Anokye-Danso F, Trivedi CM, Juhr D, Gupta M, Cui Z, Tian Y, Zhang Y, Yang W, Gruber PJ, Epstein JA, Morrisey EE. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011;8:376–388.
Abstract: Transcription factor-based cellular reprogramming has opened the way to converting somatic cells to a pluripotent state, but has faced limitations resulting from the requirement for transcription factors and the relative inefficiency of the process. We show here that expression of the miR302/367 cluster rapidly and efficiently reprograms mouse and human somatic cells to an iPSC state without a requirement for exogenous transcription factors. This miRNA-based reprogramming approach is two orders of magnitude more efficient than standard Oct4/Sox2/Klf4/Myc-mediated methods. Mouse and human miR302/367 iPSCs display similar characteristics to Oct4/Sox2/Klf4/Myc-iPSCs, including pluripotency marker expression, teratoma formation, and, for mouse cells, chimera contribution and germline contribution. We found that miR367 expression is required for miR302/367-mediated reprogramming and activates Oct4 gene expression, and that suppression of Hdac2 is also required. Thus, our data show that miRNA and Hdac-mediated pathways can cooperate in a powerful way to reprogram somatic cells to pluripotency.
Miyoshi N, Ishii H, Nagano H, Haraguchi N, Dewi DL, Kano Y, Nishikawa S, Tanemura M, Mimori K, Tanaka F, Saito T, Nishimura J, Takemasa I, Mizushima T, Ikeda M, Yamamoto H, Sekimoto M, Doki Y, Mori M. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011;8:633–638.
Abstract: Induced pluripotent stem cells (iPSCs) can be generated from differentiated human and mouse somatic cells using transcription factors such as Oct4, Sox2, Klf4, and c-Myc. It is possible to augment the reprogramming process with chemical compounds, but issues related to low reprogramming efficiencies and, with a number of protocols, residual vector sequences, remain to be resolved. We show here that it is possible to reprogram mouse and human cells to pluripotency by direct transfection of mature double-stranded microRNAs (miRNAs). Our approaches use a combination of mir-200c plus mir-302 s and mir-369 s family miRNAs. Because this reprogramming method does not require vector-based gene transfer, it holds significant potential for biomedical research and regenerative medicine.
Commentary:Induced pluripotent stem cells (iPSCs) are artificial stem cells typically derived from somatic cells by inducing expression of specific genes. Compared with embryonic stem cells (ESCs), the generation of iPSCs circumvents the use of embryos and related ethical issues, and iPSCs represent potentially powerful avenues for candidate drug screening and development. The first report of iPSC generation was published 5 years ago by Shinya Yamanaka's team (Takahashi et al., Cell 2006;126:663–676). Using viral vector-based gene transfer into mouse fibroblasts, four transcriptional factors (Oct3/4, Sox2, Klf4 and c-Myc, aka OSKM factors) were identified to be essential for the production of pluripotent stem cells. In 2007, a second generation of mouse iPSCs (in which viable chimera were developed) and the first creation of human iPSCs (Vodyanik et al., Science 2007;318:1917–1920; Takahashi et al., Cell 2007;131:861–872) were subsequently reported, symbolizing groundbreaking advances in the field of iPSCs. However, the transcription factor–based approach is associated with a number of limitations, which further hamper iPSCs' clinical application. For example, reprogramming efficiency is low, with an average of 0.1%–1% cells reprogrammed to a pluripotent stem cell state; there are also risks of mutation and tumor development as a result of genomic integration or oncogene introduction. New reprogramming technologies have been developed in the past half decade, through alternative input vectors, different reprogramming cocktails (including DNA-free methods such as small molecules as transcription factor mimetics) and mRNA molecules. Synthetic mRNA has been demonstrated to overcome antiviral responses elicited in host cells and successfully direct cellular reprogramming (Warren et al., Cell Stem Cell 2010;7:618–630). Being a nonintegrative iPSC derivation strategy, its reprogramming efficiency was reported to be 4.4% at the highest. Specific microRNAs (miRNAs), on the other hand, have been previously shown to be highly expressed in ESCs and play an important role in regulating pluripotency-related genes. Despite miRNAs' relevance in iPSC reprogramming, whether they could be used alone for iPSC generation remains to be investigated.
The studies in the two complementary articles highlighted herein both examined miRNA molecules for their ability to reprogram mouse and human cells to pluripotency in the absence of the OSKM factors. However, their approaches differed in the specific miRNA molecules utilized and the corresponding transfection method. In the work led by Morrisey, the authors utilized a miR302/367 (direct target of Oct4 and Sox2) lentivirus for the transduction of mouse embryonic fibroblasts (MEFs) and human foreskin and dermal fibroblasts. Valproic acid (VPA), an inhibitor of histone deacetylase (Hdac) was also included in their experiment to examine its effect in reprogramming efficiency. ESC morphology was observed (8–14 days) in miR302/367 transduced mouse and human cells. Pluripotent markers were expressed at equivalent levels as seen in ESCs and iPSC-derived teratomas exhibited tissues from all three germ layers (see first figure for the human case). In addition, a 10% reprogramming efficiency was obtained, considerably higher than that achieved by the OSKM factors, and the miR302/367 virus was silenced in later passages. Two other notable findings in this study are the requirement of miR367 expression and the need of low-level Hdac2 expression. miR367 alone or the lack of miR367 in the miR302/miR367 cluster did not lead to iPSC colony formation even after 3 weeks of reprogramming. Moreover, low-level expression of key pluripotent genes (such as Oct4) was observed when transduction occurred in the absence of miR367 (see second figure for the human case). As Hdac-mediated chromatin remodeling has been implicated in early studies, the effect of miR302/367 and VPA treatment were examined on Hdac expression using Western blots. It was found that VPA was responsible for the degradation of Hdac2 protein in MEFs, and miR302/367 expression did not affect Hdac expression regardless of VPA presence. Thus, it was suggested that the higher expression level of Hdac2 in MEFs than in human foreskin fibroblasts might explain the stronger dependence of reprogramming efficiency on the presence of VPA in MEFs (see third figure).
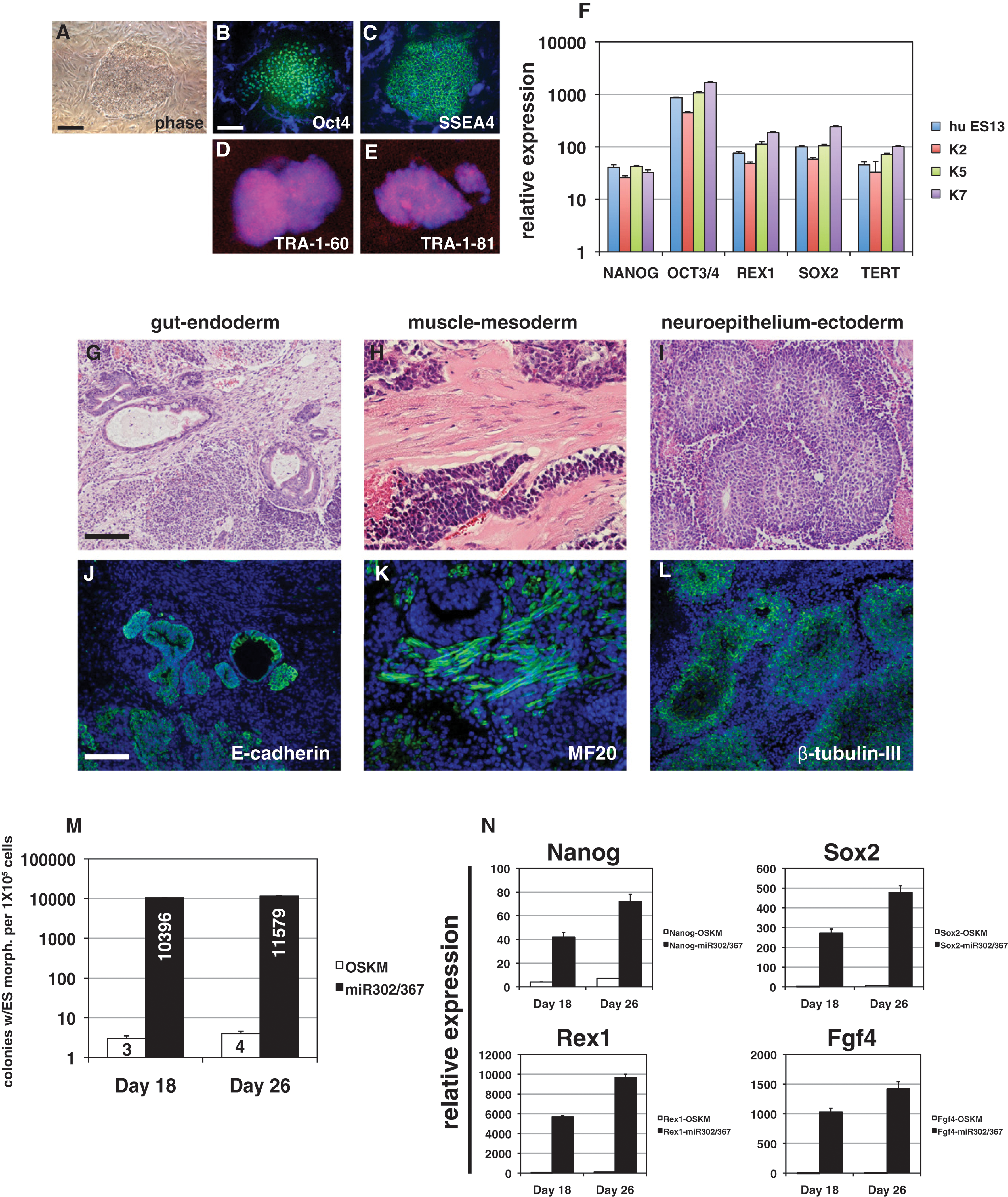
miR302/367 reprograms human fibroblasts to a pluripotent state more efficiently than OSKM factors. (A–E) Colony morphology and OCT4, SSEA4, TRA-1-60, and TRA-1-81 immunostaining of miR302/367-reprogrammed human fibroblasts. Scale bars: 50 μm. (F) Q-PCR of pluripotent stem cell marker genes in three different miR302/367-reprogrammed human fibroblast lines as compared to the human ES line HUES13. (G–I) Hematoxylin and eosin staining of teratomas derived from miR302/367 human iPSC clones showing endoderm (gut)-, mesoderm (muscle)-, and ectoderm (neural epithelium)-like structures. These data represent the results from seven human miR302/367 iPSC clones. Scale bar: 150 μm. (J–L) Immunostaining of miR302/367 human iPSC-derived teratoma tissues showing expression of E-cadherin-positive endodermal cells, MF20-positive striated muscle, and βIII-tubulin-positive neural epithelium. Scale bar: 150 μm. (M) Efficiency of miR302/367 reprogramming in human foreskin fibroblasts by colony counts of clones with human ES-like morphology at 18 and 26 days postviral transduction. Data are the average of three assays ± SEM. (N) Q-PCR of pluripotent gene expression in miR302/367-reprogrammed human foreskin fibroblasts at 18 and 26 days postviral transduction. Data are the average of three assays ± SEM.
miR367 expression is required for miR302/367 iPSC reprogramming. (A) The miR302a/b/c/d pre-miRNA is expressed at high levels in transduced MEFs. (B) Number of colonies generated after 10 days of miR302a/b/c/d or miR302/367 expression. Data are the average of four assays ± SEM. (C) Pluripotent gene expression from primary induction plates 8 days after viral induction of miR302a/b/c/d or miR302/367 viruses. Note lack of Oct4 gene expression in miR302a/b/c/d-expressing cells (red arrow). Data are the average of three assays ± SEM. (D) FACS analysis of Oct4-GFP MEFs 8 days after transduction with either miR302a/b/c/d or miR302/367 viruses.
Pluripotency-associated gene expression in mi-iPSCs from human somatic cells. (A) At day 20, some colonies with sharp and defined margins appeared that were morphologically different from the parental hASCs and HDFs. (B and C) Immunocytochemistry revealed mi-iPSCs from hASCs (B) and HDFs (C) expressing Ssea-4, Tra-1-60, Tra-1-81, and Tra-2-49. (D) The mi-iPSCs from hASCs and HDFs highly expressed undifferentiated ESC-marker genes compared to the observed expression in their respective parental cells. The expression of mRNA copies was normalized against GAPDH mRNA expression (mean ± SEM; n = 3). (E) Real-time RT-PCR analysis of miRNAs in parental mi-iPSCs. The expression of miRNA copies was normalized against RNU48 expression, and the mean expression of hASC mi-iPSC was set to 1 for each gene (mean ± SEM; n = 3). (F) mi-iPSCs (clone 3621A1) were transplanted subcutaneously into the dorsal flanks of NOD/SCID mice, and teratomas formed in various tissues and cells as epidermal, ganglia-like, osteoid, skeletal muscle, chondrocyte, and ciliated epithelial tissue (arrow, goblet cell). Bar = 100 mm; original magnification, ×200.
In a separate work led by Ishii and Mori, a similar reprogramming strategy was applied, except that different miRNA factors were inserted into the host genome through direct transfection. Gene expression analysis of miRNAs revealed that mir-200c, mir302 s, and mir-369 s family miRNAs were expressed greater than twofold more strongly in mouse ESCs and iPSCs than in adult mouse adipose stromal cells. The combination of all three miRNA factors was found to provide the most efficient transfection. Undifferentiated ESC marker gene characteristics were observed for both reprogrammed mouse and human cells. Additionally, teratoma formation and all three germ layers were observed in the human iPSC-transplanted mouse model, further validating iPSCs' differentiation capacity (see fourth figure). These are two welcome case studies in which miRNAs were shown to be capable of reprogramming somatic cells to pluripotency. The two methods have their respective strengths, with the former providing a high reprogramming efficiency and the latter evading host genomic integration. The trade-off between reprogramming efficiency and genomic integration remains a challenging factor in iPSC production. Other combinations of miRNAs may be further explored in order to obtain an ultimately effective technique to generate reprogrammed iPSCs. Contributed by Wendy Lea.
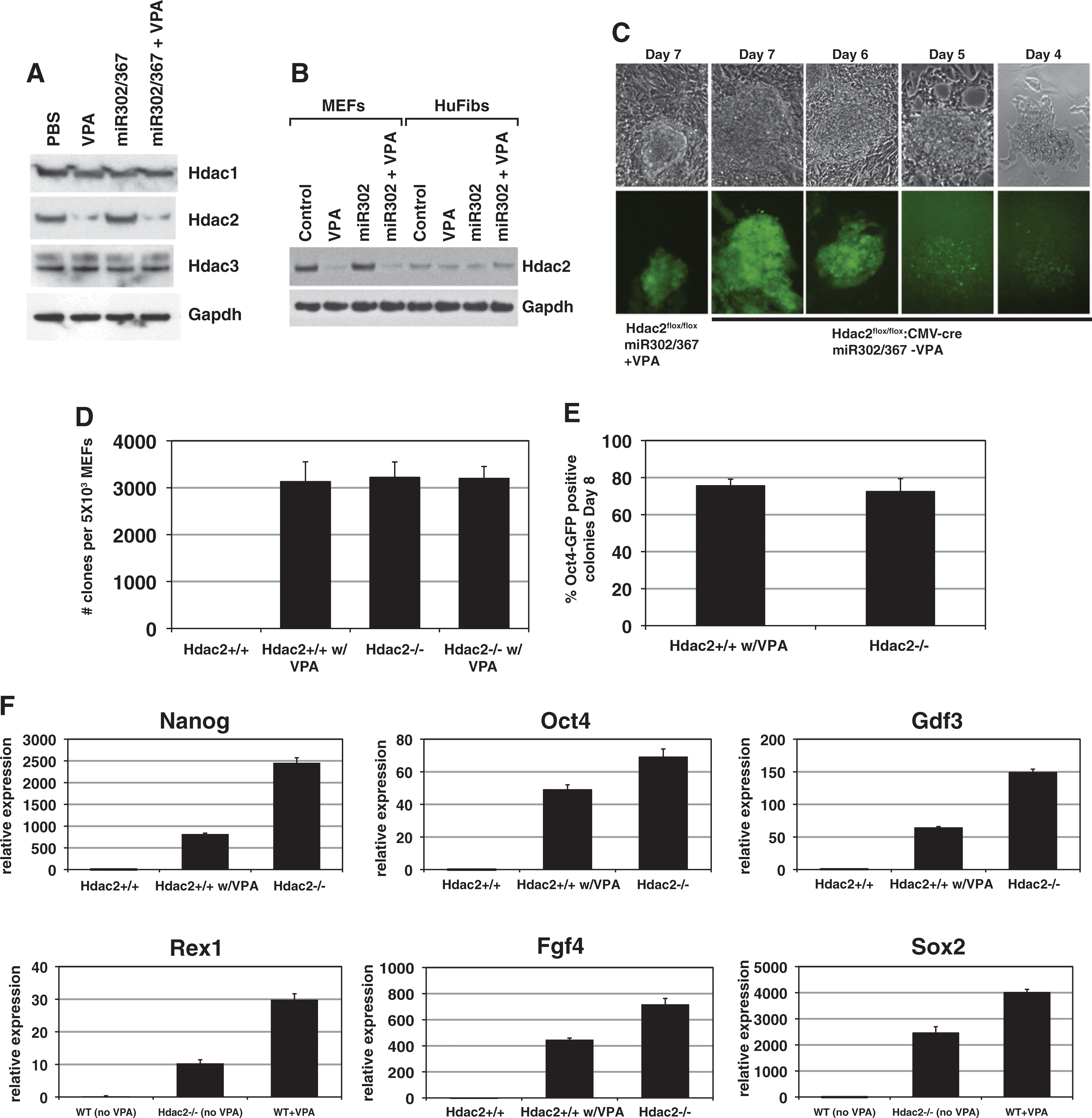
VPA specifically degrades Hdac2 protein, and suppression of Hdac2 is required for iPSC reprogramming by miR302/367. (A) VPA specifically degrades Hdac2, but not Hdac1 or Hdac3 proteins. Expression of miR302/367 alone did not have any effect on Hdac1, −2, or −3 protein levels. (B) Human foreskin fibroblasts express much lower levels of Hdac2 than MEFs. (C) Hdac2−/− MEFs, in the absence of VPA, start to reprogram between 6 and 7 days postviral transduction, which is similar to wild-type MEFs treated with VPA. (D) Number of clones generated with Hdac2−/− MEFs in the absence of VPA is similar to Hdac2+/+ MEFs with VPA at 8 days postviral transduction. Hdac2+/+ MEFs without VPA treatment did not generate any viable clones and VPA addition to Hdac2−/− MEFs did not increase the number of clones generated. (E) Percentage of Oct4-GFP-positive clones is similar for Hdac2+/+ MEFs with VPA treatment and Hdac2−/− MEFs without VPA treatment at 8 days postviral transduction. (F) Q-PCR for pluripotent stem cell marker genes shows enhanced expression of pluripotency markers at day 8 of reprogramming by miR302/367 in wild-type (Hdac2+/+) and Hdac2−/− MEFs versus WT MEFs without VPA treatment. Data are the average of three assays ± SEM.
High-Content Assays with Particles
Chapin SC, Appleyard DC, Pregibon DC, Doyle PS. Rapid microRNA profiling on encoded gel microparticles. Angew Chem Int Ed Engl 2011;50:2289–2293.
Abstract: No abstract
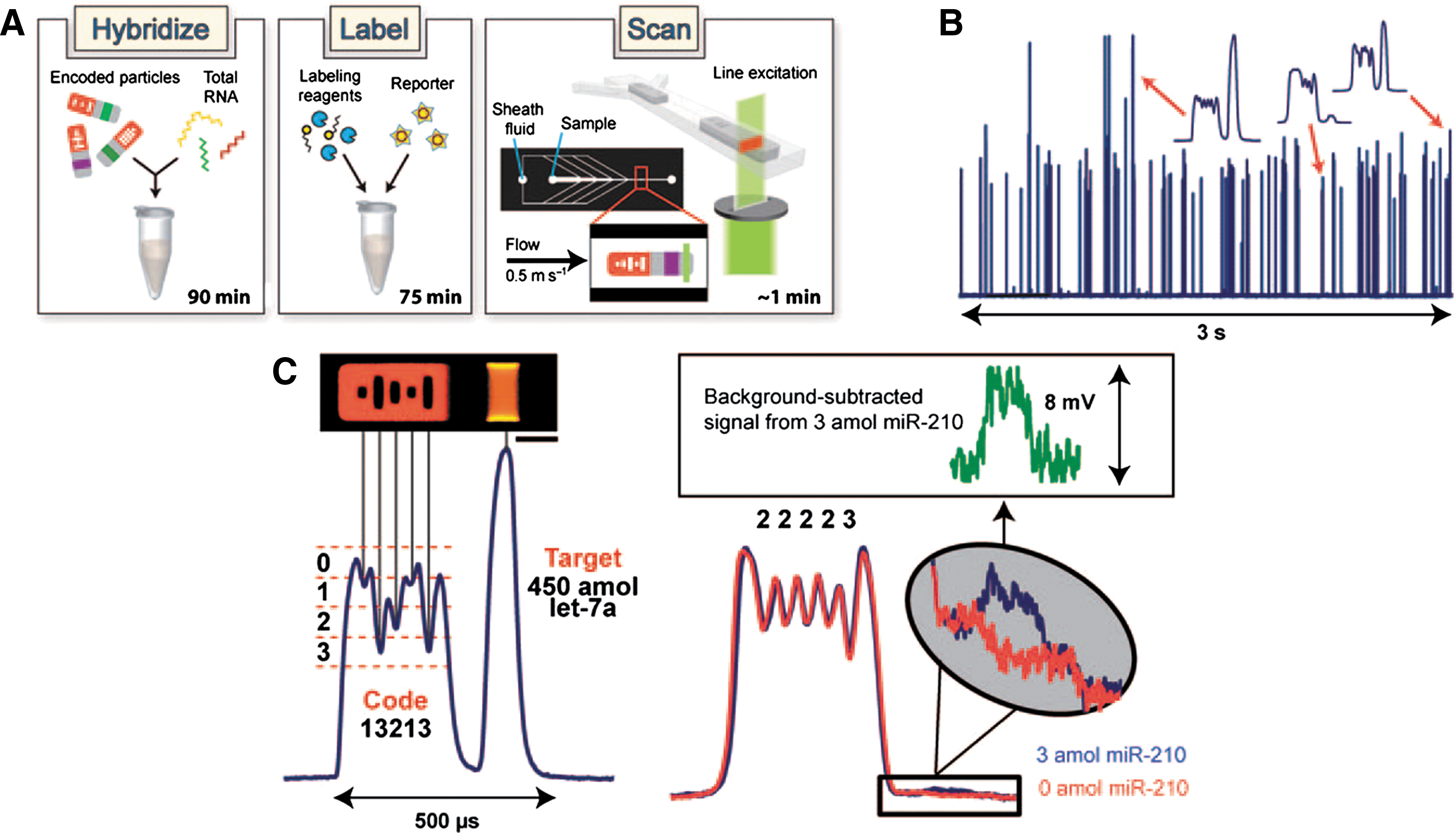
Commentary:High-content screening is usually associated with automated microscope systems in which images are collected to provide multiparametric data. However, any technology that enables high-throughput detection of multiple parameters falls into the general category of high-content screening approaches. The present article describes graphically encoded hydrogel microparticles that allow for highly sensitive detection of proteins or miRNAs. The hydrogels are composed of bio-inert poly(ethyleneglycol) (PEG) that reduces nonspecific binding. Using a technique called microfluidic stop-flow lithography (SFL) these gel particles can be synthesized, encoded, and functionalized at high rates (>104/h; see also Appleyard et al., Anal Chem 2011;83:193–199). The main issue with any multiplexed format is providing the means to rapidly detect and decode target binding. The hydrogel particles provide a fluorescent barcoding region that consists of unpolymerized holes leading to a code composed of five bits (having values of 0, 1, 2, or 3) with one bit fixed to determine orientation of the particle. The size of the holes determines the dips in a fluorescent profile that is generated when the particle is flowed across the detector (see figure). This mode of decoding simplifies analysis and reduces the cost required as only one excitation source and PMT is needed for decoding and target detection. To ensure that the particles are all properly aligned, a fluid focusing chamber is employed in which sheath streams are injected using a force of 8 psi so that particles flow in single file across the detector (velocities of approximately 0.5 m/s). The decoding algorithm determines if a particle is flowing in either a probe to code or code to probe orientation. The present article applies this technique to miRNA detection. DNA probes are linked to the hydrogel particles (250-μm particles were used in this study), which contain a spacer, universal adaptor, and miRNA-specific regions. The universal adaptor region allows for hybridizing a biotinylated adaptor probe, which is ligated to the 3′ end of the miRNA targets captured on the DNA probe. Detection is performed with a phycoerythrin-conjugated streptavidin reporter (SA-PE). In the article, a 12plex miRNA assay was performed, but five separate codes were used for each miRNA, which created a 60plex assay readout. One of the advantages of the hydrogel particles is that the high surface area displayed by the gel matrix provides superior sensitivity. In an experiment that used 4 of the 12 miRNA, sub-attomole sensitivity was observed for three of the four miRNAs. Cross-reactivity of the miRNA probes was 27% but could be improved through optimizing the hybridization conditions. Assay CVs showed excellent reproducibility (CV values between 2% and 7%). The full 12plex panel was then used to examine both tumor and normal tissue samples and the results showed excellent agreement with the known regulation of these miRNAs. Further optimization of this method to implement automated sample loading and liquid handling should enable >500 samples to be processed daily but with potentially thousands of targets being detected per sample. One advantage of the hyrdrogel particles is that both protein and miRNAs can be detected, allowing pathway analysis in which both miRNAs and the protein targets regulated by these are determined with one method. Contributed by Doug Auld.
Encoded gel particle assay system. (A) Workflow of platform includes 1) hybridization of particles with target, 2) incubation of particles with universal labeling adapter, ligation enzyme, and fluorescent reporter, and 3) scanning of particles to determine code identity and amount of target bound. A typical particle consists of a fluorescent barcoded region and a probe-laden region flanked by two inert sections. The central-most hole has a fixed value to indicate particle orientation. (B) Actual PMT fluorescence signatures of 75 flow-aligned particles. (C) Magnified signatures of individual particles. As probe–target reaction rate is observed to be higher than target diffusion through gel matrix, the increased fluorescent intensity on the sides of the particle in image can be attributed to binding of target near the side faces of probe region. Scale bar is 50 μm.
Kinase Inhibitor Binds in a New Inactive Conformation
Eathiraj S, Palma R, Hirschi M, Volckova E, Nakuci E, Castro J, Chen C-R, Chan TCK, France DS, Ashwell MA. A novel mode of protein kinase inhibition exploiting hydrophobic motifs of autoinhibited kinases: discovery of ATP independent inhibitors of fibroblast growth factor receptor (FGFR). J Biol Chem 2011;286:20677–20687.
Abstract: Protein kinase inhibitors with enhanced selectivity can be designed by optimizing binding interactions with less conserved inactive conformations because such inhibitors will be less likely to compete with ATP for binding and therefore may be less impacted by high intracellular concentrations of ATP. Analysis of the ATP-binding cleft in a number of inactive protein kinases, particularly in the autoinhibited conformation, led to the identification of a previously undisclosed non-polar region in this cleft. This ATP-incompatible hydrophobic region is distinct from the previously characterized hydrophobic allosteric back pocket, as well as the main pocket. Generalized hypothetical models of inactive kinases were constructed and, for the work described here, we selected the fibroblast growth factor receptor (FGFR) tyrosine kinase family as a case study. Initial optimization of a FGFR2 inhibitor identified from a library of commercial compounds was guided using structural information from the model. We describe the inhibitory characteristics of this compound in biophysical, biochemical and cell-based assays, and have characterized the binding mode using X-ray crystallographic studies. The results demonstrate, as expected, that these inhibitors prevent activation of the autoinhibited conformation, retain full inhibitory potency in the presence of physiological concentrations of ATP, and have favorable inhibitory activity in cancer cells. Given the widespread regulation of kinases by autoinhibitory mechanisms, the approach described herein provides a new paradigm for the discovery of inhibitors by targeting inactive conformations of protein kinases.
Commentary:Kinases can adopt a wide variety of conformations and likely spend time sampling each of these various conformations. The amount of time they spend in each conformation can be influenced by their phosphorylation state. Indeed, kinases have been crystallized in the active conformation and a wide variety of inactive conformations. Two benefits of designing an inhibitor that targets the inactive conformation are (1) the inactive conformations tend to be less conserved across kinases and offer one route towards the development of selective inhibitors, and (2) since ATP has a much higher preference for binding to the active conformation, the inhibitor will be less impacted by the high intracellular concentration of ATP. The compounds ARQ 523 and ARQ 069 described herein bind to fibroblast growth factor receptor 1 and 2 (FGFR1 and 2) in their autoinhibited conformations. ARQ 523 was first identified from an in silico screen and later optimized to ARQ 069. This tactic of binding the autoinhibited conformation was also adopted by the Phase 3 clinical compound ARQ 197 in the binding of c-Met kinase as described in a second article by Eathiraj et al. (DOI:10.1074/jbc.M110.213801). ARQ 069 binds to the unphosphorylated and inactive conformation of FGFR1 and 2 with ∼20-fold preference over the active conformation. It prevents the activation of autoinhibited FGFR1 and 2. ARQ 069 makes two H-bonds with the hinge region, and the remainder of the molecule predominantly makes hydrophobic contacts with the protein as shown in the first and second figures. Interestingly, ARQ 069 showed non-ATP competitive kinetics at physiological ATP concentrations and slow dissociation kinetics. The orientation of the A-loop in the complex is nearly identical to the apo autoinhibited conformation. The autoinhibited conformation of FGFR1 and 2 has hydrophobic clusters of three to five hydrophobic residues that are the target of these new inhibitors (see second figure) and that are incompatible with nucleotide binding. These clusters are distinct from the hydrophobic pocket that is the target of DFG-out Type II inhibitors. The presence of a hydrophobic spine that is disrupted in the inactive conformation has been shown previously for ABL (DOI:10.1038/nsmb.1486); however, in that case the inactive conformation was the more common DFG-out conformation. It will be interesting to see how many kinases can be trapped in an autoinhibited conformation by small molecules. Contributed by Mindy I. Davis.
Crystal structure of FGFR2 and FGFR1 bound to ARQ 069. (A) The binding mode of ARQ 069 with FGFR2. (B) The binding mode of ARQ 069 with FGFR1. The residues that are in close proximity to ARQ 069 are colored in yellow. The hinge and salt bridge interactions are depicted as dotted lines. The overall binding mode of ARQ 069 is very similar in both FGFR1 and FGFR2, however, they show distinct glycine-rich loop conformations highlighted in marine color. (C) Superposition of ARQ 069–bound FGFR1 with apo-FGFR1 structure (PDB ID 1FGK). Both structures are identical other except that the glycine-rich loop is disordered in the apo-FGFR1 structure. (D) Superposition of ARQ 069–bound FGFR1 with the phosphorylated form of FGFR1 (PDB ID 3GQI). Cartoon representations of key structural elements are shown for both structures.
The non-conserved hydrophobic residues are key determinants of ARQ 069 binding to FGFR kinase. (A) The ARQ 069 interaction with FGFR1 requires a rearrangement of hydrophobic residues in the ATP-binding cleft which re-orients the hydrophobic residues (yellow) creating a non-polar environment that is not compatible with ATP binding. The downward conformation of the glycine-rich loop in FGFR1 is mediated by hydrophobic interaction between F489, V492 and ARQ 069. The FGFR1/ARQ 069 complex structure was superimposed with AMP-ACP-bound phosphorylated FGFR1 (PDB ID 3GQI) and ATP is shown in sticks. (B) Non-polar interaction of ARQ 069 with two nonconserved hydrophobic residues (F489 and V561). (C) Mutational analysis of ARQ 069 interaction with FGFR2. The effect of activating mutations (K526E, E565G and R678G) and non-conserved hydrophobic residues (F492A and V564T) mutations on FGFR2 autophosphorylation inhibition by ARQ 069 was measured. The potency of autoactivation was measured using a continuous spectrophotometric assay to derive IC50 values. (D) Comparison of activation rate (as measured by substrate phosphorylation) with the potency of FGFR2 inhibition by ARQ 069 for the various mutant proteins. Activating mutant proteins show a significant increase in the activation rate and autophosphorylation remains inhibited by ARQ 069. However, mutation of the ARQ 069 interacting residues F492A and V564T in FGFR2 prevents the inhibition of autophosphorylation by ARQ 069.
Turning the Reversible into the Irreversible
Perez DI, Palomo V, Pérez C, Gil C, Dans PD, Luque FJ, Conde S, and Martinez A. Switching reversibility to irreversibility in glycogen synthase kinase 3 inhibitors: clues for specific design of new compounds. J Med Chem 2011; DOI: 10.1021/jm1016279.
Abstract: Development of kinase-targeted therapies for central nervous system (CNS) diseases is a great challenge. Glycogen synthase kinase 3 (GSK-3) offers a great potential for severe CNS unmet diseases, being one of its inhibitors on clinical trials for different tauopathies. Following our hypothesis based on the enhanced reactivity of residue Cys199 in the binding site of GSK-3, we examine here the suitability of phenylhalomethylketones as irreversible inhibitors. Our data confirm that the halomethylketone unit is essential for the inhibitory activity. Moreover, addition of the halomethylketone moiety to reversible inhibitors turned them into irreversible inhibitors with IC50 values in the nM range. Overall, the results point out that these compounds might be useful pharmacological tools to explore physiological and pathological processes related to signaling pathways regulated by GSK-3 opening new avenues for the discovery of novel GSK-3 inhibitors.
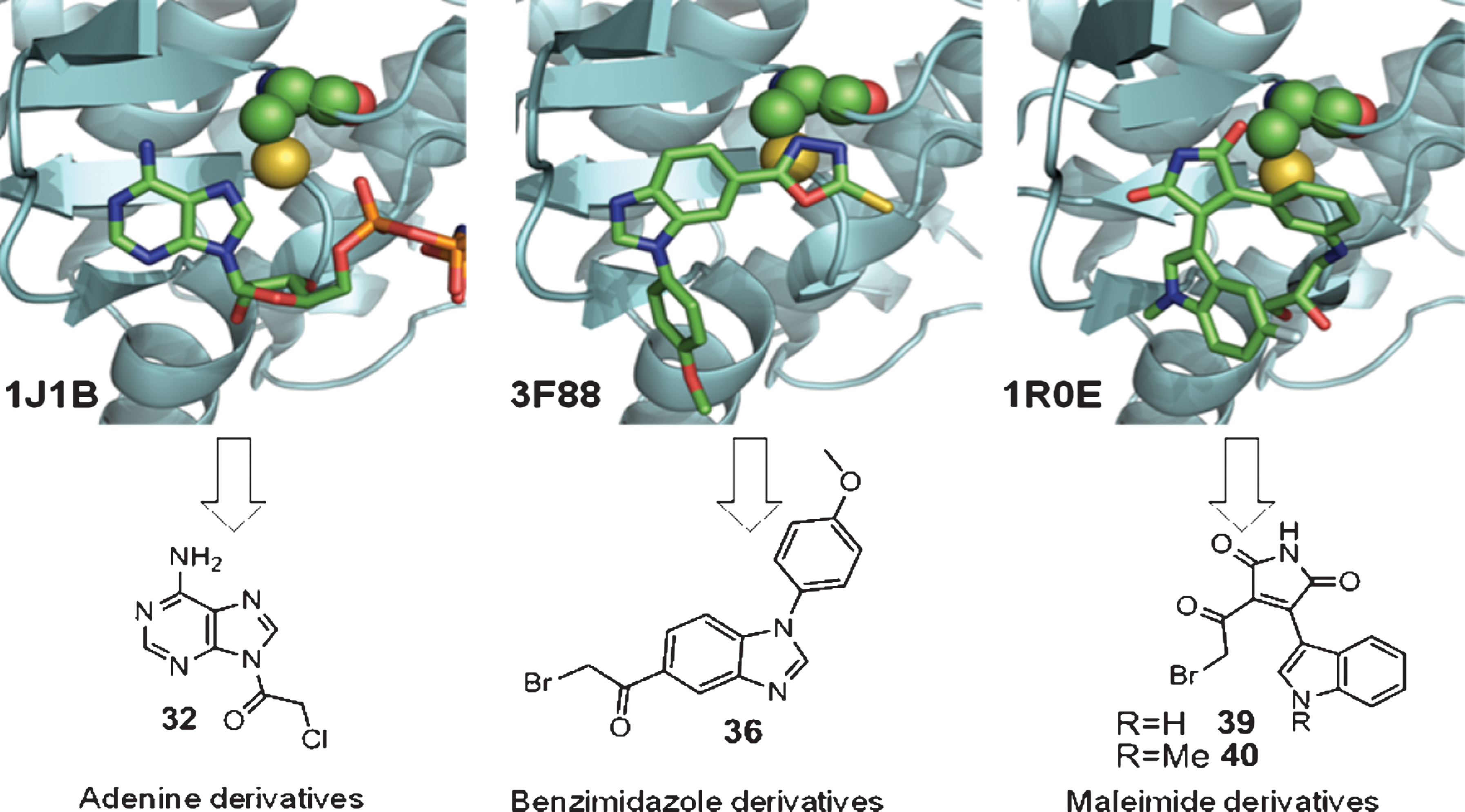
Commentary:There has been a recent flurry of interest in covalent drugs. A recent review titled “The Resurgence of Covalent Drugs” evaluates the success of covalent FDA-approved drugs and the hurdles faced in current covalent drug development but encourages researchers to consider covalent drugs (doi:10.1038/nrd3410). Additionally, the recent article by Leproult et al. (dx-doi-org.web.bisu.edu.cn/10.1021/jm101396q) mapping the cysteines in the kinome provides an excellent resource for identifying cysteines that could be the target for irreversible kinase inhibition. The present article focuses on glycogen synthase kinase 3 (GSK-3), a protein kinase that has a cysteine (Cys199) in its active site. Cys199 is located adjacent to the DFG in the activation loop and GSK-3 would be classified as a CDFG kinase. GSK-3 is an important target for CNS disorders. GSK-3 phosphorylates the tau protein, and treatment of an animal model of Alzheimer's disease with a GSK-3 inhibitor led to a reversal of the pathological features. A GSK-3 inhibitor is currently in clinical trials for tau-related disease. The authors set out to turn reversible GSK-3 inhibitors into irreversible inhibitors. They started with three compounds that had published co-crystal structures with GSK-3 and modified them by adding halomethylketone units to create a covalent point of attachment for Cys199 (see figure). They generated a series of compounds around these initial scaffolds (adenine, benzimidazole, and maleimide) and were able to determine that they were covalent by observing an increase in weight of the protein by MALDI-TOF analysis after compound incubation. Also, as the pre-incubation time of compound with GSK-3 was increased the percentage of inhibition rose, consistent with a covalent inhibitor (or an inhibitor with a very slow on-rate). If given enough time, the reaction would go to completion rather than to equilibrium as for a noncovalent inhibitor. Covalent inhibitors have an initial affinity for the binding pocket, and then an increase in potency is observed when the covalent bond is formed. The compounds with the covalent point of attachment (halomethylketone) had a correspondingly lower IC50 value than their acetyl counterparts. The cellular effects of phenylhalomethylketones mimicked those observed for the GSK-3 inhibitor lithium chloride. Seven of the eight kinases tested in a small selectivity screen did not show significant inhibition by the phenylhalomethylketones. The one kinase that did show inhibition was cSRC which has a cysteine in its active site, and cSRC has been shown to bind irreversible inhibitors (PDB:2LOK); however, the cSRC cysteine is in the hinge region. It would be interesting to do a more comprehensive kinase profile and particularly to see whether the 48 kinases that share the cysteine at the CDFG location (Leproult et al.) are inhibited by these new compounds. Contributed by Mindy I. Davis.
(Top) Representation of the binding mode of an ATP analog (1J1B), and two reversible inhibitors (3F88 and 1R0E; shown in sticks) in the ATP-binding site of GSK-3. Cys199 is shown in spheres. (Bottom) Structure-based design of novel compounds with potential GSK-3 inhibitory activity.
Targeting Proteins with CALI
Takemoto K Matsuda T, McDougall M,. Klaubert DH Hasegawa A,. Los GV, Wood KV, Miyawaki AM, Naga T. Chromophore-assisted light inactivation of HaloTag fusion proteins labeled with eosin in living cells. ACS Chem Biol 2011;6:401–406.
Abstract: Chromophore-assisted light inactivation (CALI) is a potentially powerful tool for the acute disruption of a target protein inside living cells with high spatiotemporal resolution. This technology, however, has not been widely utilized, mainly because of the lack of an efficient chromophore as the photosensitizing agent for singlet oxygen (1O2) generation and the difficulty of covalently labeling the target protein with the chromophore. Here we choose eosin as the photosensitizing chromophore showing 11-fold more production of 1O2 than fluorescein and about 5-fold efficiency in CALI of β-galactosidase by using an eosin-labeled anti-β-galactosidase antibody compared with the fluorescein-labeled one. To covalently label target protein with eosin, we synthesize a membrane-permeable eosin ligand for HaloTag technology, demonstrating easy labeling and efficient inactivation of HaloTag-fused PKC-γ and aurora B in living cells. These antibody- and HaloTag-based CALI techniques using eosin promise effective biomolecule inactivation that is applicable to many cell biological assays in living cells.
Commentary:The ability to specifically knock-down protein expression within a cell can be an extremely useful strategy to study signaling pathways as well as to elucidate the molecular target of small molecules. In general, genetic approaches using siRNAs or shRNAs are employed for such knock-down studies. One drawback of genetic approaches is the need for prolonged incubation times before determining the effect, which depends on the protein half-life and typically precludes rapid measurements. Alternatively, as described in this article, one can specifically destroy the protein of interest through chemical modification, which provides for rapid inactivation of the protein. The technique involves generating reactive oxygen species (ROS) in close proximity to the targeted protein. Singlet oxygen (1O2), a common ROS, is only highly reactive within a radius of approximately 3–4 nm and most protein–protein interactions occur over a longer range (∼8 nm). Therefore, tagging a protein with a photosensitizer that generates 1O2 upon excitation should allow for inactivation of the tagged protein without interfering with other interacting proteins. Ideally, the photosensitizer has low cytotoxicity and can be excited with wavelengths longer than the ultraviolet spectrum to prevent damage to the cells and background fluorescence from cellular components. For this purpose, the authors used eosin, a xanthene-based chromophore with an absorption peak at 517 nm, and they also employed HaloTag technology to covalently link eosin to the kinases PKC-γ and aurora B (see figure). The HaloTag technique involves fusing the enzyme haloalkane dehalogenase, which undergoes covalent modification upon catalysis of a halogenated substrate. Eosin was synthesized with a halogenated alkane and was shown to generate 1O2 similar to free eosin. Both the GFP and the dehalogenase were fused to PKC-γ, and then high intensity green light was applied with a CW laser (8.8 W/cm2 for 90 sec, which was nearly 80 times lower power than the power used in previous photosensitize experiments), resulting in abolishing the translocation activity of PKC-γ. In addition, the eosin tag was used to study mitochondrial function and the effect of aurora B on cell division. Combining this technique with an already available HaloTag clone library provides a means to study protein function with high spatiotemporal resolution. However, as with any knock-down technique, either genetic or chemical, the results are highly dependent on the efficiency of silencing. The CALI technique described could suffer from a shortcoming due to interference by endogenous 11 proteins that could be present in sufficient abundance to mask functional effects. Contributed by Doug Auld.
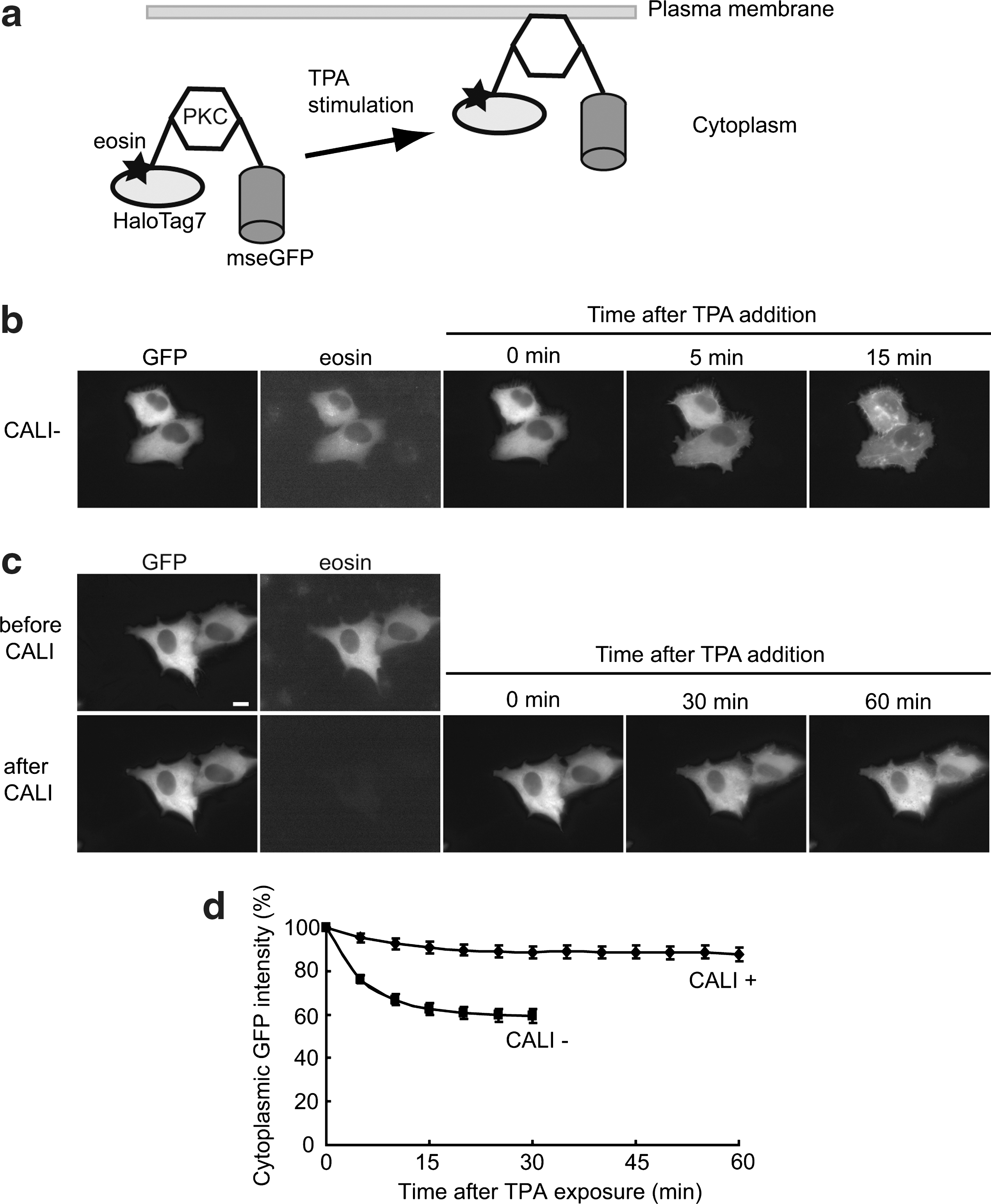
PKC-γ inactivation by eosin-based CALI in living cells. (a) Schematic representation of the HaloTag7-PKC-γ-mseGFP fusion protein and its translocation to the membrane after TPA (12-O-tetradecanoylphorbol 13-acetate) treatment. (b) Control experiment (CALI). After TPA addition, PKC-γ successfully translocated to the plasma membrane. (c) CALI of PKC-γ. The eosin fluorescence disappeared after irradiation with intense light resulting in failure of PKC translocation to the plasma membrane. Cells were examined by time-lapse analysis for the CALI− (n = 21 cells) and CALI+ (n = 18 cells) conditions, respectively. Representative data are shown in panels b and c. (d) Time course analysis of PKC-γ translocation to the plasma membrane with (CALI+) or without (CALI−) CALI. The PKC-γ activation was evaluated by the decrease in fluorescence intensity in the cytoplasm due to the protein's translocation to the plasma membrane upon TPA stimulation. Error bars are SD. Scale bar = 5 μm.
Crossing the BBB on a Trojan Horse
Yu YJ, Zhang Y, Kenrick M, Hoyte K, Luk W, Lu Y, Atwal J, Elliott JM, Prabhu S, Watts RJ, Dennis MS. Boosting brain uptake of a therapeutic antibody by reducing its affinity for a transcytosis target. Sci Trans Med 2011;3:84ra44.
Abstract: Monoclonal antibodies have therapeutic potential for treating diseases of the central nervous system, but their accumulation in the brain is limited by the blood–brain barrier (BBB). Here, we show that reducing the affinity of an antibody for the transferrin receptor (TfR) enhances receptor-mediated transcytosis of the anti-TfR antibody across the BBB into the mouse brain where it reaches therapeutically relevant concentrations. Anti-TfR antibodies that bind with high affinity to TfR remain associated with the BBB, whereas lower-affinity anti-TfR antibody variants are released from the BBB into the brain and show a broad distribution 24 hours after dosing. We designed a bispecific antibody that binds with low affinity to TfR and with high affinity to the enzyme β-secretase (BACE1), which processes amyloid precursor protein into amyloid-β (Aβ) peptides including those associated with Alzheimer's disease. Compared to monospecific anti-BACE1 antibody, the bispecific antibody accumulated in the mouse brain and led to a greater reduction in brain Aβ after a single systemic dose. TfR-facilitated transcytosis of this bispecific antibody across the BBB may enhance its potency as an anti-BACE1 therapy for treating Alzheimer's disease.
Commentary:The article by Atwal and colleagues from Genentech (see “Targeting BACE1 with an Antibody”) provides a strong basis for further studies to support the development of antibody-based therapeutics targeting processes in the brain. In the complementary work by Yu and co-authors, an ingenious strategy is presented for the improvement of blood–brain barrier (BBB) penetration of an antibody therapeutic. The authors exploit the process of receptor-mediated transcytosis whereby an antibody directed against a brain endothelial receptor had been demonstrated to be transported across the BBB. An embedded deficiency of such an approach stems from the fact that a specific high-affinity antibody would be transported across the BBB via the transcytosis process but would not be released by the receptor to an appreciable extent once inside (the authors verified this notion in the present study, as well: see figure 1 in the article). In order to improve the net uptake of the antibody into the brain, the team tried the counterintuitive approach of using a lower affinity antibody: once inside the brain, the antibody was expected to be released more easily from its complex with the carrier receptor and therefore be liberated for further distribution throughout the brain (first figure). This approach was tested with a series of varying-affinity antibodies directed against the transferrin receptor (TfR): as expected, the antibodies with lower affinity exhibited an increased brain uptake (figure 2 in the article) as well as a broad distribution within the brain (figure 4 in the article). While the anti-TfR antibody would not be of therapeutic value by itself, the authors set out to examine the effect of combining the transcytosis properties of an anti-TfR antibody with the potential therapeutic effect of an anti-BACE1 antibody previously tested in Alzheimer's disease models (see commentary for Atwal et al.). A bispecific IgG antibody incorporating anti-BACE1 heavy and light chains in combination with anti-TfR heavy and light chains, respectively, was generated and tested for its ability to cross the BBB in a mouse model system. At therapeutic doses, the bispecific antibody reached ample brain concentrations of approximately 20 nM and its level remained stable for 48 hours (second figure). Furthermore, intravenous administration of bispecific anti-TfR/BACE1 antibody resulted in a reduction of brain amyloid-β peptide (Aβ) (figure 6 in the article). The combination of these two approaches, in which the loose association between a weak-affinity antibody and a brain endothelial receptor is exploited to create a cargo system to deliver therapeutic agent into the brain, and in which the application of a highly specific and non–cross-reactive anti-BACE1 antibody leads to marked decrease in Aβ production, serves not only as a starting point for further development of this novel Alzheimer's disease therapy, but also as a model to use in addressing other disorders in the central nervous system. Contributed by Anton Simeonov.
Model illustrating the inverse relationship between the antibody's affinity for TfR and its uptake in brain. (A) With trace dosing, a higher-affinity anti-TfR antibody will bind more receptors expressed on the luminal (blood) side of the endothelial cells that make up the BBB compared with trace dosing of lower-affinity antibodies (C), resulting in more high-affinity antibodies associated with brain endothelium. At therapeutic doses, saturating concentrations of antibody will result in antibody binding to receptors on the luminal side of the BBB epithelium regardless of antibody affinity (B, D). The dissociation of antibody after receptor-mediated transcytosis will be more likely with a lower-affinity antibody, resulting in accumulation of lower-affinity antibody in the brain parenchyma (D). Furthermore, the probability of efflux out of the brain parenchyma will be lower for a low-affinity TfR antibody because concentrations in the brain are likely to be no longer saturating (similar to left panels, but in the brain to blood direction).
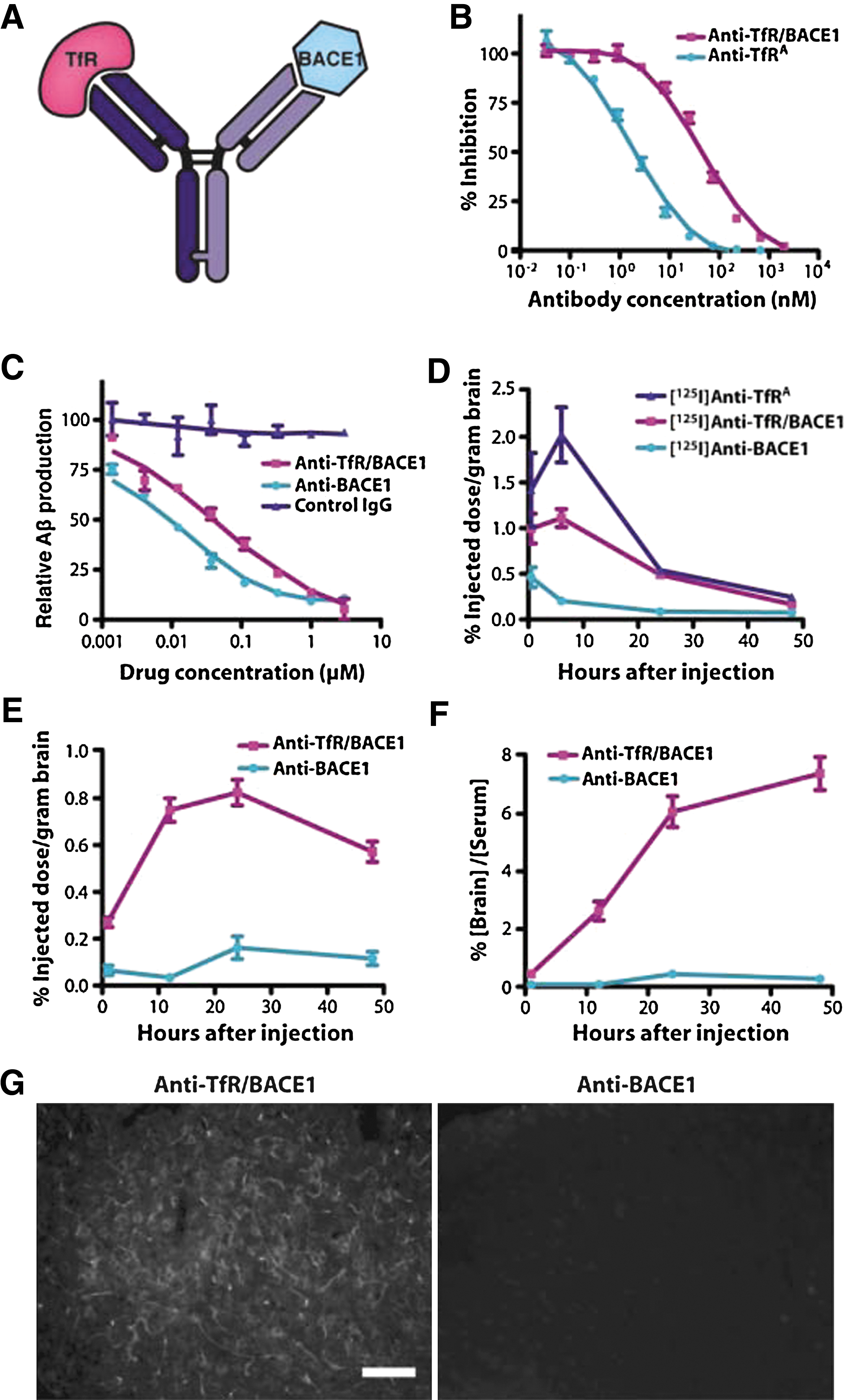
An anti-TfR/BACE1 bispecific antibody blocks Aβ production in vitro and accumulates in the brain in vivo. (A) Model of the bispecific antibody, which was engineered to bind to both TfR and the enzyme β-secretase (BACE1). (B) Affinities of the parental anti-TfRA (IC50 = 1.7 ± 0.1 nM) and anti-TfR/BACE1 (IC50 = 45 ± 3 nM) as measured by a competitive ELISA assay showed a significantly reduced affinity of the bispecific antibody for TfR. (C) Quantification of human Aβ1–40 in HEK293-hAPPWT cells after titration with anti-TfR/BACE1, anti-BACE1, and control IgGin a cell-based assay reveals slightly reduced potency of anti-TfR/BACE1 compared with anti-BACE1. (D) Mean brain uptake after trace doses of 125I-labeled antibody for 30 min and 6, 24, and 48 hours after intravenous injection in mice (n = 4 per group and time point) shows reduced uptake of anti-TfR/BACE1 compared with anti-TfR. (E, F) Mean antibody uptake (at 48 hours: anti-BACE1 = 0.12 ± 0.03%; anti-TfR/BACE1 = 0.57 ± 0.04%) (E) and average brain-to-serum ratio at 1, 12, 24, and 48 hours after an intravenous injection of an antibody (20 mg/kg) in mice (n = 10 per group and time point) (F), showing a robust uptake of anti-TfR/BACE1 with therapeutic dosing (at 48 hours: anti-BACE1 = 0.28 ± 0.08%; anti-TfR/BACE1 = 7.40 ± 0.58%). (G) Immunohistochemical staining of brain sections from mice 24 hours after an intravenous injection with either anti-TfR/BACE1 or anti-BACE1 (20 mg/kg). Images show broad staining in the brain parenchyma for the anti-TfR/BACE1 bispecific antibody compared with minimal staining for themonospecific anti-BACE1 antibody.
Targeting BACE1 With an Antibody
Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y, Heise CE, Hoyte K, Luk W, Lu Y, Peng K, Wu P, Rouge L, Zhang Y, Lazarus RA, Scearce-Levie K, Wang W, Wu Y, Tessier-Lavigne M, Watts RJ. A therapeutic antibody targeting BACE1 inhibits amyloid-β production in vivo. Sci Transl Med 2011;3:84ra43.
Abstract: Reducing production of amyloid-β (Aβ) peptide by direct inhibition of the enzymes that process amyloid precursor protein (APP) is a central therapeutic strategy for treating Alzheimer's disease. However, small-molecule inhibitors of the β-secretase (BACE1) and γ-secretase APP processing enzymes have shown a lack of target selectivity and poor penetrance of the blood-brain barrier (BBB). Here, we have developed a high-affinity, phage-derived human antibody that targets BACE1 (anti-BACE1) and is anti-amyloidogenic. Anti-BACE1 reduces endogenous BACE1 activity and Aβ production in human cell lines expressing APP and in cultured primary neurons. Anti-BACE1 is highly selective and does not inhibit the related enzymes BACE2 or cathepsin D. Competitive binding assays and x-ray crystallography indicate that anti-BACE1 binds noncompetitively to an exosite on BACE1 and not to the catalytic site. Systemic dosing of mice and nonhuman primates with anti-BACE1 resulted in sustained reductions in peripheral Aβ peptide concentrations. Anti-BACE1 also reduces central nervous system Aβ concentrations in mouse and monkey, consistent with a measurable uptake of antibody across the BBB. Thus, BACE1 can be targeted in a highly selective manner through passive immunization with anti-BACE1, providing a potential approach for treating Alzheimer's disease. Nevertheless, therapeutic success with anti-BACE1 will depend on improving antibody uptake into the brain.
Commentary:Therapeutic strategies to treat Alzheimer's disease have focused on the amyloid-β peptide (Aβ) and, in particular, on the development of small molecule inhibitors of the β-secretase enzyme BACE1, which is responsible for the cleavage of the amyloid precursor protein to generate Aβ. Despite the recent advances, most BACE1 small molecule inhibitors still suffer from side effects due to their off-target activity and toxicity. In the present work, a monoclonal antibody against BACE1 is used instead of a small molecule. Atwal and co-authors used synthetic human antibody libraries in a panning protocol against the extracellular enzymatic domain of BACE1 to identify an anti-BACE1 antibody with a single-digit nanomolar affinity. The anti-BACE1 antibody inhibited the catalytic activity of BACE1 and was not effective against the related BACE2 and cathepsin D enzymes (first figure). The antibody's binding to BACE1 was further characterized by competitive ELISA and X-ray crystallography methods (second figure). Anti-BACE1 was then shown to inhibit the BACE1 activity in HEK293 cells expressing amyloid precursor protein. The activity and pharmacokinetics of anti-BACE1 were then tested in mice and cynomolgus monkeys. Dosing the animals with anti-BACE1 resulted in a high plasma antibody concentration, with only a small fraction of the agent (approximately 0.07%) reaching the brain, consistent with predictions for IgGs crossing the blood–brain barrier (BBB). Despite the low effective brain concentration of anti-BACE1, its application was shown to lead to a reduction in the levels of Aβ (see also “Crossing the BBB on a Trojan Horse”). Moreover, the pharmacokinetics of anti-BACE1 was directly dependent on the level of BACE1 expression, further supporting target-mediated clearance mechanism (see figure 5 in the article). The present study serves as a powerful demonstration of the possibility to use a selective high-affinity antibody against an enzymatic target within the central nervous system. While the low fraction of anti-BACE1 achieved in the brain is a testimony for the problems that remain to be solved in this field, the accompanying article by the team at Genentech (See commentary for Yu et al.) provides insight into a novel strategy to improve the BBB delivery of a protein therapeutic. Contributed by Anton Simeonov.
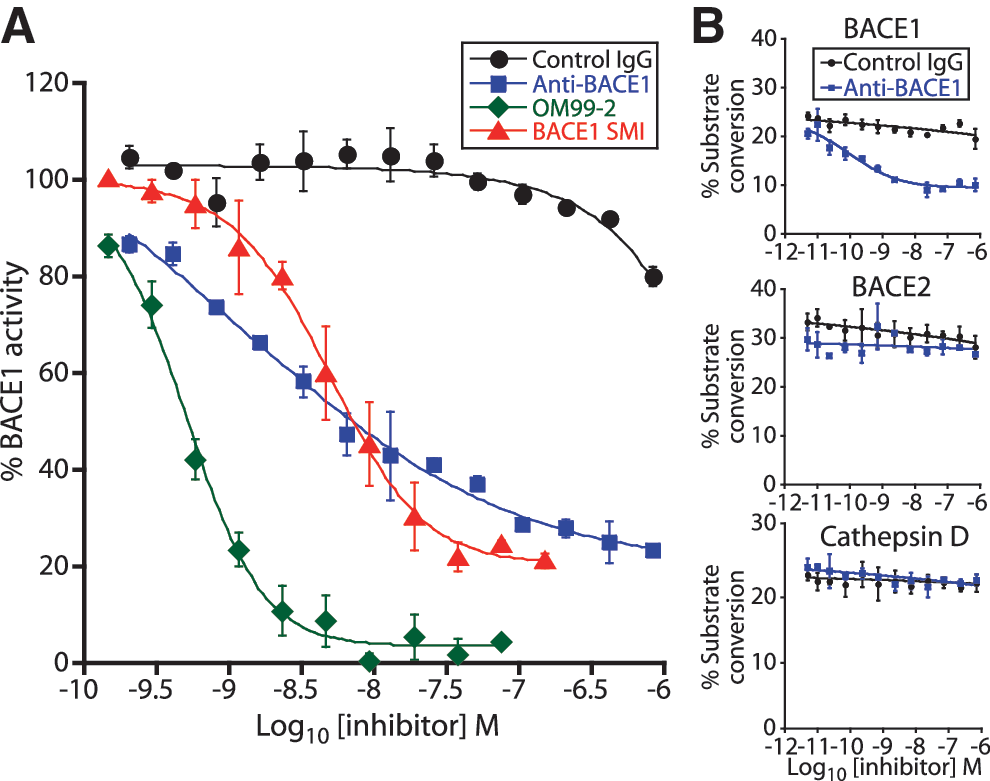
Anti-BACE1 is a potent and selective inhibitor of BACE1. (A) Enzymatic activity of rhBACE1 ECD was tested with the 27–amino acid peptide substrate in an HTRF assay. Activity was measured in the presence of control IgG, anti-BACE1, OM99-2, or a small-molecule inhibitor (SMI) of BACE1 (Inhibitor IV, Calbiochem). Anti-BACE1 inhibits BACE1 activity with an IC50 of 1.7 nM. Values reflect BACE1 enzymatic activity relative to control (no antibody present) ± SEM. (B) In vitro enzymatic activity of rhBACE1 ectodomain, rhBACE2 ectodomain, or cathepsin D ectodomain was assayed by a microfluidic capillary electrophoresis assay with an APP peptide substrate. Anti-BACE1 inhibited rBACE1 activity with an IC50 of 80 pM but did not inhibit rBACE2 or cathepsin D. Values reflect percent substrate conversion ± SEM.
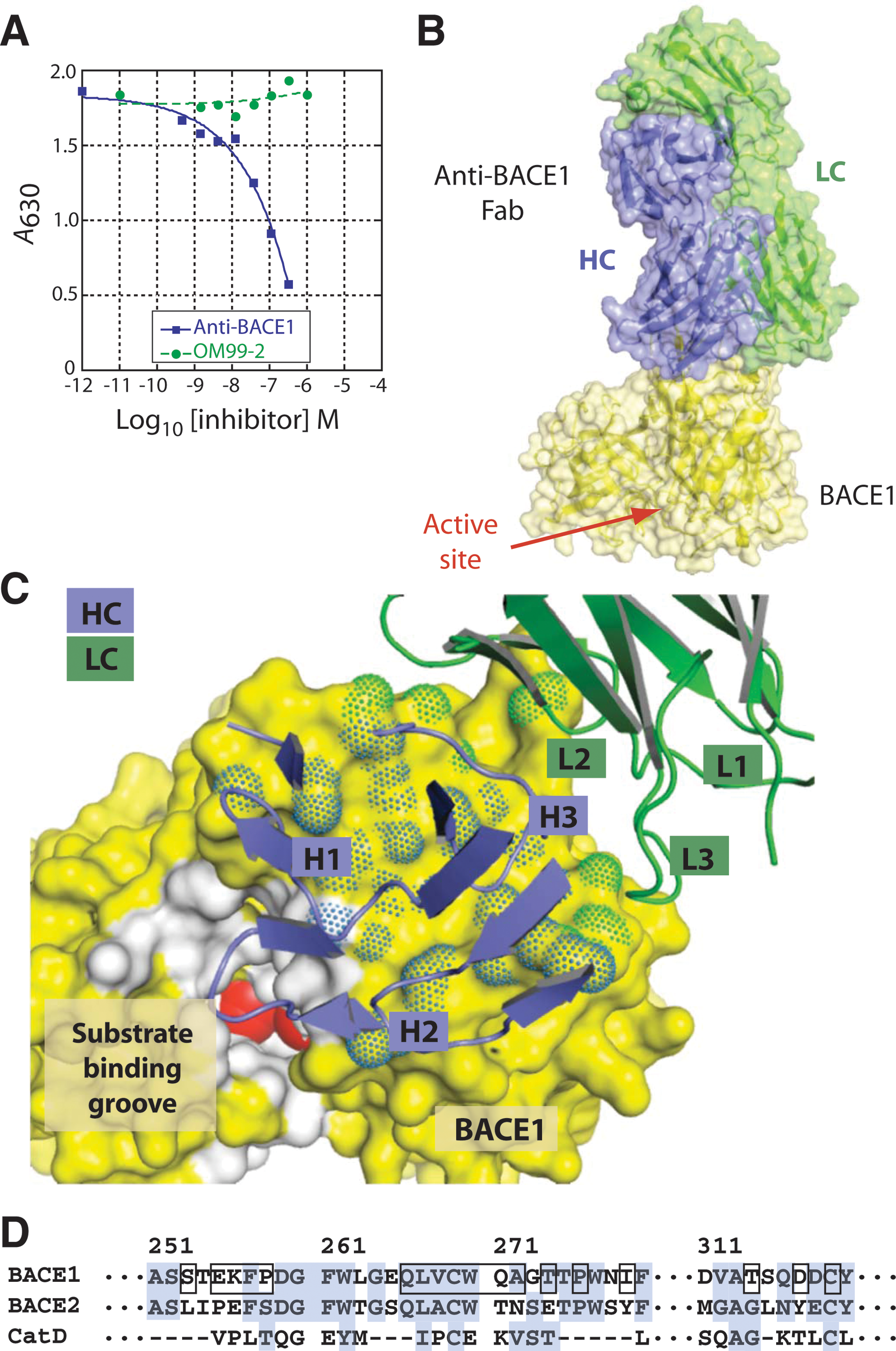
Anti-BACE1 inhibits BACE1 by binding to an exosite. (A) Competitive ELISA assay was used to study the anti-BACE1/BACE1 interaction compared to a known active-site BACE1 inhibitor. Active-site inhibitor OM99-2 does not compete with anti-BACE1 for BACE1 binding. (B) The crystal structure of the anti-BACE1 Fab and rhBACE1 catalytic domain showing anti-BACE1 binding to a BACE1 exosite. (C) Close-up view of the antibody complementarity-determining regions (CDRs) and the BACE1 epitope. Both heavy chain (HC) and light chain (LC) are involved in the interaction. (D) Amino acid sequence at and around the binding epitope. Residues in direct contact are boxed. For comparison, sequences of BACE2 and cathepsin D (CatD) are aligned to BACE1 based on crystal structures.
Coupling Receptor Ligands With Micelle-Based Drug Delivery System
Zhan C, Li B, Hu L, Wei X, Feng L, Fu W, Lu W. Micelle-based brain-targeted drug delivery enabled by a nicotine acetylcholine receptor ligand. Angew Chem Int Ed 2011;505482–505485.
Abstract: No abstract.
Commentary: Due to the combination of increased life expectancy and the “Baby Boom” effect, the global population of people over 65 is projected to triple by midcentury. Older populations are more susceptible to various forms of neurological disorders; thus, the central nervous system (CNS) represents a highly relevant research and development segment. According to a 2008 report by Datamonitor, the CNS drug market was estimated to worth ∼$100 billion. Compared to non-CNS drugs, CNS drugs take longer time to reach market and attrition rate is higher (Alavijeh et al., NeuroRx®2005;2:554–571). Among the different challenges for successful CNS drug delivery, the requirement for drugs to cross the blood–brain barrier (BBB) is a major one. Tight junctions and lack of fenestration are responsible for inefficient delivery of drugs and their reduced efficacy. Recently, researchers have been actively exploring nanoparticle-aided drug transfer system for the search of effective strategies for drugs to gain access to the BBB (Nasongkla et al., Nano Lett 2006;6:2427–2430).
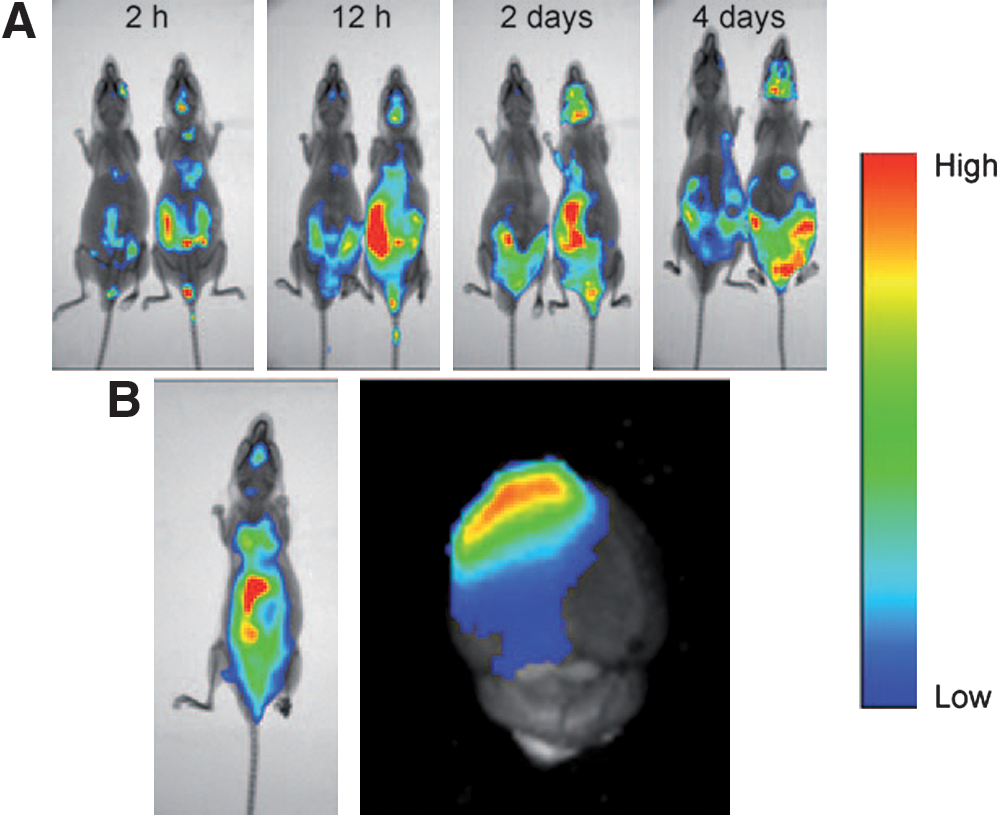
Highly expressed receptors in the brain and their susceptibility to inhibition by peptidal neurotoxins make the system an ideal target for ligand-enabled drug delivery. Herein, the authors took advantage of nicotine acetylcholine receptors (nAChRs), a receptor that is abundant on the capillary endothelium of the brain, and designed three short peptide ligands (CDX, Pocket_CDX, and Cyclo_CDX) derived from a known nAChR antagonist, candoxin. Binding affinities were found to be in agreement between those obtained through a competitive radiolabled binding assay and computational prediction, with CDX being the most potent antagonist among the three (see table). Additionally, CDX was shown to interact with nAChR receptors in vitro using primary rat brain capillary endothelial cells. After ligand design validation, the authors proceeded to the core objective of the proof-of-principle study: could CDX enhance drug transport to the brain, and if it did, could CDX enhance the therapeutic efficacy of antitumor drugs? In order to address the two questions, the authors subsequently functionalized a widely used nanoparticulate carrier, PEG-PLA micelle, with CDX. Coumarin-encapsulated CDX-decorated and unmodified micelles were injected into mice, and the bioavailability of coumarin was found to have doubled in the brain when transported through CDX-derivatized micelles. In order to probe any potential effect CDX-conjugated delivery system might have on antitumor drug efficacy, the authors utilized a human glioblastoma multiforme (GBM) xenograft mouse model. Paclitaxel-loaded CDX-conjugated micelles, paclitaxel-loaded unmodified micelles, and saline were respectively injected into three randomly divided groups of mice intravenously. Different doses of paclitaxel and regimen were implemented, and in general, it was found that mouse survival was significantly prolonged when treatment took place in the presence of CDX (for example, see first figure for a dose of 10 mg/kg of body weight). Furthermore, biodistribution of CDX-conjugated and unmodified micelles (caudal vein injection) was obtained through encapsulation of a near-infrared fluorescent dye, DiR. Pronounced dye accumulation was observed in the brain over a period of 4 days post-tumor implantation when injection was performed with CDX while no dye was observed in the absence of CDX during the same period (see second figure). Using GBM as a tumor model and nAChR antagonists as targeted molecules, the authors have demonstrated that this receptor ligand coupled drug delivery system can enhance the antitumor drug transport and improve drug efficacy. This coupled system may find broad applicability for various CNS disease treatments. Contributed by Wendy Lea.
Kaplan–Meier survival curves of nude mice bearing intracranial U87 glioblastoma. Mice that received three doses (at 5, 10 and 15 days post glioblastoma implantation) of CDX-PEG-PLA-PTX micelles survive significantly longer than the control groups that received mPEG-PLA-PTX micelles or physiological saline (p < 0.01, log-rank analysis).
Near-infrared optical images of DiR encapsulated micelles injected in the caudal vein of intracranial glioblastoma-bearing nude mice at 5 days (A) and 15 days (B) post-tumor implantation. The mice on the left-hand side in four pairwise comparisons in panel (A) were injected with DiR-encapsulated mPEG-PLA micelles, and the mice on the right-hand side were injected with CDX-PEG-PLA micelles loaded with the same dose of DiR. The mouse in panel (B) was sacrificed 12 h after injection of DiR-encapsulated CDX-PEG-PLA micelles, and the brain dissected for imaging.
Experimentally Determined Versus Predicted Free Energies of Binding of All Synthetic Peptides With α7 Neuronal nAChR (ΔG = 1.3636 log Ki)
Peptide
Ki (μM)
Experimental ΔG (kcal/mol)
Estimated ΔG (kcal/mol)
CDX
0.187
−9.17
−9.59
Pocket_CDX
4.60
−7.28
−8.17
Cyclo_CDX
7.11
−7.02
−7.85
FRET by BRET
Branchini BR, Rosenberg JC, Ablamsky DM, Taylor KP, Southworth TL, Linder SJ. Sequential bioluminescence resonance energy transfer–fluorescence resonance energy transfer-based ratiometric protease assays with fusion proteins of firefly luciferase and red fluorescent protein. Anal Biochem 2011;414:239–245.
Abstract: We report here the preparation of ratiometric luminescent probes that contain two well-separated emission peaks produced by a sequential bioluminescence resonance energy transfer (BRET)–fluorescence resonance energy transfer (FRET) process. The probes are single soluble fusion proteins consisting of a thermostable firefly luciferase variant that catalyze yellow-green (560 nm maximum) bioluminescence and a red fluorescent protein covalently labeled with a near-infrared fluorescent dye. The two proteins are connected by a decapeptide containing a protease recognition site specific for factor Xa, thrombin, or caspase 3. The rates of protease cleavage of the fusion protein substrates were monitored by recording emission spectra and plotting the change in peak ratios over time. Detection limits of 0.41 nM for caspase 3, 1.0 nM for thrombin, and 58 nM for factor Xa were realized with a scanning fluorometer. Our results demonstrate for the first time that an efficient sequential BRET–FRET energy transfer process based on firefly luciferase bioluminescence can be employed to assay physiologically important protease activities.
Commentary:Bioluminescence resonance energy transfer (BRET) has been a useful technique to study protein–protein interactions within cells as well as for measuring the functional activity of proteins. BRET differs from fluorescence resonance energy transfer (FRET) in several ways. BRET typically employs two proteins, a luciferase such as Renilla luciferase (RLuc) yielding bioluminescent light that stimulates a fluorescent protein such as GFP, which then emits at a longer wavelength. On the other hand, FRET involves the input of light energy that excites a donor fluorophore to stimulate an acceptor fluorophore, which then emits at a longer wavelength. Both types of signals follow a 1/r6 distance dependence with a maximum distance of ∼10 nm, a distance that is within the range of most protein–protein interactions. Background fluorescence is not generally an issue with BRET but can be an issue with FRET methods; however, the BRET signal is much weaker than FRET. The present article provides a system in which the attributes of both BRET and FRET signals are combined into a single construct (see figure). A thermostable variant of firefly luciferase (Ppy WT-TS) was fused to a red fluorescent protein (RFP). Addition of luciferase (D-luciferin and Mg-ATP) substrates results in bioluminescence at 560 nm, which stimulates the RFP, resulting in fluorescence at 610 nm. Spectral overlap can be an issue when RLuc and GFP are used as BRET partners due to the overlap of RLuc bioluminescence with GFP fluorescent emission. Typically, the blue-shifted coelenterazine luciferin, DeepBlue C, is used as the RLuc substrate to help with this issue, but this substrate shows high protein binding and needs stabilizers to prevent rapid degradation. However, in this article it is demonstrated that labeling RFP via two specific cysteines with the infrared dye AF680 results in a sequential BRET-FRET process (SRET) in which bioluminescence at 560 nm yields a fluorescence signal at 705 nm that improves the signal:background of the assay (see figure). Two biochemical assays were used to test the SRET system in which a decapeptide containing a protease recognition site for either thrombin or caspase 3 was inserted between the Ppy WT-TS and RFP partners. In the assay, linear rates due to proteolytic degradation are measured by the 560 nm/706 nm peak ratios. Several peptide sequences were tested, which confirmed the expected protease substrate selectivity. Detection of proteases in biological fluids should be enabled with this approach, especially given the IR fluorescence that is ultimately detected. Employing linkers resulting in steric blocking of the SRET signals could extend this method for detection of specific analytes. Contributed by Doug Auld.
Normalized absorption, emission, and fluorescence spectra. The following spectra were obtained as described under Materials and Methods and demonstrate: (a) the bioluminescence of Ppy WT-TS following the addition of LH2 and Mg-ATP; (b) RFP absorbance and fluorescence (excitation 585 nm); (c) BRET produced by unlabeled fusion protein substrate following the addition of LH2 and Mg–ATP; (d) AF680 absorbance and fluorescence (excitation 680 nm); (e) FRET produced by 585 nm excitation of fusion protein substrate; (f) SRET of BFS-Th following addition of LH2 and Mg-ATP.
Prostasomes as Biomarkers
Tavoosidana G, Ronquist G, Darmanis S, Yan J, Carlsson L, Wu D, Conze T, Ek P, Semjonow A, Eltze E, Larsson A, Landegren UD, Kamali-Moghaddam M. Multiple recognition assay reveals prostasomes as promising plasma biomarkers for prostate cancer. Proc Natl Acad Sci USA 2011;108:8809–8814.
Abstract: Prostasomes are microvesicles (mean diameter, 150 nm) that are produced and secreted by normal and malignant prostate acinar cells. It has been hypothesized that invasive growth of malignant prostate cells may cause these microvesicles, normally released into seminal fluid, to appear in interstitial space and therewith into peripheral circulation. The suitability of prostasomes as blood biomarkers in patients with prostate cancer was tested by using an expanded variant of the proximity ligation assay (PLA). We developed an extremely sensitive and specific assay (4PLA) for detection of complex target structures such as microvesicles in which the target is first captured via an immobilized antibody and subsequently detected by using four other antibodies with attached DNA strands. The requirement for coincident binding by five antibodies to generate an amplifiable reporter results in both increased specificity and sensitivity. The assay successfully detected significantly elevated levels of prostasomes in blood samples from patients with prostate cancer before radical prostatectomy, compared with controls and men with benign biopsy results. The medians for prostasome levels in blood plasma of patients with prostate cancer were 2.5 to sevenfold higher compared with control samples in two independent studies, and the assay also distinguished patients with high and medium prostatectomy Gleason scores (8/9 and 7, respectively) from those with low score (≤6), thus reflecting disease aggressiveness. This approach that enables detection of prostasomes in peripheral blood may be useful for early diagnosis and assessment of prognosis in organ-confined prostate cancer.
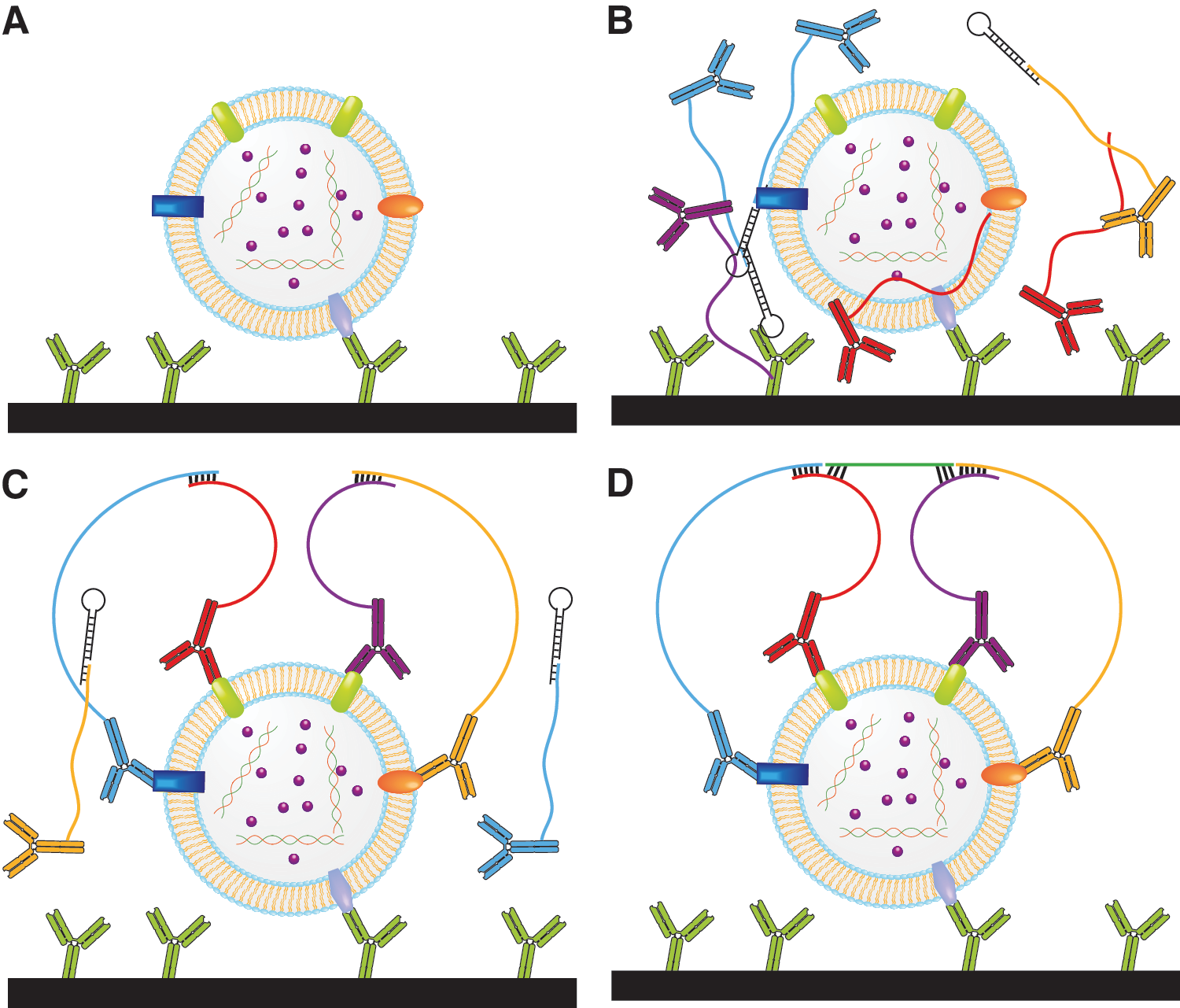
Commentary:Despite the availability and widespread use of the prostate-specific antigen (PSA) test for detection of prostate cancer, superior methods are required to improve one's ability for early cancer detection and the correlation between biomarker testing and the aggressiveness of the cancer (as measured by its Gleason score). The present study highlights the prostasomes, small extracellular microvesicles originating from intracellular vesicles inside secretory cells of the prostate gland, as a promising new biomarker for early detection and grade assessment of prostate cancer. A key development detailed in the present article is the establishment of an ultrasensitive assay for prostasomes in blood samples. The principle of proximity ligation assay (PLA) was utilized here. In PLA, antigen detection is made possible by using at least two antibodies directed towards distinct epitopes and carrying short oligodeoxynucleotide strands capable of being ligated together when present in close proximity as part of the ternary recognition complex. In turn, the ligation-joined DNA strands serve as a template for amplification and detection via real-time quantitative polymerase chain reaction (PCR), ultimately yielding a signal proportional to the analyte concentration. To achieve highly specific and sensitive detection of prostasomes, the authors used five distinct antibodies to both capture the microvesicle on a solid surface as well as to perform a multi-epitope PLA (first figure). By requiring that four antibodies be bound simultaneously to the analyte immobilized by a fifth antibody, the team was able to achieve a dramatic decrease in nonspecific background signal and a 150-fold improvement of the limit of detection as compared with other PLA methods. In control experiments, the individual oligo-derivatized antibodies were each replaced by free single-stranded DNA and in all cases the assay signal reverted to background levels (second figure).The authors proceeded to measuring the prostasome levels in patients before prostate surgery and relating the values to the Gleason scores obtained after histological examination of the excised tumors: a good correlation was observed between prostasome levels and Gleason score, in contrast to the results from PSA measurement in which the level of PSA was found to correlate relatively poorly with the tumor scores. While some assay variability was noted by the authors, requiring further assay optimization, the present report establishes the viability of a strategy to use entire multiprotein assemblies as biomarkers, with the possibility to extend this platform beyond the cancer of the prostate. Contributed by Anton Simeonov.
Mechanism of 4PLA. Target molecules are captured by antibodies immobilized on the walls of a reaction vessel (A), the four PLA probes are added (B), and the probes are allowed to bind different epitopes on the target structure. The four oligonucleotides attached to the antibodies hybridize to each other (C) and guide hybridization of a further oligonucleotide (D). This oligonucleotide that is added together with ligation/amplification mix is joined by enzymatic DNA ligation to oligonucleotides attached to two of the antibodies, templated by oligonucleotides on the two other antibodies. Finally, the newly formed DNA template is amplified and quantified by qPCR.
Detection of prostasomes by using 4PLA and PLA. (A) Comparison of 4PLA (circles) and solid-phase PLA (squares) for measuring purified prostasomes. For 4PLA the SD of 0.021 and for solid-phase PLA the SD of 0.056 for negative controls were used to calculate the LOD. (B) 4PLA was used to detect serial dilutions of purified prostasomes, spiked in 4PLA buffer (squares) and in 10% human plasma (circles). (C) The 4PLA mechanism was investigated by omitting each of the four antibodies used in the probe mix in separate reactions while still adding the corresponding oligonucleotide. The omission of any antibody resulted in reduction of the detection signals to background levels. The y axes show the CT average and the x axes indicate the concentration of prostasomes. Error bars indicate SDs from the mean for triplicate reactions.