells multiplied by MFI, given as arbitrary units. All experiments were performed twice in duplicates. Data represent the mean ± s.e.m.
Rescue of SP-B deficient mice with repeated doses of modified SP-B mRNA. (a) Application scheme of control and SP-B mRNA. (b) Life expectancy of SP-B−/− mice withdrawn from doxycycline at day 0 that were treated with modified EGFPLuc control mRNA (green line, n = 10), s2U(0.25)m5C(0.25) SP-B mRNA twice (orange line, n = 10), or continuously, twice a week, for 28 d (blue line, n = 13). Kaplan-Meier survival curves were plotted and statistical analysis (Wilcoxon-Gehan test) was performed. (c) Cell-free BALF supernatant (10 μg of total protein per lane) from mice on Doxy, continuously treated with SP-B mRNA, and control mRNA was blotted against specific SP-B antibody. Two representative blots are shown. (d) Typical expression of SP-B (brown) in lung tissue from mice described in c. Scale bar = 20 μm. (e,f) Typical lung histology and BALF cytospin preparations (stained with May-Grünwald/Giemsa) of the same mice described in c. Scale bar = 20 μm. (g) Cytokine levels at the time of necropsy were quantified in mice BALF by ELISA (mean ± s.e.m.); *P < 0.05 versus the untreated group. (h) Lung compliance over time. (i) Early cytokine levels were quantified in mice BALF by ELISA 8 h after administration (mean ± s.e.m.); 20 μg (50 μl) unmodified, s2U(0.25)m5C(0.25) SP-B mRNA and SP-B pDNA was delivered to the mice lungs (n = 4 each); Doxy, doxycycline; Pfc, perfluorocarbon. *P < 0.05 versus the untreated group.
Ozonide Antimalarial
Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FC, Chollet J, Craft JC, Creek DJ, Dong Y, Matile H, Maurer M, Morizzi J, Nguyen T, Papastogiannidis P, Scheurer C, Shackleford DM, Sriraghavan K, Stingelin L, Tang Y, Urwyler H, Wang X, White KL, Wittlin S, Zhou L, Vennerstrom JL. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci U S A 2011; doi:10.1073/pnas.1015762108.
Abstract: Ozonide OZ439 is a synthetic peroxide antimalarial drug candidate designed to provide a single-dose oral cure in humans. OZ439 has successfully completed Phase I clinical trials, where it was shown to be safe at doses up to 1,600 mg and is currently undergoing Phase IIa trials in malaria patients. Herein, we describe the discovery of OZ439 and the exceptional antimalarial and pharmacokinetic properties that led to its selection as a clinical drug development candidate. In vitro, OZ439 is fast-acting against all asexual erythrocytic Plasmodium falciparum stages with IC50 values comparable to those for the clinically used artemisinin derivatives. Unlike all other synthetic peroxides and semisynthetic artemisinin derivatives, OZ439 completely cures Plasmodium berghei-infected mice with a single oral dose of 20 mg/kg and exhibits prophylactic activity superior to that of the benchmark chemoprophylactic agent, mefloquine. Compared with other peroxide-containing antimalarial agents, such as the artemisinin derivatives and the first-generation ozonide OZ277, OZ439 exhibits a substantial increase in the pharmacokinetic half-life and blood concentration versus time profile in three preclinical species. The outstanding efficacy and prolonged blood concentrations of OZ439 are the result of a design strategy that stabilizes the intrinsically unstable pharmacophoric peroxide bond, thereby reducing clearance yet maintaining the necessary Fe (II)-reactivity to elicit parasite death.
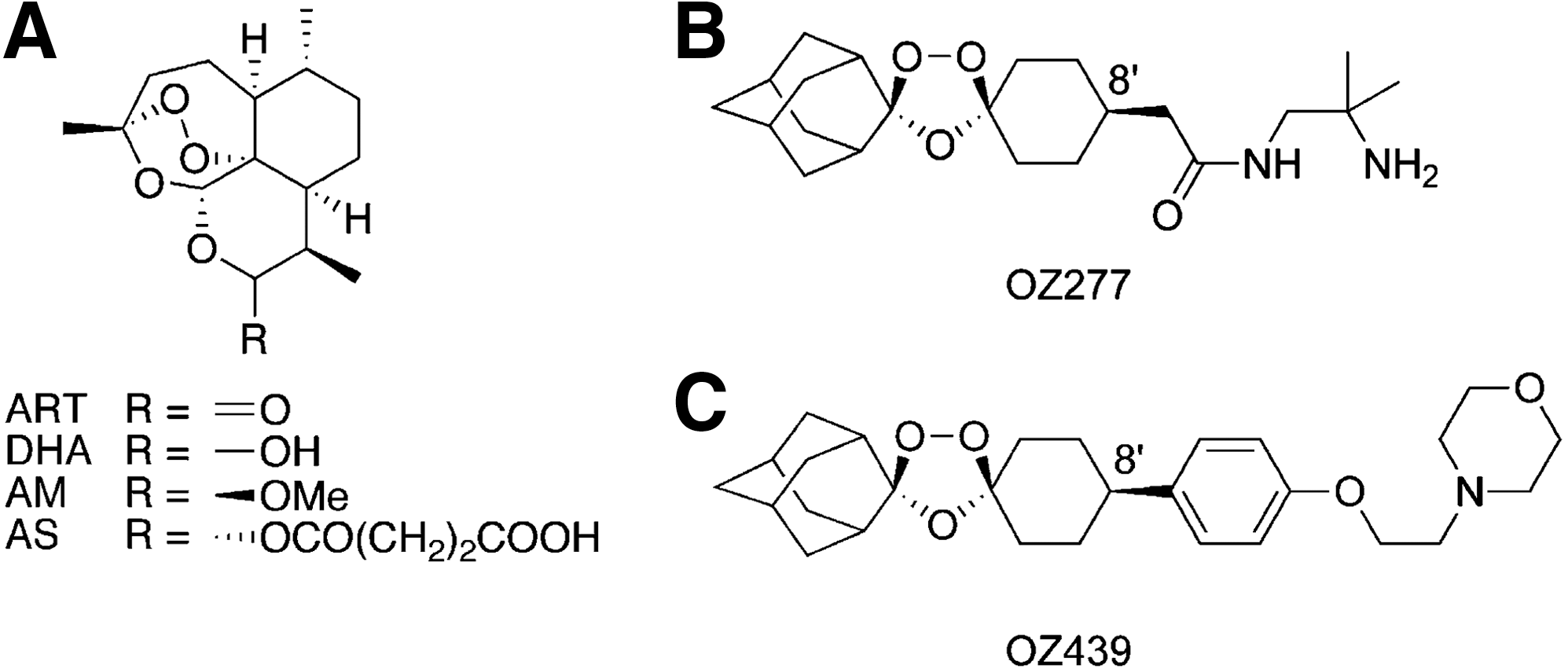
Commentary:Artemisinin and its close derivatives have enjoyed a great degree of popularity due to their clinical effectiveness in treating various stages of malaria. The high and fluctuating price, coupled with uncertainties surrounding artemisinin supply and the drug's short half-life, have prompted research efforts aimed at developing alternatives that nevertheless share its attractive features. A key property of artemisinin relates to its somewhat unique and not fully understood mechanism of action: its peroxide functionality is thought to react with ferrous [Fe(II)] species and heme or heme derivatives produced upon hemoglobin digestion by the parasite, and this reaction is thought to generate reactive moieties that in turn destroy key parasite proteins. The present study reports on the development of an ozonide-based antimalarial agent OZ439 (see figure) acting in a manner similar to that of artemisinin but possessing superior stability and improved route of production. Earlier research by the team had led to the generation and testing of OZ277, an ozonide with antimalarial action that exhibited relatively high reactivity with ferrous species and had a correspondingly lower half-life in the bloodstream. The new generation ozonide, represented by OZ439, was shown to be approximately 15-fold more stable than OZ277 when tested in rabbit blood at 37°C. Furthermore, when the two leads were tested in plasmodium-infected blood, the relative stability of OZ439 increased to 20-fold, primarily due to the increased degradation of OZ277 in the presence of parasite-driven release of heme derivatives. The new lead was superior to the early ones also with respect to biodistribution after an oral delivery (figure 3 in the article). When tested in a murine model of Plasmodium berghei infection, OZ439 exhibited efficacy that exceeded most of the existing antimalarials (see table); moreover, the drug was effective as a prophylactic agent when delivered 24 hours prior to the challenge infection. The authors attribute the streamlined development of this new lead to the enhanced collaboration afforded by the Product Development Partnership, under the Medicines for Malaria Ventures private-public consortium. Contributed by Anton Simeonov.
Structures of artemisinin derivatives and synthetic ozonides. (A) Artemisinin (ART), dihydroartemisinin (DHA), artemether (AM), and artesunate (AS); (B) OZ277; and (C) OZ439.
In Vivo Efficacy of Comparator Antimalarial Drugs, OZ277 and OZ439, Against Murine P. berghei
Data from Vennerstrom JL, Arbe-Barnes S, Brun R, et al.: Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 2004;430:900–904.
Anti-VEGFs Before Chemotherapy
Zhang D, Hedlund E-ME, Lim S, Chen F, Zhang Y, Sun B, Cao Y. Antiangiogenic agents significantly improve survival in tumor-bearing mice by increasing tolerance to chemotherapy-induced toxicity. Proc Natl Acad Sci U S A 2011; doi:10.1073/pnas.1016220108.
Abstract: Chemotherapy-induced broad toxicities are the leading cause of the drug-induced mortality in cancer patients. Antiangiogenic drugs (ADs) in combination with chemotherapy are widely used as frontline therapy for the treatment of various human cancers. However, the beneficial mechanisms underlying combination therapy are poorly understood. Here we show that, in several murine tumor models, administration of sunitinib markedly reduced chemotherapy-induced bone marrow toxicity. Intriguingly, in a sequential treatment regimen, delivery of ADs followed by chemotherapy demonstrated superior survival benefits compared with simultaneous administration of two drugs. In murine tumor models, we show that VEGF increased chemotoxicity by synergistically suppressing bone marrow hematopoiesis with cytostatic drugs. These findings shed light on molecular mechanisms by which ADs in combination with chemotherapy produce survival benefits in cancer patients and provide conceptual information guiding future designs of clinical trials, current practice, and optimization of ADs for the treatment of cancer.
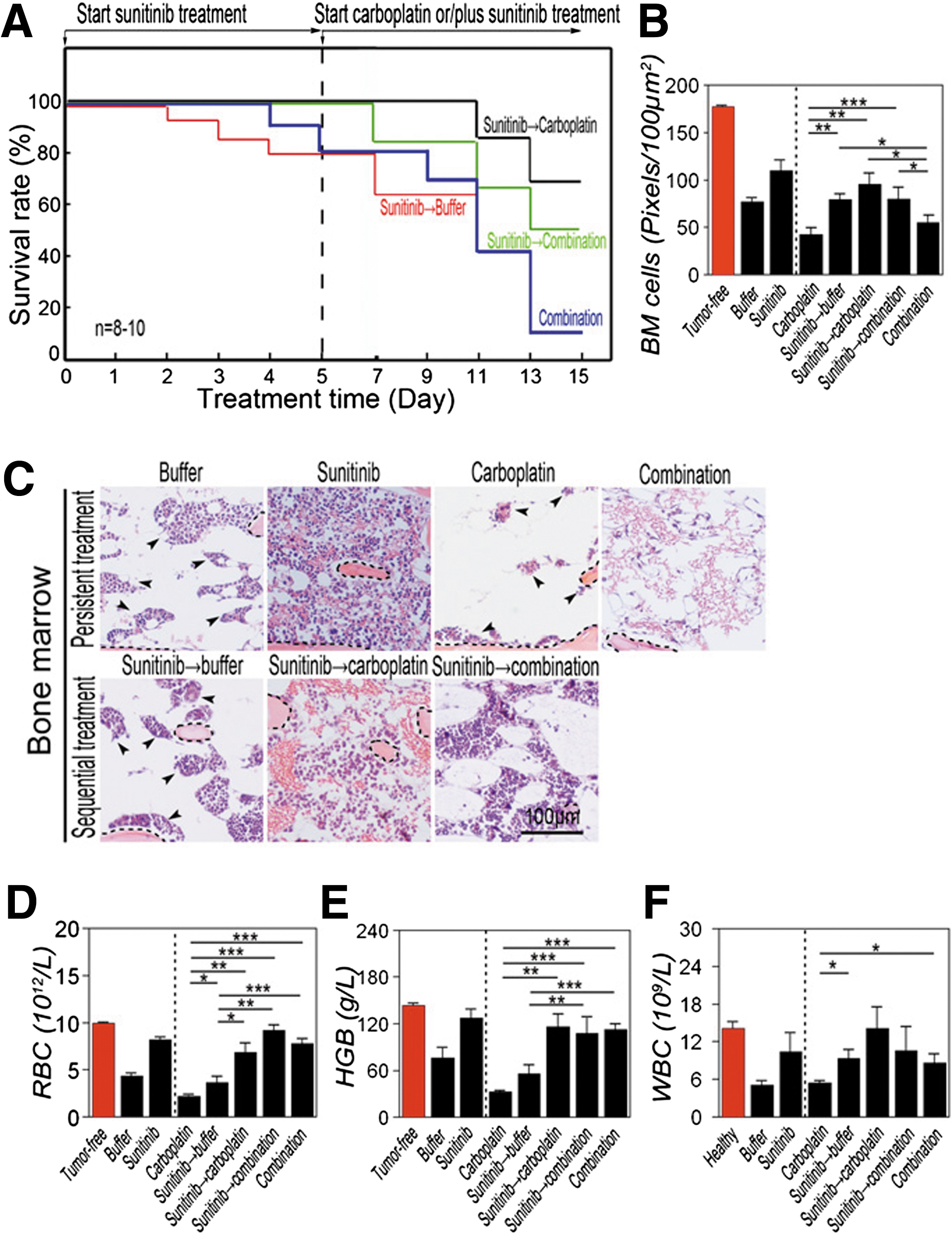
Commentary:Antiangiogenic therapies, targeting the vascular endothelial growth factor (VEGF) by the use of anti-VEGF antibodies like avastin or small molecule agents acting on receptor tyrosine kinases, have been used as either stand-alone treatments or in combination therapies along with DNA-damaging drugs in order to treat a range of cancers. The basic rationale for use of anti-VEGF agents is that when VEGF is blocked the growing tumors are deprived of new blood vessels and corresponding depletion of nutrients and oxygen can lead to tumor starvation and eventual tumor cell death. In the clinic, anti-VEGF agents are often used in combination therapies together with DNA-damaging agents such as cisplatin and related chemotherapy drugs. While the overall rationale for this dual therapy is clear—the anti-VEGF agent causes tumor starvation while the DNA-damaging drug exerts a direct cytotoxic effect on the dividing tumor cells—the drug-induced mortality stemming from the multiple side effects of the chemotherapy, particularly its harsh effect on the ability of the patient's body to regenerate its blood cells, has led to a search of novel treatment strategies. Here, a simple shift in the order of application of anti-VEGF agent relative to DNA-agent chemotherapy is suggested to produce a profound effect on the chemotherapy-induced patient mortality. The authors used the anti-VEGF drug sunitinib, a receptor tyrosine kinase inhibitor, along with carboplatin in a series of carefully designed treatment protocols using murine models of melanoma and fibrosarcoma. Based on earlier studies pointing to the causal role of elevated VEGF on the development of hematopoietic defects in the bone marrow of tumor-bearing animals, the authors tested the hypothesis that suppression of VEGF prior to initiation of chemotherapy may yield a beneficial effect by reducing the chemotherapy's side effects. Treatment using carboplatin was shown to result in significant animal mortality with the attendant strong suppressive effects on the bone marrow and blood cell regeneration (see first figure). Separately, the authors demonstrated that in the same models an increased level of VEGF production correlates with enhanced tumor angiogenesis and severe hematopoietic defects (figure 2 in the article). Combination treatment in which both carboplatin and sunitinib were delivered simultaneously from the onset of the treatment was also shown to be associated with high animal mortality and bone marrow suppression; thus, the inclusion of the anti-VEGF agent once the chemotherapy was started did not appear to provide a significant advantage with respect to relief of the bone marrow toxicity of carboplatin. However, in both the melanoma and fibrosarcoma models, administration of sunitinib for 5 days prior to initiation of carboplatin treatment had a profound effect on the animals' survival and their ability to regenerate blood cells (see first and second figures). These studies, based on two murine models, provide strong initial evidence for the protective effect that anti-VEGF treatment can provide when initiated prior to chemotherapy and should serve as a rationale for designing future clinical studies with human patients. It is worth noting that while the present work produced conclusive results, it remains to be evaluated whether other types of agents acting along the VEGF axis, such as anti-VEGF monoclonal antibodies, are capable of providing a similar type and degree of protection. Furthermore, a separate study published less than a week apart from the present report (Keunen et al. Proc Natl Acad Sci U S A Early Edition doi:10.1073/pnas.1014480108) serves as a reminder that the real biological picture is always more complicated: Keunen and colleagues performed detailed investigation of the anti-VEGF antibody avastin in a glioblastoma model in which they found that the use of the antibody as a single treatment led to a reduction in tumor vascularization, as expected, but that this process was also accompanied by the highly unwanted side effect of increase in the circulating tumor cells, likely as a result of the induced tumor metabolism change to its new hypoxic environment. The moral of the work highlighted here, along with Keunen's study, appears to be that no single anticancer agent acts in isolation and that the precise regimen of target-based therapy remains crucial to its success. Contributed by Anton Simeonov.
Improvement of survival in a murine melanoma model. (A) Treatment of B16 melanoma-bearing mice (n = 12–15 mice/group) with sunitinib, carboplatin, or vehicle was started when the average tumor size reached 0.4 cm3. Survivals of mice were closely monitored several times per day. (B) Tumor growth rates of sunitinib-, carboplatin-, or vehicle-treated groups (n = 12–15 mice/group). (C) When the average tumor size reached 0.4 cm3, tumor-bearing mice (n = 12–15 mice/group) were treated with sunitinib. At day 6 after treatment, sunitinib-treated mice received carboplatin or carboplatin plus sunitinib until the end of experiments. Vehicle-pretreated group followed by carboplatin was used as a control. (D) Kaplan–Meier survival curve of various treated groups under the regimen described in (C). Dashed line marks pretreatment endpoint. (E and F) BM histology of various treated groups described in (A–D). Dashed lines enclose bone matrix. Scale bar = 100 μm. (G) Quantification of density of BM cells (20 × magnification, eight randomized fields per group). (H–J) RBC (H), hemoglobin (I), and WBC (J) in peripheral blood. *P < 0.05, **P < 0.01, ***P < 0.001. Data are shown as mean ± SEM.
Sequential treatment of T241 tumor-bearing mice with sunitinib followed by carboplatin significantly improves survival. (A) Kaplan–Meier survival curve of VEGF-T241 tumor-bearing mice (n = 8–10 mice/group) that received sequential therapy of sunitinib followed by carboplatin or by combination. Simultaneous delivery of both drugs and sunitinib followed by vehicle were used as controls. Sequential regimen of delivery sunitinib followed carboplatin markedly improved survival rates relative to rates in the group that received simultaneous combination therapy. (B) Quantification of density of BM cells (20 × magnification, eight randomized fields per group). (C) BM histology of various treated groups described in (A). Dashed lines enclose bone matrix. Arrowheads point to residual hematopoietic islets attached to bone matrix. Scale bar = 100 μm. (D–F) RBC (D), hemoglobin (E), and WBC (F) in peripheral blood. *P < 0.05, **P < 0.01, ***P < 0.001. Data are shown as mean ± SEM.