Stefan Duhr studied biochemistry at the University Witten/Herdecke, Germany, with a specialization in physical biochemistry and molecular biology. His research focused on the mechanism of ribosomal protein biosynthesis. After receiving the Diploma Degree in 2004, he began his PhD studies in the Physics Department of Ludwig-Maximilians-University, Munich, working in the Systems Biophysics group of Professor Dieter Braun on the principles of thermophoresis. The results were the basis for patent applications that cover biomolecule analytics using thermophoresis induced by infrared laser heating. Dr. Duhr and colleagues evaluated possible applications of thermophoresis and founded the spin-off company NanoTemper Technologies GmbH in May 2008. Dr. Duhr is CEO of NanoTemper. The company received an award in the Munich Businessplan Competition, won the nationwide Cyber-One Award, and was one of three finalists for the European Fast Start Award in 2009. The company develops, produces, and sells instruments based on the proprietary Microscale Thermophoresis technology.
Moran Jerabek-Willemsen studied Biology at the Heinrich-Heine University in Düsseldorf, Germany, with a specialization in cell and molecular biology. He worked in the laboratory of Professor Claus Pfeffer at the Institute of Medical Microbiology. After obtaining his Master's degree in 2005, he began his PhD studies at the Institute of Cell Biology at the University Hospital of Essen in the DNA repair group of Professor Jürgen Thomale. He also worked on the development of new strategies for in vivo chemo-protection of hematopoietic stem cells from DNA damage caused by methylating agents in Professor David William's lab at Cincinnati Children's Hospital and Medical Center in Ohio. At the beginning of 2010, Dr. Jerabek-Willemsen joined NanoTemper Technologies and the Systems Biophysics group of Professor Braun at Ludwig-Maximilians-University in Munich. He has been responsible for the development of new applications and the improvement of existing life science-related applications of MST.
What are the most common methods used today to measure the interaction of a ligand and its target or receptor? What are the advantages and limitations or barriers to the routine use of these methods?
Stefan Duhr (SD): The most common methods are isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR). Thermal titration calorimetry, which measures the heat produced or consumed in a binding event, allows you to work label-free and to measure binding in free solution. So, you do not have to couple anything to a surface or to a carrier material; however, a very high amount of sample is consumed by ITC. Surface plasmon resonance has the disadvantage that you have to couple proteins or samples covalently to a gold surface, which might change the properties of the sample. Another disadvantage of SPR is that because it takes so long to establish an assay, people may be more reluctant to use this technology. On the other hand, SPR allows you to get information on the rate of a reaction—the on-rate and off-rate—which you cannot get from thermal titration calorimetry.
We view Microscale Thermophoresis (MST) as an additional method that allows measurements that are not possible with either of these well-established methods, since it is very sensitive to conformational changes of molecules. It can replace some parts of what these other instruments do, and there are some experiments that are much better obtained by MST (e.g., liposome and membrane receptors). In addition to enabling certain experiments, the technology is also very fast. You can label your protein in about 30 minutes, prepare a serial dilution in about 10 minutes, load the sample into our instrument, and obtain an affinity measurement in about 10 minutes.
Moran Jerabek-Willemsen (MJ-W): An important advantage of MST is that it can work with much less material than other technologies, with sample sizes as low 5 μL and a concentration of 1 nM.
How would you describe the principle behind MST technology?
MJ-W: With MST, we measure the motion of molecules in a microscopic temperature gradient that the instrument generates using an infrared laser. The motion of a molecule is dependent on parameters such as size, charge, and hydration shell. On binding of one molecule to another, whether protein to protein, protein to DNA, or protein to small molecule, at least one of the parameters that affect thermophoresis will always change, making it possible to detect the binding event. This method differs from others in that it is not dependent on size; even without a change in size, it can detect an interaction.
How does this method differ from electrophoresis?
MJ-W: Electrophoresis mainly depends on the charge of molecules, whereas thermophoresis also depends on their size and hydration shell.
SD: Anything that changes the shape or conformation of a molecule will affect the hydration shell. Therefore, this method is very sensitive and different from other methods.
How is the hydration shell of a molecule affected by a binding event?
SD: You can think of it as a water molecule being released from a binding site, or the structural elements of a protein being reorganized upon binding. These events change the interaction of a protein with water molecules.
Is the change in the hydration shell of a molecule predictable for a given type of interaction?
SD: We do not predict how many water molecules are released from a surface or what the energetics of the changes are; we instead look at relative changes. Imagine that we want to measure an interaction between two proteins. We label one of the proteins, use a constant concentration of the labeled protein, and add an increasing concentration of the binding partner. If the proteins bind to each other we will observe a change in the thermophoretic mobility. We compare the thermophoretic mobility of the bound to the unbound state of the protein.
MJ-W: Binding always results in a change in the hydration shell, so we can always compare binding to nonbinding situations. Thermophoresis can also distinguish between different types of binding interactions, such as binding to different epitopes.
What types of interactions can be measured? Are there limits to the mass of the ligand?
SD: MST is an equilibrium method. To measure binding affinity, you prepare 10 samples. The first sample contains only one of the binding partners. In the second sample you add a small amount of the second binding partner, and you gradually increase the amount of the second binding partner in the subsequent samples. You then measure the thermophoresis of the labeled protein in each of these samples. You will observe that the thermophoretic mobility of the labeled protein changes when there is a molecule bound to it. When you have measured all 10 samples you can generate a binding curve. For each sample you will know what fraction of the molecule is bound and what fraction is still free in solution without a binding partner. Any interaction that takes place in an aqueous liquid can be measured. We have measured the binding of two vesicles to each other, of nucleic acid to protein, of nucleic acid binding to another nucleic acid to form a triple helix, as well as protein–protein and protein–small molecule binding.
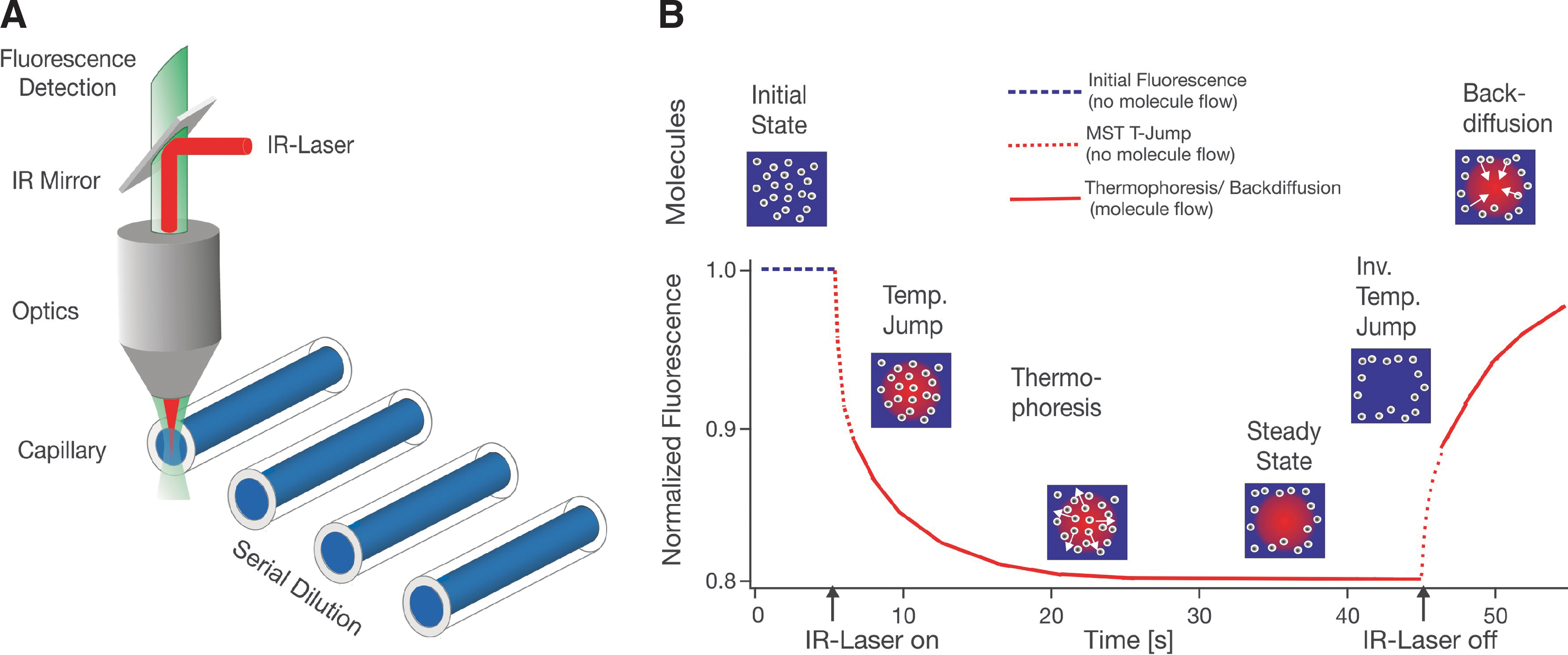
Experimental setup and MST signal. (A) MST is measured in a capillary with a total volume of 4 μL. The fluorescence within the capillary is excited and measured through the same optical element. An IR-Laser is used to locally heat the sample volume that is observed by fluorescence. In this way the T-Jump and thermophoresis is directly observed as a change in fluorescence at different time scales. (B) A typical MST signal for a given capillary is shown on the right. Initially, the molecules are homogeneously distributed and a constant “initial fluorescence” is measured. As soon as the IR-Laser is turned on, a fast T-Jump is observed, followed by thermophoretic molecule motion. The fluorescence decrease is measured for about 30 s. When the IR-Laser is turned off, an inverse T-Jump is observed, followed by the “backdiffusion” of molecules, which is purely driven by mass diffusion, allowing to deduce information on the molecule size. MST, Microscale Thermophoresis; IR, infrared; T-Jump, temperature jump.
With regard to the limits of the mass of the ligand, so far we have measured binding of a calcium ion to a labeled protein, fragments with molecular weight of 120 Da binding to proteins, and also whole ribosomes binding to proteins, as well as vesicle-containing membrane receptors binding to proteins. There is no real upper limit to the mass of the binding events we can detect. The binding partner could be a 100-nm vesicle or a 2.5-MDa ribosome.
Is there a theoretical upper limit?
MJ-W: Maybe with a binding partner bigger than 1 μm it would be more difficult.
What are the advantages and limitations of this technology? What types of measurements can/cannot be readily made (e.g., dissociation constant [Kd], on/off rates, enthalpy, etc.)?
MJ-W: Because MST can detect binding even without any change in charge or the size of the analyzed molecules, we are not restricted to labeling the smaller binding partner in an interaction. We can easily label the bigger one as well. For example, when performing a screen of compounds, you do not want to modify the small molecule with a dye; it is more convenient to label the much bigger protein. In addition, MST is very user friendly compared with other types of biophysical measurement techniques. After 2 or 3 days of training, MST users can work independently with our technology.
SD: Because this is an equilibrium method we are not measuring the rate constant of an interaction. However, it is possible to measure the kinetics of some biochemical processes: enzyme kinetics, for example, such as phosphorylation of a protein or peptide or methylation of a substrate. We can also measure slow binding reactions, such as protein–protein interactions in which binding takes place on the order of 10 minutes. In that type of reaction we can collect rate information. But kinetic measurements are not possible in the same way they are with surface plasmon resonance, with which you can measure on- and off-rates of fast binding events.
Thermophoresis can not only measure the binding affinity (dissociation constant) but also stoichiometry—how many binding sites a molecule has and how many molecules are bound to a target. By measuring at different temperatures we can also assess the energetics of an interaction, such as the delta H (change in enthalpy) and delta S (change in entropy), by doing a van't Hoff type of analysis.
MJ-W: Another important advantage of our technology is that it can detect aggregation. For example, it can be used to rule out artifacts and be sure that a binding event is not merely an artifact of aggregation. Also, this method is independent of the buffer system. We can measure in cell extracts in whole blood, serum, or even milk. This is an advantage, because the technique can be used to measure binding in an environment that is very similar to where a drug would be targeted. In some cases, binding of a small molecule takes place in an in vitro system but then does not take place in vivo. This technique could help shorten the time to new drug development by avoiding these types of discrepancies.
SD: It can work in almost any aqueous system, whether a complex bioliquid or a buffer, and even in solutions with a high percentage of dimethylsulfoxide or glycerol. Since we are creating a temperature gradient through infrared laser heating, there needs to be some water in the sample so the infrared energy is absorbed and heat is generated in the sample.
The term “immobilization free” is used to describe this method. What does that mean and what is its significance?
SD: Immobilization free has one really practical advantage, and that is the ability to set up assays and obtain affinities very quickly. With our method you only have to mix your two samples in a capillary and put them into the instrument. If you had to immobilize one of the binding partners before taking a measurement the experiment could take a few days to set up.
The other advantage is that when you couple a molecule to a surface you alter its properties and you may change its activity. Although we are still working with a fluorescent dye coupled to our sample, we do the labeling in a way that, in most cases, avoids any negative effect of the label on the activity and properties of the molecule.
MJ-W: In the past we have tested whether the affinity of a binding event is affected by the labeling. The results have shown that the deduced KD is not affected by fluorescent labeling in all systems we have analyzed, whereas it is well accepted that immobilization efficiency is highly dependent on the molecule you are using. Some proteins can be immobilized very well without affecting their activity, whereas others are difficult to immobilize.
What other types of applications do you envision for this technology?
SD: Our system can easily work with liposomes and this is an important advantage. Most drugs today target membrane proteins, and these proteins are difficult to purify and work with. Our system allows you to incorporate these membrane proteins in their natural environment, in a vesicle, and then measure the binding of small molecules or proteins to the membrane-bound receptors. Sometimes people only work with the soluble part, or they may use complex liquids containing detergents to keep the molecule in a folded state.
Please describe the instrument, its size, and its mode of operation. How are samples prepared? How are data collected and analyzed?
SD: The instrument is about 40 by 40 by 50 cm. It has a tray onto which you load the samples; you then insert the tray into the machine and push the “Close” and “Start” buttons, and the machine takes all the measurements and the software collects and analyzes the data automatically. You can put up to 16 capillaries onto each tray, and each capillary can accommodate a sample volume of less than 4 μL. The machine measures one capillary after the other. It takes about 30 seconds to capture the data from each capillary and less than 10 minutes to process an entire tray of samples.
MST is an equilibrium method and the samples are prepared the following way to measure binding affinities: You prepare 10 samples. The first sample contains only one of the binding partners. In the second sample you add a small amount of the second binding partner, and you gradually increase the amount of the second binding partner in the subsequent samples. You then measure the thermophoresis of the labeled protein in each of these samples. You will observe that the thermophoretic mobility of the labeled protein changes when there is a molecule bound to it. When you have measured all 10 samples you can generate a binding curve. For each sample you will know what fraction of the molecule is bound and what fraction is still free in solution without a binding partner.
MJ-W: The instrument can detect almost any fluorescent dye—red, blue, or green. It can also regulate the strength of the thermophoretic field, which allows you to detect various kinds of interactions.
Since some proteins bind to glass surfaces, we have developed capillaries with either a hydrophilic or hydrophobic treated surface. This has allowed us to measure interactions that would be very difficult to do with other technologies.
Can you use more than one substrate to measure a competitive binding type of event, or if a co-factor were needed for binding?
SD: You can do that. You can even measure binding reactions in which more than two binding partners are involved. Imagine that you label a DNA molecule with a fluorescent dye and you want to determine how many proteins bind to and form a complex with the DNA. We call these multicomponent reactions. You can also determine the order of binding events—whether protein A or B binds first. To do this you first take measurements with both protein A and B present and then with only one of the binding partners present. Another type of assay that is useful in drug discovery is to identify a compound that can interrupt a protein–protein interaction. You label one protein, add a certain amount of a nonlabeled protein, and allow them to form a complex. You then keep the concentration of the complex constant and add an increasing amount of the drug candidate. You will get the expected thermophoresis signal only if the compound interrupts the interaction between the two proteins. In this way you not only get information that the drug candidate is binding, but also that it is capable of interrupting the protein–protein interaction. These types of competition experiments are easily doable and have a high potential for future applications.
Do you plan to develop a device that can utilize the common microtiter plate format? Will this be 384-well and 1536-well capable? When do you expect that device to be on the market?
SD: Yes, we are working on that and we envision that a system that utilizes a microwell format will be available in 2 to 3 years. We want to design it in a way that it will be able to handle plates with up to 1536 wells.
How and when was NanoTemper founded? What are the backgrounds of the individuals who started the company?
SD: The technology was based on PhD research done in the Department of Biophysics at Ludwig-Maximilians-University. The company was founded in May 2008 by Philipp Baaske, PhD, a biophysicist, and me. Moran, who has a background in biology, joined shortly thereafter. The company initially rented rooms from the university and at first provided services to the biotech and pharmaceutical industries. In the very beginning we had only a self-built instrument available that we could not sell to customers. It looked a lot like a fluorescent microscope. In early 2010 we finished the Monolith instrument, which we have described here, and so far we have sold 45 units, mainly in Europe and some in the United States.
MJ-W: A few instruments are currently in use in the United States. One of these instruments is located in the Division of Experimental Hematology and Cancer Biology at Cincinnati Children's Hospital in Professor Yi Zheng's lab. They use the technology to characterize novel small GTPase inhibitors, which could play a role in the treatment of some malignant diseases.
We hope that in 2011 many more U.S. scientists will recognize the advantages of NanoTemper technology and join the MST community, which is growing on a daily basis.