Typing Protein–Metabolite Interactions on Nitrocellulose
Roelofs KG, Wang J, Sintim HO, Lee VT. Differential radial capillary action of ligand assay for high-throughput detection of protein-metabolite interactions. Proc Natl Acad Sci USA 2011;108:15528–15533.
Abstract: Interactions of proteins with low-molecular-weight ligands, such as metabolites, cofactors, and allosteric regulators, are important determinants of metabolism, gene regulation, and cellular homeostasis. Pharmaceuticals often target these interactions to interfere with regulatory pathways. We have developed a rapid, precise, and high-throughput method for quantitatively measuring protein–ligand interactions without the need to purify the protein when performed in cells with low background activity. This method, differential radial capillary action of ligand assay (DRaCALA), is based on the ability of dry nitrocellulose to separate the free ligand from bound protein–ligand complexes. Nitrocellulose sequesters proteins and bound ligand at the site of application, whereas free ligand is mobilized by bulk movement of the solvent through capillary action. We show here that DRaCALA allows detection of specific interactions between three nucleotides and their cognate binding proteins. DRaCALA allows quantitative measurement of the dissociation constant and the dissociation rate. Furthermore, DRaCALA can detect the expression of a cyclic-di-GMP (cdiGMP)-binding protein in whole-cell lysates of Escherichia coli, demonstrating the power of the method to bypass the prerequisite for protein purification. We have used DRaCALA to investigate cdiGMP signaling in 54 bacterial species from 37 genera and 7 eukaryotic species. These studies revealed the presence of potential cdiGMP-binding proteins in 21 species of bacteria, including 4 unsequenced species. The ease of obtaining metabolite-protein interaction data using the DRaCALA assay will facilitate rapid identification of protein-metabolite and protein-pharmaceutical interactions in a systematic and comprehensive approach.
Commentary:Proteomics and metabolomics methods almost invariably require the use of sophisticated separation and detection equipment (typically electrophoretic or liquid chromatography separation combined with high-sensitivity mass spectrometry detection), which limits their widespread use. Furthermore, these approaches, while allowing quantitation of proteins and metabolites, do not readily permit the study of the interactions between the proteome and the metabolome; that is, the spectrum of binding affinities of a metabolite against most/all proteins in the cell. Most studies (using techniques such as calorimetry, surface plasmon resonance, and equilibrium dialysis) have consisted of testing only one purified protein at a time or, in the case of protein arrays, have been strictly dependent on one's ability to prepare and maintain a large number of active protein samples. Here, the authors introduce a technique dubbed DRaCALA (differential radial capillary action of ligand assay) in which advantage is taken of the well-known property of nitrocellulose membranes to retain proteins at nearly 100% efficiency at the physical point of spotting on the surface, while at the same time allowing smaller molecules to spread away from the spot by capillary action. The authors reasoned that if a mixture of protein and a radiolabeled version of its cognate small molecule ligand (which by default will consist of equilibrium amounts of free and protein-bound ligand) is spotted onto nitrocellulose membrane and the spot is dried and imaged after capillary diffusion, the radial distribution of radioactive signal in relation to the center of the spot should produce a quantitative measure of the binding affinity of the ligand towards the protein. The proportion of ligand retained on the spot by virtue of being bound to the protein will be a function of the association constant for the particular interaction (
first figure
). To test the concept, the authors used several model systems of nucleotide ligands and their known cognate proteins of bacterial origin: the cdiGMP binder Alg44PilZ from Pseudomonas aeruginosa, the cyclic AMP receptor protein from Escherichia coli, and ATP binder nitrogen regulator B, also from E. coli. The radiolabeled version of the nucleotides were incubated with the proteins in various combinations and concentration ranges. These tests not only showed that the DRaCALA technique allowed detection of the specific protein–ligand interaction (with nonbinding controls failing to produce response), but also demonstrated that with the proper selection of the experimental conditions (such as the use of unlabeled/cold ligand as a competitor in an interaction with a preformed protein–radioligand complex) it was possible to determine the basic biophysical parameters of the interaction, such as Kd and koff (
second figure
here andFigure 3in the article). In the case of the cdiGMP-Alg44PilZ interaction, the Kd derived in the present work agreed very well with previously published data. Lastly, the authors demonstrated the detection of known and previously unreported protein–ligand interactions in whole-cell lysates in which the interactions were studied without sample purification (
third figure
). The DRaCALA method described in this article is fairly simple, requiring only the use of a PhosphorImager or a similar device, and its general principle can be adopted for testing other combinations of ligand and receptor classes in which a solid surface is available to strongly and selectively bind only one of the two categories of molecules. Contributed by Anton Simeonov.
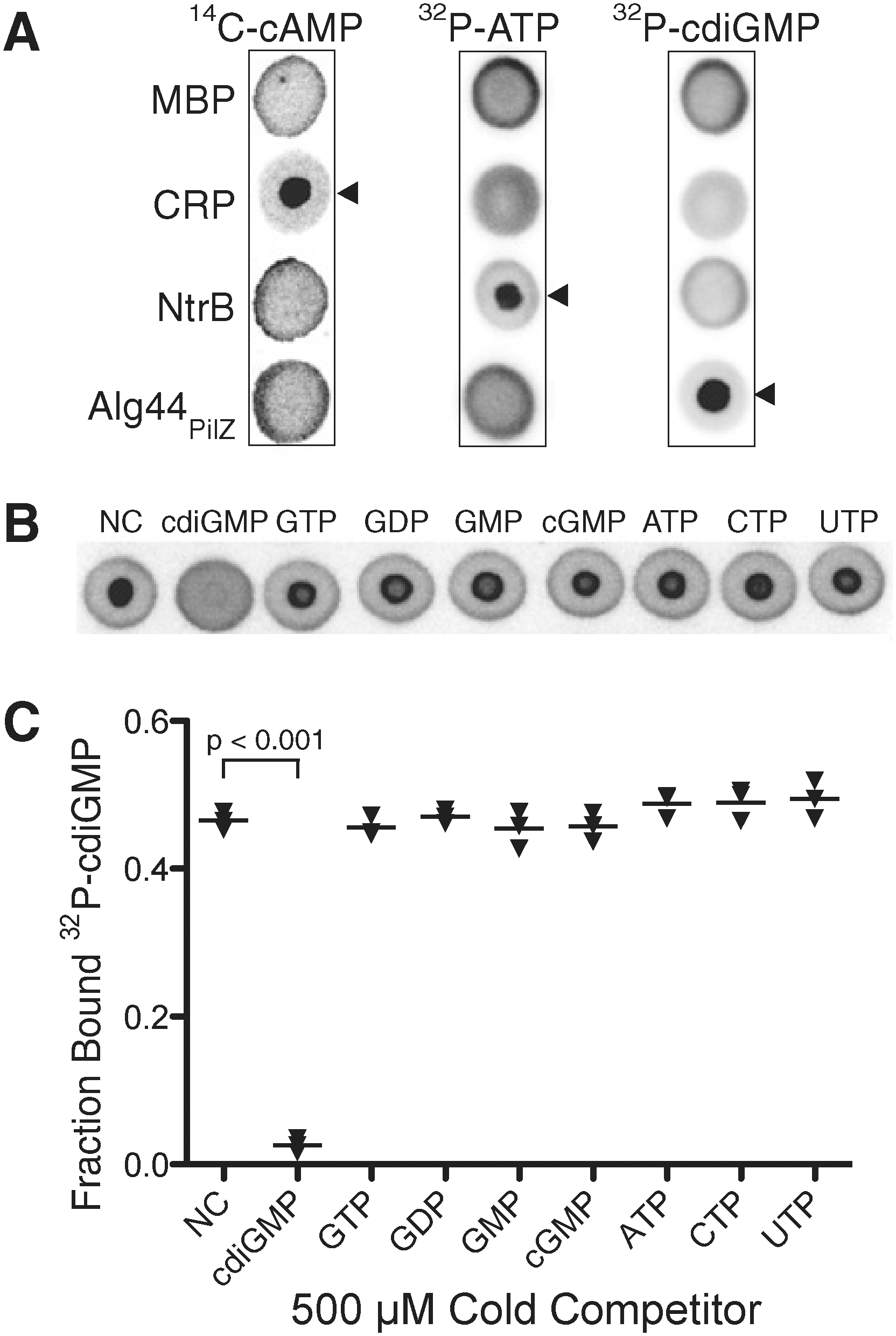
Principle of DRaCALA. (A) Schematic representation of DRaCALA assay on application of protein-ligand mixture onto nitrocellulose and capillary action. Protein (P), ligand (L), and protein–ligand complex (P•L) distribution during the assay is shown. (B) Equations are used to analyze DRaCALA data for FB for purified proteins. An explanation of the apparent edge effect at the capillary migration front is provided in Fig. S3 of the article's Supporting Information.
Detection of specific protein-ligand interactions by DRaCALA. (A) DRaCALA images of interactions between purified proteins (20 μM) incubated with 500 nM 14C-cAMP, 4 nM [32P]ATP, or 4 nM 32P-cdiGMP. Protein–ligand mixtures were spotted on nitrocellulose and allowed to dry before imaging using a Fuji FLA7100 PhosphorImager. Cognate protein–nucleotide combinations are indicated by arrowheads. MBP was used as a negative control. (B) DRaCALA images of competition assays assessing the ability of 1 mM indicated cold nucleotides to compete with binding interactions between 4 nM 32P-cdiGMP and 2.5 μM HisMBP-Alg44PilZ. (C) Graph of FB for each sample in (B), with averages indicated by a horizontal bar. P values were determined by a Student t test for significant differences compared with the NC control for three independent experiments. The Itotal of each DRaCALA spot in (A) and (B) is provided in Tables S1 and S2 of the article's Supporting Information. NC, no competitor.
Detection of specific protein-ligand interaction in whole-cell lysates by DRaCALA. (A) Images of Alg44PilZ interaction with 4 nM 32P-cdiGMP and either 1 mM cold cdiGMP or GTP with purified proteins or when expressed in E. coli BL21(DE3). (B) Graph of 32P-cdiGMP binding by whole-cell lysate samples (○) and purified proteins (▾) in (A), with the average indicated by a horizontal bar. P values were determined by a Student t test for significant differences compared with the no-competitor control for three independent experiments. The Itotal of each DRaCALA spot in (B) is provided in Tables S5 and S6 of the article's Supporting Information. (C) Graph of 32P-cdiGMP binding by purified MBP-Alg44PilZ, purified MBP-Alg44PilZ added to BL21 whole-cell lysates, and whole-cell lysates of BL21(DE3) overexpressing MBP-Alg44PilZ. Protein concentrations were determined by separation on SDS/PAGE and staining with Coomassie blue (Fig. S4 in the article's Supporting Information). C, cdiGMP; G, GTP.
Shopping for New Treatments at a High-End Department Store
Li YY, An J, Jones SJM. A computational approach to finding novel targets for existing drugs. PLoS Comput Biol 2011;7:e1002139.
Abstract: Repositioning existing drugs for new therapeutic uses is an efficient approach to drug discovery. We have developed a computational drug repositioning pipeline to perform large-scale molecular docking of small molecule drugs against protein drug targets, in order to map the drug-target interaction space and find novel interactions. Our method emphasizes removing false positive interaction predictions using criteria from known interaction docking, consensus scoring, and specificity. In all, our database contains 252 human protein drug targets that we classify as reliable-for-docking as well as 4621 approved and experimental small molecule drugs from DrugBank. These were cross-docked, then filtered through stringent scoring criteria to select top drug-target interactions. In particular, we used MAPK14 and the kinase inhibitor BIM-8 as examples where our stringent thresholds enriched the predicted drug-target interactions with known interactions up to 20 times compared to standard score thresholds. We validated nilotinib as a potent MAPK14 inhibitor in vitro (IC50 40 nM), suggesting a potential use for this drug in treating inflammatory diseases. The published literature indicated experimental evidence for 31 of the top predicted interactions, highlighting the promising nature of our approach. Novel interactions discovered may lead to the drug being repositioned as a therapeutic treatment for its off-target's associated disease, added insight into the drug's mechanism of action, and added insight into the drug's side effects.
AuthorSummary: Most drugs are designed to bind to and inhibit the function of a disease target protein. However, drugs are often able to bind to “off-target” proteins due to similarities in the protein binding sites. If an off-target is known to be involved in another disease, then the drug has potential to treat the second disease. This repositioning strategy is an alternate and efficient approach to drug discovery, as the clinical and toxicity histories of existing drugs can greatly reduce drug development cost and time. We present here a large-scale computational approach that simulates three-dimensional binding between existing drugs and target proteins to predict novel drug-target interactions. Our method focuses on removing false predictions, using annotated ‘known’ interactions, scoring and ranking thresholds. Thirty-one of our top novel drug-target predictions were validated through literature search, and demonstrated the utility of our method. We were also able to identify the cancer drug nilotinib as a potent inhibitor of MAPK14, a target in inflammatory diseases, which suggests a potential use for the drug in treating rheumatoid arthritis.
Commentary:There has been a wave of interest in drug repurposing recently. Drugs often have multiple targets, and these secondary targets may provide opportunities to rapidly expand the use of a drug approved by the Food and Drug Administration (FDA) to alternative indications. One major benefit of drug repurposing is that the drug has already been evaluated for safety and has been approved for clinical use. With the extremely high cost of developing a drug and moving it through the preclinical and clinical pipeline, researchers are exploring ways, such as drug repurposing, to speed up the process and decrease the cost of drug development, while maintaining high safety standards. New resources have been developed to enable drug repurposing, such as the NIH Chemical Genomics CenterPharmaceutical Collection (Huang et al., Sci Transl Med 2011;3:1; seehttp://tripod.nih.gov/npc). In addition to in vitro screening of targets against a collection of FDA-approved drugs, virtual screening is also a viable way to identify potential drug–target interactions. Earlier this year, a computational approach was used to determine from the X-ray crystallographic structures of pairs of targets with a common ligand, whether a new ligand that bound only one of the two targets would be expected to bind the other (Kalinina et al., PLoS Comput Biol 2011;7:e1002043). One major benefit of this method was that the ability to predict new protein–chemical interactions grows more rapidly than the number of available structures. The authors of the new computational study described herein used large-scale molecular docking of 252 human protein drug targets with 4621 approved and experimental small molecule drugs. The authors point out the importance of the ranking and scoring thresholds in limiting the number of false positives that can plague virtual screening campaigns. They were able to reproduce known binding conformations with good RMSD values in most cases. The 252 targets were selected because one or more of their known inhibitors scored well in the docking function used here. A total of 426 targets were excluded from the analysis because drugs known to interact with them did not dock well with the docking function used here, indicative of the current limitations in docking methodology. Binding site flexibility is an important aspect in many proteins, such as kinases. The authors used multiple static structures for docking when they were available (the 252 targets were represented by 2923 binding pockets) to represent the flexibility of the protein and to provide a greater chance of finding a docking pose for a given compound. For in vitro testing of selectivity, usually related proteins would be tested; i.e., a new kinase inhibitor would be tested for selectivity against the kinome. A potential advantage of the approach here would be the testing of the drug against not just the functional relatives but rather a wider swatch of the proteome, ideally the entire druggable proteome. The
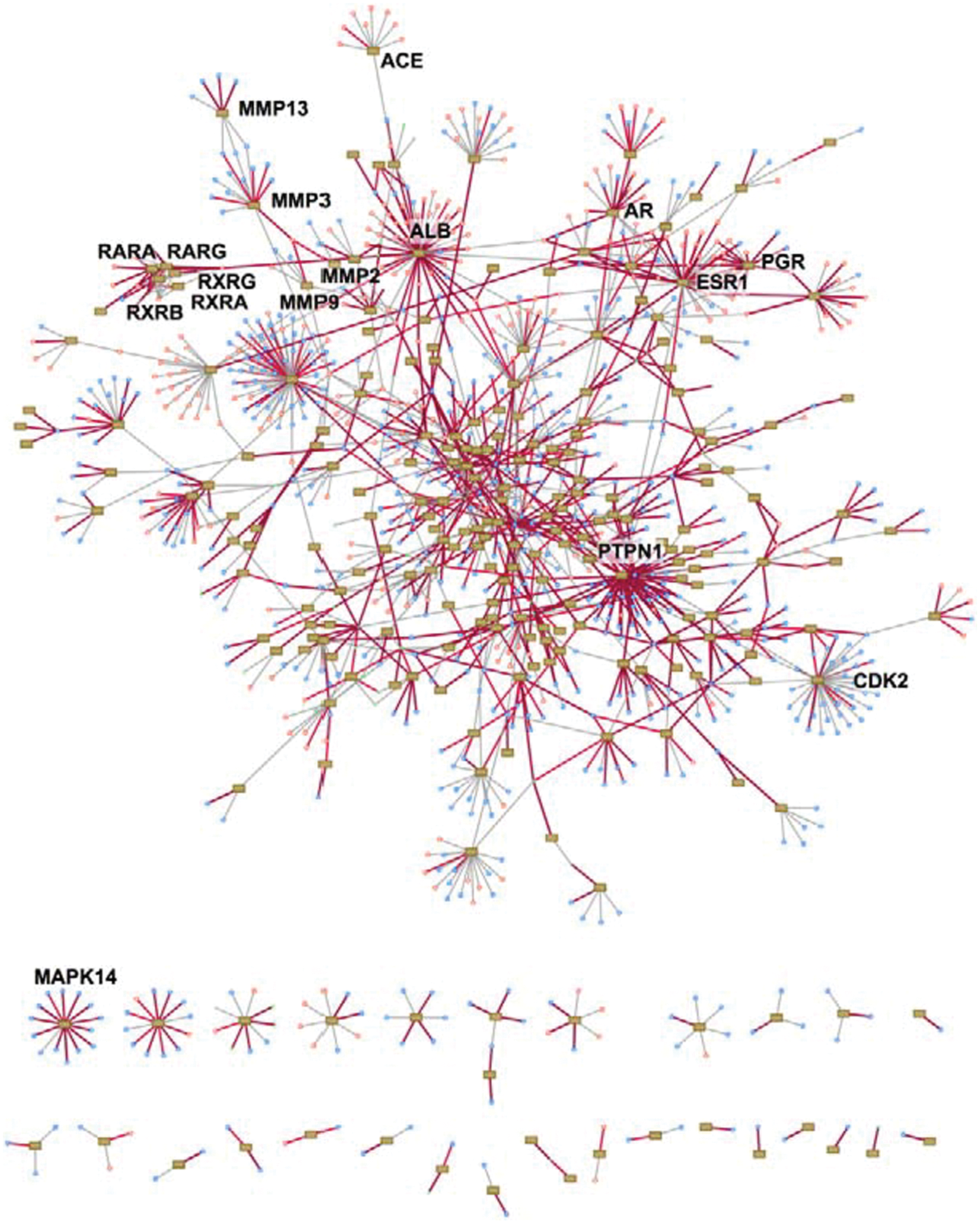
first figure
shows the drug–target network, with targets such as the phosphatase PTPN1 having the most known drug interactions. Their work identified a drug–target interaction of nilotinib and MAPK14 (also known as p38 alpha), which was confirmed in vitro (IC50=40 nM). MAPK14 is a target for the development of anti-inflammatory drugs for the treatment of arthritis. The
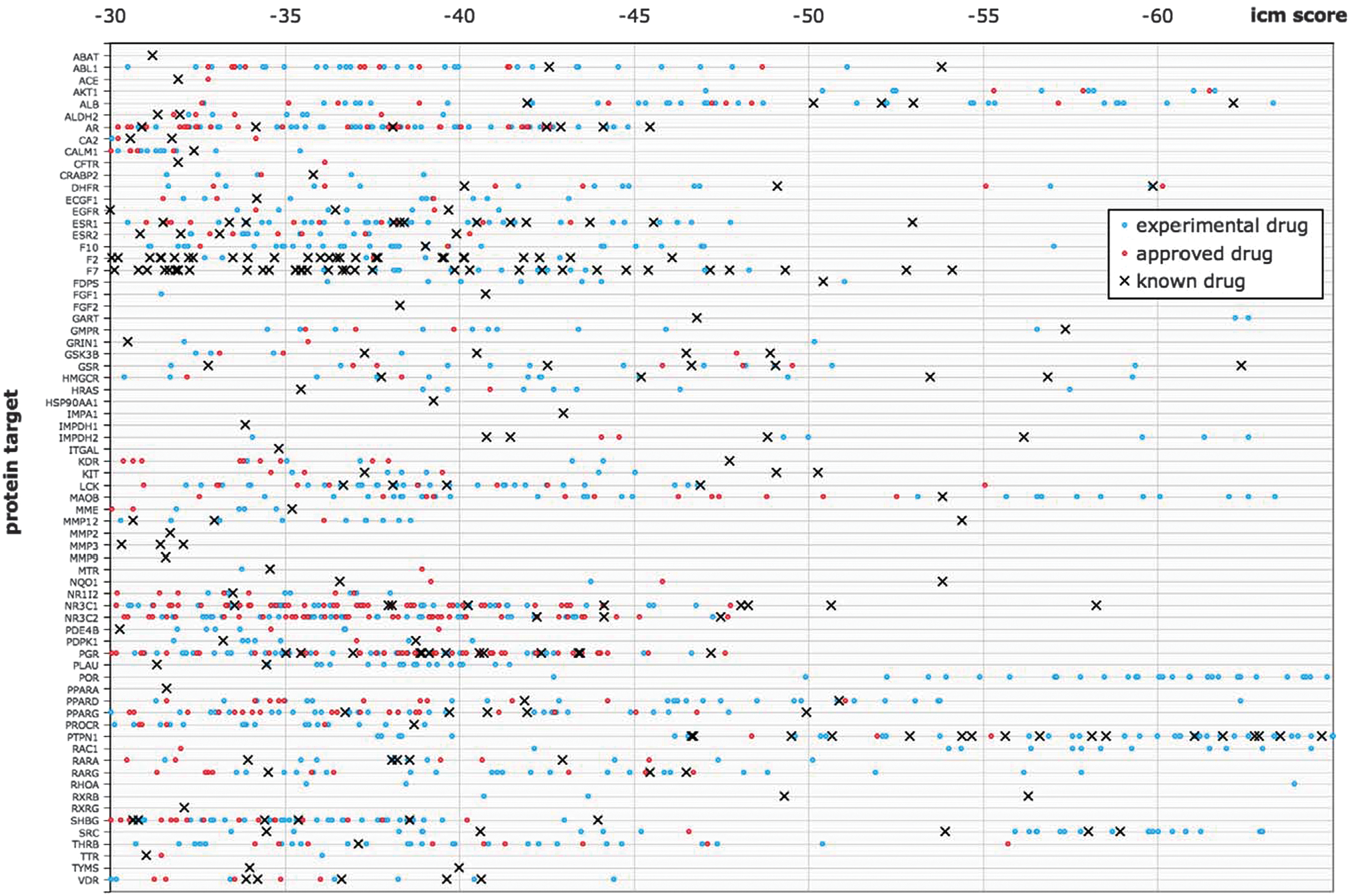
second figure
shows the predicted protein–drug interactions. There were many interactions predicted. Some could be confirmed from the literature but others would require in vitro testing to validate the docking prediction. One difficulty in using experimental drugs as part of this project was that these compounds were often not readily available, and so when these scored as well as or better than known drugs, the result was not validated in vitro. Ideally, in vitro screening and in silico screening will complement each other and will enable rapid drug repurposing. Contributed by Mindy I. Davis.
Network of known protein–drug interactions. Proteins are shown as rectangular boxes (nodes), drugs are shown as pink (approved) and blue (experimental) circles, and edges represent known interactions annotated by DrugBank. Edges colored red denote known interactions that were docked with a good internal coordinate modeling (ICM) score. Here we show only the 252 proteins for which at least one known drug docked well—the “reliable-for-docking” set. The proteins at the bottom of the graph are not connected to other proteins through shared binding drugs.
Quantitative interaction map of drugs docked to protein targets, according to their ICM docking score. Each protein is represented by a column, on which a black cross denotes a known drug docked to the target, a red dot denotes an approved drug docked to the target, and a blue dot denotes an experimental drug docked to the target. Only the top predictions for established drug targets (at least one known approved drug) that docked with a score passing the consensus threshold and had a protein-rank ≤5 are shown.
A Little Mass Spec Goes a Long Way
Robbins DW, Hartwig JF. A simple, multidimensional approach to high-throughput discovery of catalytic reactions. Science 2011;333:1423–1427.
Abstract: Transition metal complexes catalyze many important reactions that are employed in medicine, materials science, and energy production. Although high-throughput methods for the discovery of catalysts that would mirror related approaches for the discovery of medicinally active compounds have been the focus of much attention, these methods have not been sufficiently general or accessible to typical synthetic laboratories to be adopted widely. We report a method to evaluate a broad range of catalysts for potential coupling reactions with the use of simple laboratory equipment. Specifically, we screen an array of catalysts and ligands with a diverse mixture of substrates and then use mass spectrometry to identify reaction products that, by design, exceed the mass of any single substrate. With this method, we discovered a copper-catalyzed alkyne hydroamination and two nickel-catalyzed hydroarylation reactions, each of which displays excellent functional-group tolerance.
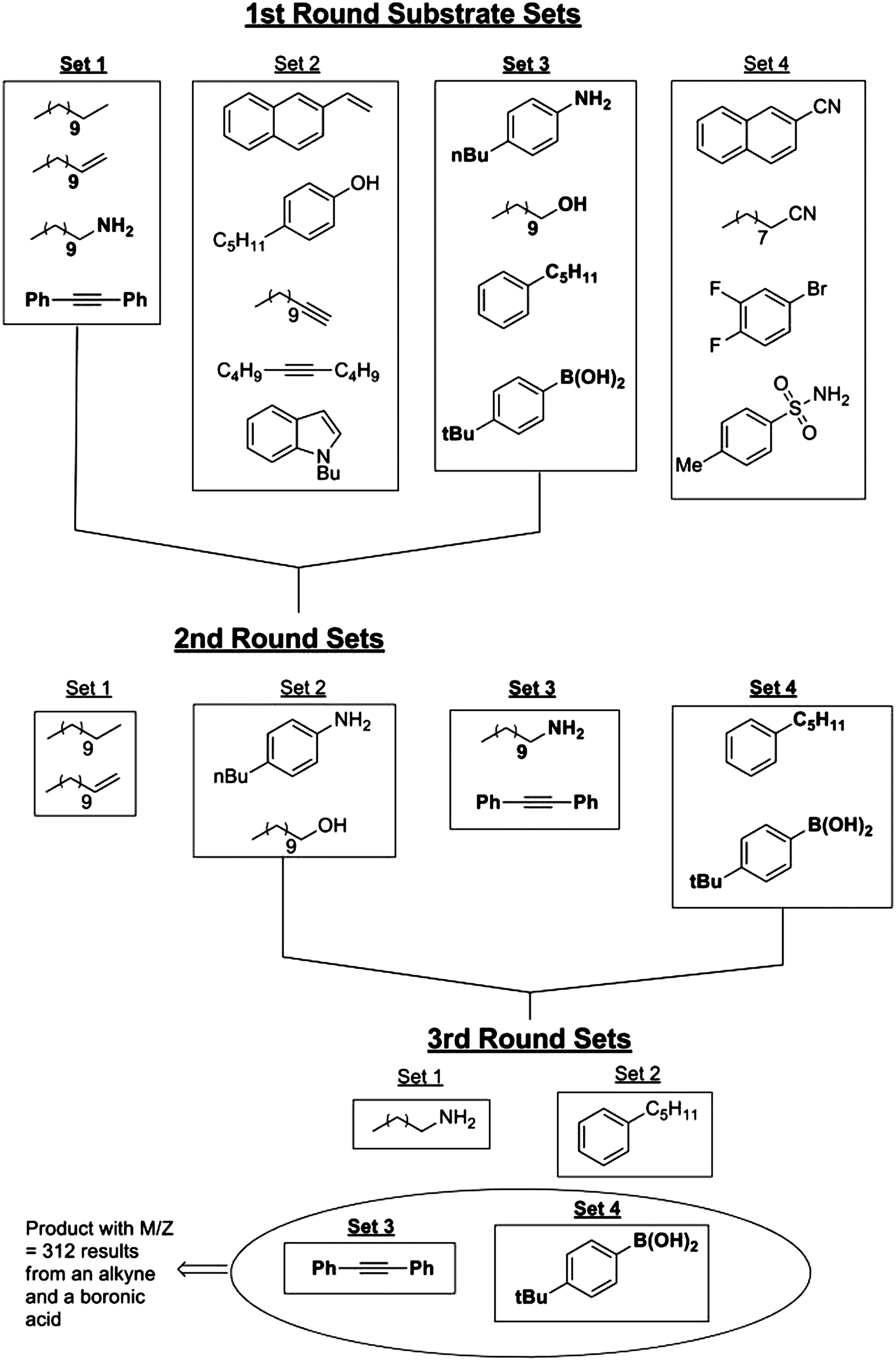
Commentary:Testing combinations of reactants and candidate catalysts with the aim of discovering a new reaction or a more optimal catalyst has been done over the past decades in both academic and industrial settings. Almost invariably this process has required the use of robotic liquid dispensing equipment (to accommodate the large number of individual samples involved in the scheme) and/or the application of complicated deconvolution steps. To make such matrix testing easily adoptable by any typical organic synthesis laboratory, Robbins and Hartwig present a dramatically simplified workflow: candidate substrates, ligands, and catalysts are mixed by simple pipetting into a 96-well plate and the outcome of the matrix test is determined by a simple mass spectrometry analysis of a small fraction of the samples on the plate. The authors achieve this by testing a pool of substrates dispensed in each reaction well and combining them with one individual ligand per column and one catalyst candidate per row; thus, in the proof-of-principle experiments, a pool of 17 substrates was tested against 12 ligands and eight metal catalysts (
first figure
), resulting in over 50,000 possible combinations. During the initial phase of the deconvolution, a total of 20 samples were tested by gas chromatography–mass spectroscopy (GC-MS) and electrospray ionization mass spectrometry (ESI-MS), the two techniques being used in order to capture both potential polar and nonpolar reaction products. The initial 20 samples corresponded to eight mixtures of aliquots from each row and 12 mixtures representing the 12 columns. The appearance of novel mass peaks from any of these 20 samples was flagged as a successful reaction. Further deconvolution involved judicious binary splitting of the reactants into smaller subsets and testing the simplified reaction pools (
second figure
). Using this strategy, the authors ended up conducting only an additional 10 reactions followed by the corresponding GC-MS and ESI-MS analyses. The study is particularly noteworthy because it not only provides proof-of-principle results to support the simplified matrix testing and deconvolution strategy but also shows novel reactions with unexpected stereochemistry outcome. Additionally, new catalysts based on the more abundant and significantly cheaper earth-abundant metals such as nickel and manganese were identified. Contributed by Anton Simeonov.
Contents of a single well in the multidimensional experiments for reaction discovery. The combination of 17 substrates was placed into each reaction well. Twelve ligands were dispensed, one into each well of a column, and eight metal catalyst precursors were dispensed, one into each well of a row. The plate was sealed and heated at 100°C for 18 hours. After this time, the contents of the wells in the plate were analyzed by mass spectrometry. The number of substrates is arbitrary; the 17 substrates contain a representative set, not a comprehensive set, of typical organic functional groups. A group of catalysts derived from Mn, Fe, Cr, Co, Cu, Ni, and W was chosen because of its abundance and low cost. In addition, we examined catalysts derived from Ru and Mo because these are inexpensive relative to the more precious metals, Yb as a representative f-block metal, and Au because of its wide range of reactivity that has recently been uncovered. The ligands we combined with these metals included common phosphines and amines, as well as less explored phosphine oxides, phosphine sulfides, and amidinates (Table S1 in the article's Supporting Online Material). Excess of the metal complexes were used in this system to alleviate poisoning all of the potential catalysts by one substrate. Reactions discovered in such a system would be rendered catalytic after initial identification of the transformation and metal-ligand combination that induces the transformation. The 17 substrates, in combination with catalysts derived from 15 metal centers and 23 ligands or the absence of a ligand, correspond to more than 50,000 reactions. These reactions were conducted in a few days, after developing our protocol. Bu, butyl; tBu, tert-butyl; Me, methyl; Ph, phenyl.
Deconvolution strategy to identify coupling partners for products observed in high-throughput reaction discovery.
Sequencing in the Dark
Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M, Leamon JH, Johnson K, Milgrew MJ, Edwards M, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011;475:348–352.
Abstract: The seminal importance of DNA sequencing to the life sciences, biotechnology and medicine has driven the search for more scalable and lower-cost solutions. Here we describe a DNA sequencing technology in which scalable, low-cost semiconductor manufacturing techniques are used to make an integrated circuit able to directly perform non-optical DNA sequencing of genomes. Sequence data are obtained by directly sensing the ions produced by template-directed DNA polymerase synthesis using all-natural nucleotides on this massively parallel semiconductor-sensing device or ion chip. The ion chip contains ion-sensitive, field-effect transistor-based sensors in perfect register with 1.2 million wells, which provide confinement and allow parallel, simultaneous detection of independent sequencing reactions. Use of the most widely used technology for constructing integrated circuits, the complementary metal-oxide semiconductor (CMOS) process, allows for low-cost, large-scale production and scaling of the device to higher densities and larger array sizes. We show the performance of the system by sequencing three bacterial genomes, its robustness and scalability by producing ion chips with up to 10 times as many sensors and sequencing a human genome.
Commentary:The field of whole-genome sequencing has been experiencing phenomenal growth and transformation. Benchtop instruments capable of sequencing entire genomes within a fraction of the time the process took a year or two ago have become reality. Most of the so-called Next-Gen sequencing instruments are still based on different versions of fluorescence detection, which in turn requires the use of specialized fluorescent reagents in combination with complex optical systems, both of which contribute to keeping the platforms relatively expensive. The instrument and detection system described by Rothberg and colleagues is unique in its avoidance of light-based detection. Instead, the team from Ion Torrent, presently Life Technologies, makes use of a complementary metal-oxide type of semiconductor sensing device. The basic premise of the technology is that upon polymerase-catalyzed primer extension, a proton is liberated as a result of the deoxynucleotide triphosphate hydrolysis; in turn, the localized change in pH triggered by the liberated proton is detected as a voltage change by the semiconductor sensor (
first figure
). The team leverages the capabilities of today's semiconductor manufacturing to deliver very cheap, yet highly reliable, complementary metal-oxide semiconductor (CMOS) integrated circuits in a wide range of sizes and features. To enable high-throughput sequencing, a multi-well chip was developed that incorporated the CMOS on the bottom, while a 3-μm-thick patterned dielectric layer on top was used to form an array of microscopic wells. The entire chip was incorporated within a polycarbonate flow cell, thus allowing facile sample loading and washing (
second figure
). The genomic DNA was fragmented to prepare for sequencing and used to make adaptor-ligated library, which was clonally amplified onto beads. The beads carrying template strands were mixed with sequencing primers and polymerase and loaded onto the chip. The size of the bead particle was chosen to enable a sufficient number of template copies to be loaded into each well in order to ensure adequate signal. During the sequencing run, the four nucleotides were fed in a sequential manner, with brief washes in between. Upon incorporation of one or more of the correct bases, one or more protons were liberated and the magnitude of the voltage output of the CMOS detector was proportional to the number of incorporated bases; the voltage changes produced by each well were then used to perform base calling (
third figure
). In multiple validation experiments performed on both prokaryotic and eukaryotic genomes, the authors noted a high degree of accuracy and routine acquisition lengths of at least 100 bases. In addition, the team sequenced the whole genome of one male donor and compared the sequence obtained with the present technology to that derived from the ABI SOLiD platform: the sequences were cross-validated at greater than 99.9%. The new “photon-free” technology is expected to be a welcome addition to the arsenal of sequencing groups and other organizations. Further improvements to the system, as noted by the authors, include optimization of the template preparation and signal processing protocols, with the aim to increase the yield of usable reads from the sensors within the chip. Contributed by Anton Simeonov.
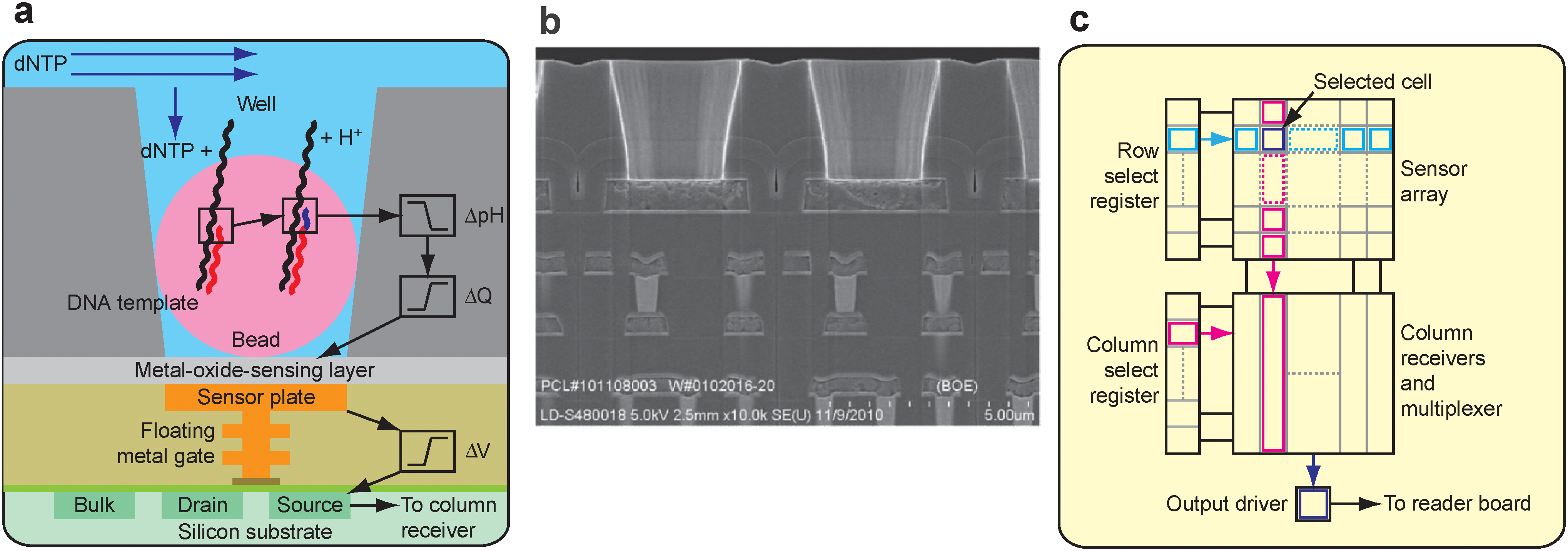
Sensor, well and chip architecture. (a) A simplified drawing of a well, a bead containing DNA template, and the underlying sensor and electronics. Protons (H+) are released when nucleotides (dNTP) are incorporated on the growing DNA strands, changing the pH of the well (ΔpH). This induces a change in surface potential of the metal-oxide-sensing layer, and a change in potential (ΔV) of the source terminal of the underlying field-effect transistor. (b) Electron micrograph showing alignment of the wells over the ISFET metal sensor plate and the underlying electronic layers. (c) Sensors are arranged in a two-dimensional array. A row select register enables one row of sensors at a time, causing each sensor to drive its source voltage onto a column. A column select register selects one of the columns for output to external electronics.
Wafer, die and chip packaging. (a) Fabricated CMOS 8′′ wafer containing approximately 200 individual functional ion sensor die. (b) Unpackaged die, after automated dicing of wafer, with functional regions indicated. (c) Die in ceramic package wire bonded for electrical connection, shown with moulded fluidic lid to allow addition of sequencing reagents.
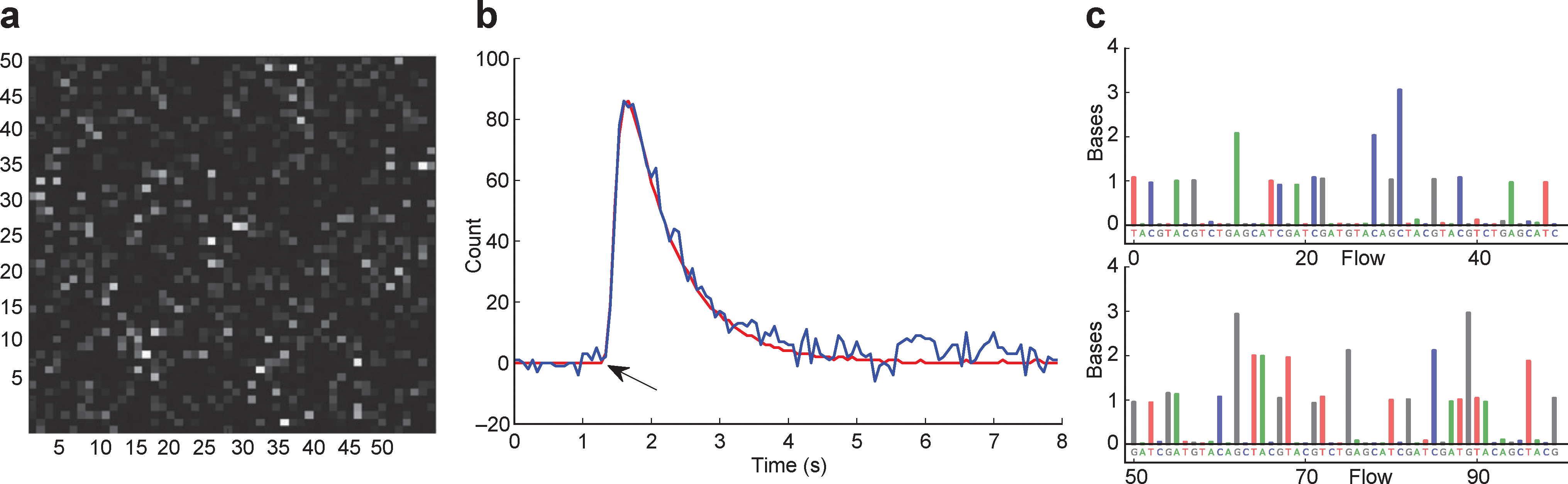
Data collection and base calling. (a) A 50×50 region of the ion chip. The brightness represents the intensity of the incorporation reaction in individual sensor wells. (b) 1-nucleotide incorporation signal from an individual sensor well; the arrow indicates start of incorporation event, with the physical model (red line) and background corrected data (blue line) shown. (c) The first 100 flows from one well. Each colored bar indicates the corresponding number of bases incorporated during that nucleotide flow.
Nuclear ROS Probe
Dickinson BC, Tang Y, Chang Z, Chang CJ. A nuclear-localized fluorescent hydrogen peroxide probe for monitoring sirtuin-mediated oxidative stress responses in vivo. Chem Biol 2011;18:943–948.
Abstract: Hydrogen peroxide (H2O2) can serve as a beneficial signaling agent or toxin depending on its concentration and location within a cell or organism. Methods to measure the localized accumulation of H2O2 in living specimens remain limited. Motivated to meet this need, we have developed a nuclear-localized fluorescent probe for H2O2, Nuclear Peroxy Emerald 1 (NucPE1), to selectively interrogate ROS fluxes within this sensitive organelle. NucPE1 selectively accumulates in the nuclei of a variety of mammalian cell lines as well as in whole model organisms like Caenorhabditis elegans, where it can respond to subcellular changes in H2O2 fluxes. Moreover, in vivo NucPE1 imaging reveals a reduction in nuclear H2O2 levels in worms overexpressing sir-2.1 compared with wild-type congeners, supporting a link between this longevity-promoting sirtuin protein and enhanced regulation of nuclear ROS pools.
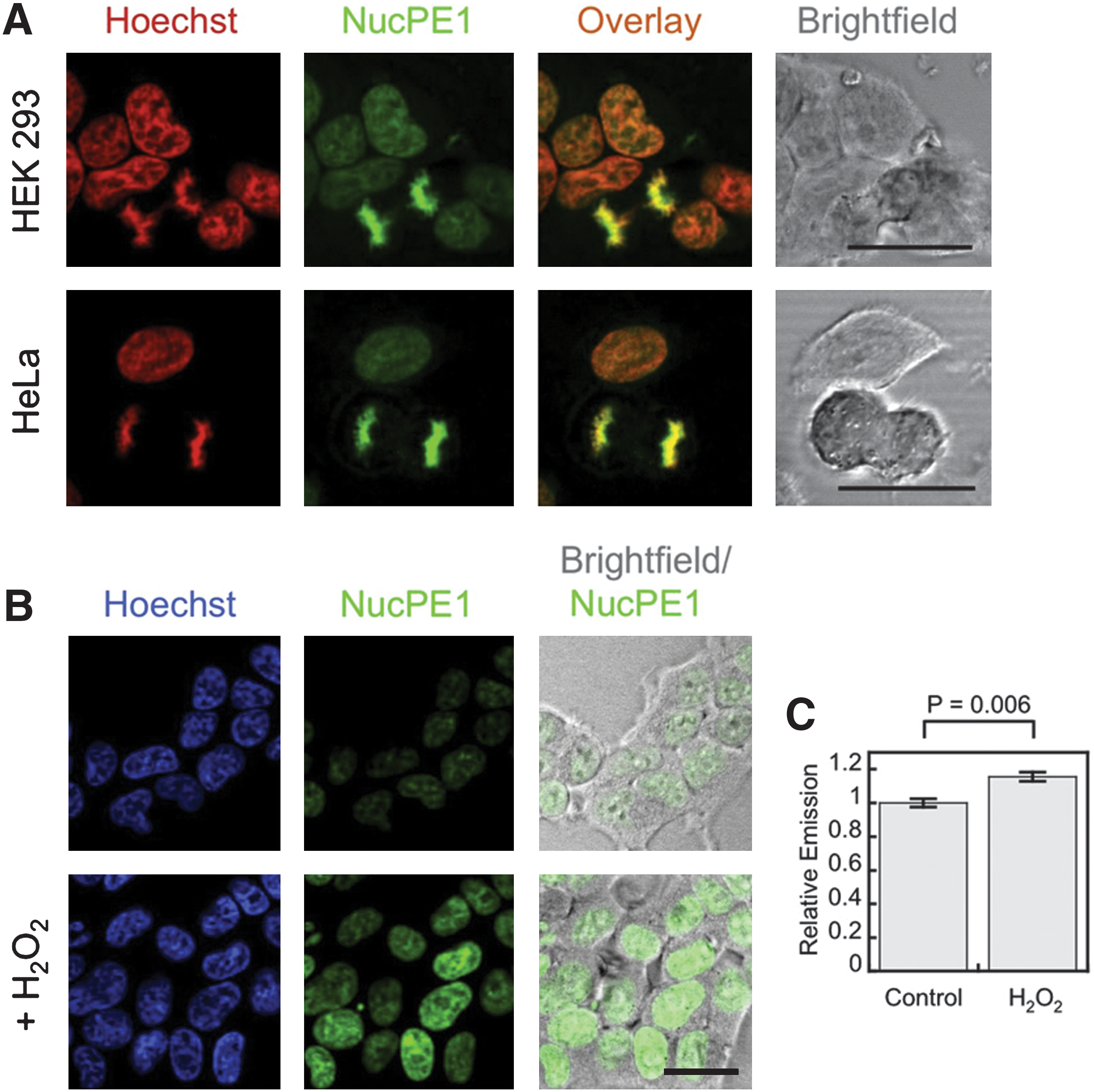
Commentary:Redox processes can be a bane of compound screening efforts employing biochemical formats due to interference with the detection technology or modification of the target protein. However, redox-based mechanisms have an important role in cellular signaling with some of these producing reactive oxygen species (ROS). While ROS such as hydrogen peroxide H2O2 can play a beneficial role in cells, H2O2 can also lead to oxidative damage of cellular components. Genomic DNA can be irreversibly damaged by H2O2 present in the nucleus, and there is a need for probes that can detect nuclear localized H2O2. The authors describe here the synthesis and application of a nuclear localized peroxide probe—Nuclear Peroxy Emerald 1 (NucPE1). Discovery of the nuclear localized characteristic of NucPE1 was serendipitous because it occurred during the screening of various rhodol compounds to act as H2O2 sensors and a boronate derivative was found to collect in nuclei of cells and co-stain with the nuclear stain Hoechst 33342 (see
figure
). NucPE1 absorbs at 468 nm and 490 nm and emits weakly at 530 nm (Φ=0.117). Reaction with H2O2 results in a large increase in fluorescence at 530 nm (Φ=0.626). This probe was found to be selective for H2O2 over a panel of six ROS including superoxide and nitric oxide. Additionally, a derivative of NucPE1 without the boronate showed that this group is required for nuclear localization, and the product of the peroxide reaction does not interfere so that only the nuclear localized signal is obtained. The mechanism of nuclear staining by NucPE1 is not completely understood; however, DNA is likely involved because it was observed that cells in anaphase still showed staining. The authors also used NucPE1 in a C. elegans aging model in which, following loading with NucPE1 and Hoescht 33342 and dosing with 10 μM H2O2, nuclear localized peroxide staining could be imaged in vivo. NucPE1 should be useful a tool for exploring the role of peroxide in biological processes. Contributed by Doug Auld.
Localization and turn-on of NucPE1 in cell culture. (A) HEK293 or HeLa cells loaded with 5 μM NucPE1 and 1 μM Hoechst 33342 for 15 min in DPBS, washed with DPBS, incubated 20 min in DPBS, and imaged. 25 μm scale bar shown. (B) HEK293 cells loaded with 5 μM NucPE1 and 1 μM Hoechst 33342 for 15 min in DPBS, washed with DPBS, incubated 20 min in DPBS with carrier or 100 μM H2O2, and imaged. 20 μm scale bar shown. (C) Quantification of NucPE1 fluorescence for experiment as shown in (B). Statistical analyses were performed with a Student's t test (n = 4) and error bars are ±SEM. See also Figures S1 and S2 in the article's Supplemental Information. DPBS, Dulbecco's phosphate-buffered saline.
Fluorescent Proteins' Latest Fruit
Subach OM, Patterson GH, Ting L-M, Wang Y, Condeelis JS, Vladislav VV. A photoswitchable orange-to-far-red fluorescent protein, PSmOrange. Nat Methods 2011;8:771–777.
Abstract: We report a photoswitchable monomeric Orange (PSmOrange) protein that is initially orange (excitation, 548 nm; emission, 565 nm) but becomes far-red (excitation, 636 nm; emission, 662 nm) after irradiation with blue-green light. Compared to its parental orange proteins, PSmOrange has greater brightness, faster maturation, higher photoconversion contrast and better photostability. The red-shifted spectra of both forms of PSmOrange enable its simultaneous use with cyan-to-green photoswitchable proteins to study four intracellular populations. Photoconverted PSmOrange has, to our knowledge, the most far-red excitation peak of all GFP-like fluorescent proteins, provides diffraction-limited and super-resolution imaging in the far-red light range, is optimally excited with common red lasers, and can be photoconverted subcutaneously in a mouse. PSmOrange photoswitching occurs via a two-step photo-oxidation process, which causes cleavage of the polypeptide backbone. The far-red fluorescence of photoconverted PSmOrange results from a new chromophore containing N-acylimine with a co-planar carbon-oxygen double bond.
Commentary:Orange or red fluorescence are desirable detection wavelengths because these spectral regions possess lower background and interferences than blue fluorescence. In this article, an orange-fluorescent protein—mOrange—was used to develop a photoswitchable fluorescent protein with stable and bright red fluorescence. mOrange exhibits fluorescence with excitation and emission peaks at 548 nm and 565 nm, respectively. Variants of mOrange were created using error-prone polymerase chain reaction (PCR) and finally a variant containing six mutations (PSmOrange) was found that showed photoswitching using blue-green light resulting in fluorescent protein with excitation and emission maxima at 636 nm and 662 nm, respectively. PSmOrange was shown to be applicable to techniques such as photoactivated localization microscopy. PSmOrange is monomeric and showed toxicity in either cells or mice that is similar to mEGFP. The red fluorescence of the photoswitched protein enables in vivo imaging. The authors tested photoswitching of PSmOrange directly in mice using a 535-nm laser. The far-red emission of the photoconverted protein was visible in mice following excitation with red light (see
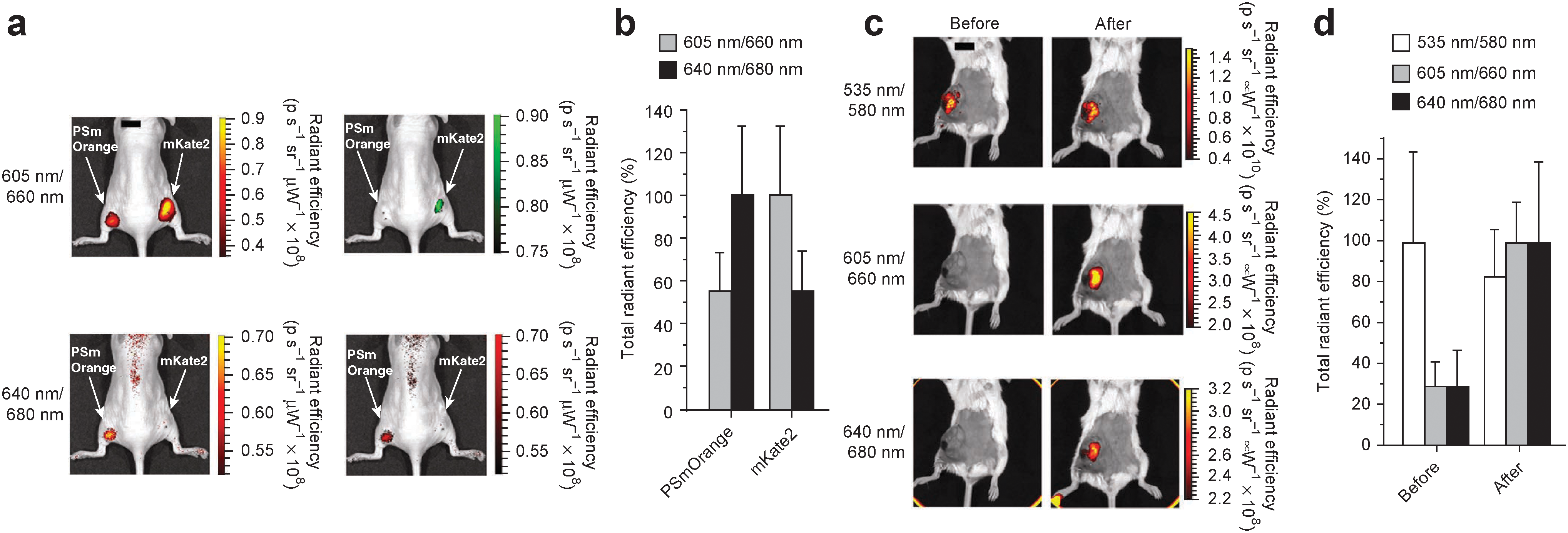
figure
). The structure of the chromophore was investigated as well. Photoswitching involves cleavage within the peptide-linked chromophore to yield 19-kDa and 9-kDa fragments resulting in the far-red chromophore. The structure of the far-red chromophore was similar to what is observed in DsRed, but unlike DsRed, the N-acylimine group (containing two double bonds: C=N-C=O, which yields the bathochromic shift) has all the bonds contained within the chromophore plane. This yields a greater bathochromic shift and provides a brighter fluorescence. A two-step photo-oxidative cleavage mechanism of the polypeptide is proposed. PSmOrange shows brighter fluorescence than other red fluorescent proteins, and the red-shifted fluorescence enables multiplexing with green and blue fluorescent proteins. PSmOrange should be a nice addition to the fluorescent protein toolbox. Contributed by Doug Auld.
Imaging of PSmOrange in vivo. (a) Whole body images of a mouse injected intramuscularly with 106 cells expressing mKate2 (right flank) or photoconverted PSmOrange (left flank). Images on the right are copies of images on the left but with fluorescence signals shown in green (605 nm excitation channel) and red (640 nm excitation channel) pseudocolors. (b) Total radiant efficiency corresponding to data in (a). Total radiant efficiency of mKate2 was set as 100% in 605/30 nm excitation channel, and total radiant efficiency of PSmOrange was set as 100% in 640/30 nm excitation channel. Error bars, s.d. (n=3). (c) Whole body images at the indicated excitation and emission wavelengths before and after photoconversion of PSmOrange in mammary tumor xenograft in a mouse. (d) Total radiant efficiency corresponding to data in (c). Maximal total radiant efficiency in each channel was normalized to 100%. Error bars, s.d. (n=4). In (a–d) the first and second numbers in 535 nm/580 nm, 605 nm/660 nm and 640 nm/680 nm indicate excitation and emission wavelengths, respectively. Scale bars (black, top of images in a,c), 1 cm.
What's New With Cell-Penetrating Peptides?
Svensen N, Díaz-Mochón JJ, Dhaliwal K, Songsak Planonth, Michael Dewar, J. Douglas Armstrong, and Mark Bradley Screening of a combinatorial homing peptide library for selective cellular delivery. Angew Chem Int Ed 2011;50:6133–6136.
No abstract.
Lättig-Tünnemann G, Prinz M, Hoffmann D, Behlke J, Palm-Apergi C, Morano I, Herce HD, Cardoso MC. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides.Nat Commun2011;2:453.
Abstract: In addition to endocytosis-mediated cellular uptake, hydrophilic cell-penetrating peptides are able to traverse biological membranes in a non-endocytic mode termed transduction, resulting in immediate bioavailability. Here we analysed structural requirements for the non-endocytic uptake mode of arginine-rich cell-penetrating peptides, by a combination of live-cell microscopy, molecular dynamics simulations and analytical ultracentrifugation. We demonstrate that the transduction efficiency of arginine-rich peptides increases with higher peptide structural rigidity. Consequently, cyclic arginine-rich cell-penetrating peptides showed enhanced cellular uptake kinetics relative to their linear and more flexible counterpart. We propose that guanidinium groups are forced into maximally distant positions by cyclization. This orientation increases membrane contacts leading to enhanced cell penetration.
Commentary:Cell-penetrating peptides (CPPs) are attractive in vitro and in vivo drug delivery vectors. CPPs usually contain less than 30 amino acids, whose compositions are often polycationic or amphipathic, and can be of different sizes, charges, and sequences. Since the discovery of the first CPP, HIV-1 TAT (Frankel et al., Cell 1988;55:1189–1193; Green et al., Cell 1988;55:1179–1188), in the late 1980s, a number of CPPs have been applied to introduce cargos of interest into living cells, either by covalent or noncovalent linkage to CPPs. Advantages of CPPs include their low immunogenicity, ease of synthesis, and compatibility in conjugation with a variety of cargos (from small molecules to proteins, from liposome to nanoparticles). One major hurdle in conventional chemotherapy is the lack of tumor selectivity for neoplastic cells, which precludes a successful therapy. Recently, CPPs have been shown to enhance the selectivity and efficient concentration of chemotherapeutics towards targeted tumors by fusing to a tumor-homing peptide (Mäe et al., Int J Pept Res Ther 2009;15:11–15). Homing peptides target either normal or pathological tissues based on their affinity to tissue-specific biomarkers/receptors (vascular “zip code”). A number of such homing peptides are available with well-documented homing specificities, with Arg-Gly-Asp (RGD) and Asn-Gly-Arg (NGR) representing the first generation of homing peptides, which are also in clinical trials (see review by Laakkonen et al., Integra Biol 2010;2:326–337). Thus, strategies to identify cell-specific homing peptide are highly desired as they could potentially help us to further decode vascular zip codes. The first article highlighted herein demonstrated a screening platform to assay for selective cellular delivery using a peptide nucleic acid (PNA)-encoded peptide library. Svensen et al. constructed a tetrapeptide library that contained almost 1300 FAM-labeled PNA-encoded peptides using five natural amino acids (Pro, Glu, Leu, Lys, and Tyr) and one peptoid monomer (N-aminohexylaminoacetic acid, Llp; tetramer of Llp as a positive control) (see
first figure
, panel b). The library was subsequently incubated with nine different cell types at 37°C before the release of internalized tagged peptides through trypsinization, followed by fluorescence detection of the FAM label through microarray analysis (see
first figure
, panel a). A consensus sequence, FAM-Ahx-Glu-Llp-Glu-Glu-NH2, was identified from the DNA microarray analysis, and was further validated by flow cytometry to have an uptake level no less than the positive control in five out of the nine cell types. Cell-selective delivery peptides were subsequently identified through computational methods (see Supporting Information of Svensen et al.). More importantly, higher selective uptake of certain peptide sequences was observed in some cell types than in others. The highlight of their findings was the sixfold higher uptake of FAM-Ahx-Leu-Lys-Lys-Pro-NH2 in the myeloid leukemia cell line K562 than in primary monocytes (both cell types are of similar phenotype and internalization selectivity could be useful for hematological cancer studies). Compared with the positive control and the general consensus peptide, all the cell-type selective peptides (hit tetramers) showed equal or greater cell-penetrating abilities. Thus, these hit tetramers have been demonstrated not only for their efficient cell-penetrating ability but also for their cell-type selectivity. The authors also performed MTT-based assays and erythrocyte hemolysis assays to further rule out the possibility of peptide- or membrane-associated toxicity. In addition to the challenge of achieving selective cell-type delivery, a second major challenge in CPP-mediated cargo delivery is our lack of understanding of their penetration mechanism. This was briefly addressed in the first article and was the main focus in the second article (by Lättig-Tünnemann and coworkers). Several different pathways for CPP penetration have been proposed, and they may transpire at the same time in a certain case, such as direct penetration, endocytosis-mediated translocation, and translocation through the formation of a transitory structure. Using LysoTracker red stain and confocal microscopy, the authors in the first article found that the hit tetramers were all compartmentalized into the lysosome, while their 4°C incubation counterparts failed to show lysosome compartmentalization. The requirement for an active cellular uptake process suggested an endocytosis-mediated translocation mechanism due to its energy-dependent nature. In the article by Lättig-Tünnemann et al., the authors conducted a study to directly compare cellular uptake characteristics among different peptides that were of different charges and structures. Their study generated three major take-home messages. First, using living mouse myoblast cells, arginine-rich CPPs (RPPs) produced a much higher transduction efficiency (analyzed in the nucleus to exclude enocytosis-driven translocation) than those of low arginine content (see
second figure
, panel a). Second, through using analytical ultracentrifugation and molecular dynamics, it was found that extended conformation (for example, a linear form of a CPP would represent a more extended conformation than its cyclic version) was not necessary for efficient CPP transduction (see
second figure
, panels b and c). Third, using two RPPs (TAT and R10) for proof-of-principle, the authors found that the cyclic versions had a more favorable transduction kinetic profile (early cell penetration and denser population inside the nucleus) than their respective linear counterparts (with R10 showing even more enhanced uptake kinetics due to its larger guanidinium group separation). The CPP penetration mechanism illustrated in the second study is of a nonendocytic mode, but is complementary to the first study and could be useful in the aspect of CPP library design. Improved/customized design in peptide library structure and composition, as suggested by these two studies, may lead to desirable cellular uptake that could be driven by a more focused mechanism. Contributed by Wendy Lea.
(a) The strategy to identify cell-selective penetrating peptides. An encoded 1296-member peptide library was incubated with cells, and any cell surface–bound peptides were released with trypsin. Cells were lysed, and intracellular PNA was extracted and hybridized onto a 44K DNA microarray. (b) General structure of the PNA-encoded 1296-member peptide library (Library 1). Each PNA quartet was designed to have a maximum of 50% similarity to each other quartet and a maximum purine content of 50%; all had uniform melting temperatures and did not include palindrome sequences or polythymine moieties.
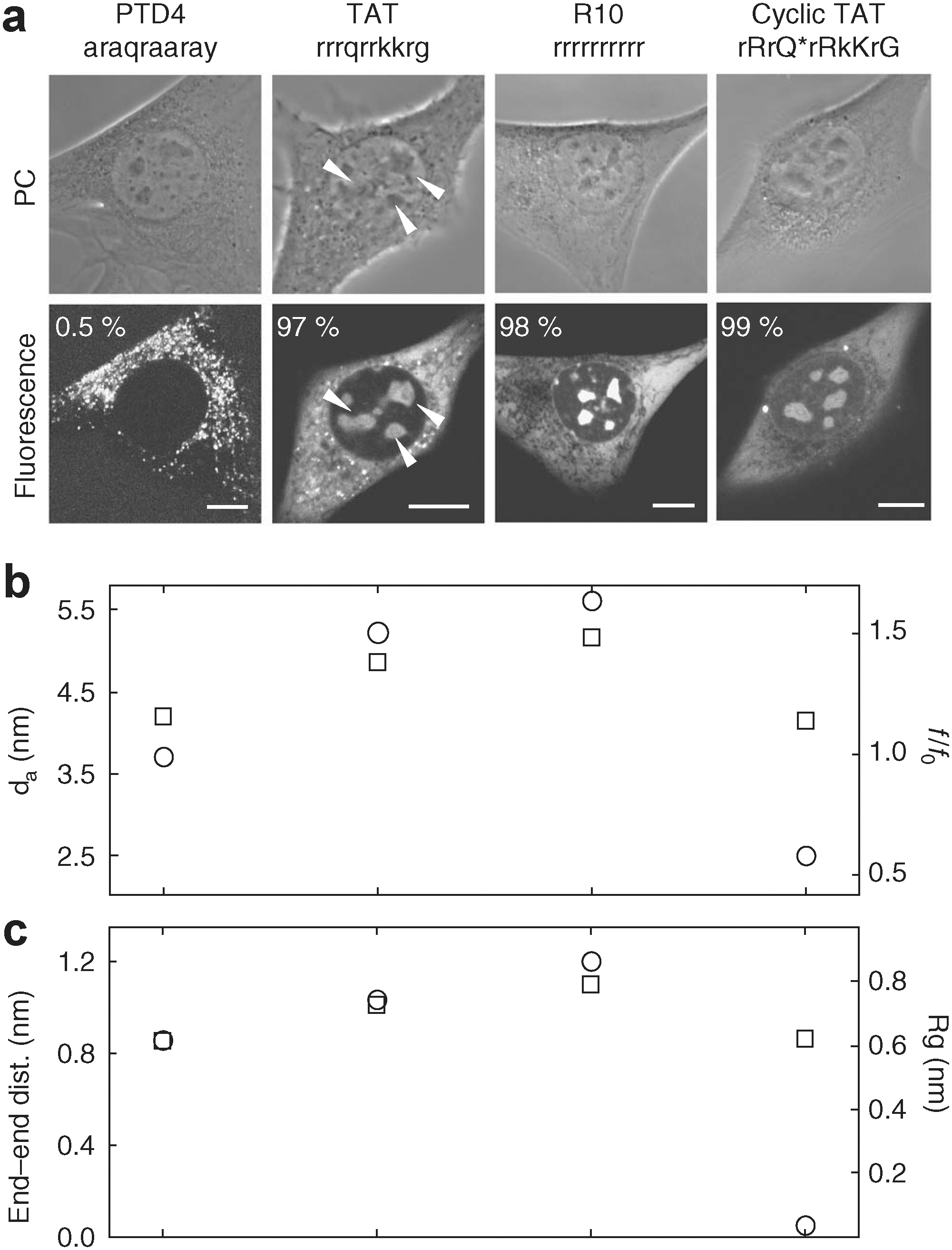
Live-cell transduction ability of CPPs and experimental and theoretical shape parameters. (a) The CPPs PTD4, TAT, R10, and cyclic TAT were transduced, or otherwise internalized, into living mouse myoblast cells in the presence of PBS. Representative transmission images of the phase contrast and confocal optical sections of the fluorescent peptides are shown. Scalebars, 10 μm. Arrows indicate the presence of fluorescent peptide inside nucleoli and percentages of the number of transduced cells are given. (b) Results from the sedimentation velocity ultracentrifugation experiments and subsequent analysis: f/f0: frictional ratio (squares), d: diameter, end–end distances (circles). (c) Results of the molecular dynamics simulations: radius of gyration (squares) and end–end distances (circles). Q* results from a decondensation reaction between a glutamic acid and a lysine during cyclization (see Supplementary Table 1 in Lättig–Tünnemann et al.).
Flow Cytometry Goes Atomic
Bendall SC, Simonds EF, Qiu P, Amir el-AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe'er D, Tanner SD, Nolan GP. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. ACS Chem Biol 2011;332:687–696.
Abstract: Flow cytometry is an essential tool for dissecting the functional complexity of hematopoiesis. We used single-cell “mass cytometry” to examine healthy human bone marrow, measuring 34 parameters simultaneously in single cells (binding of 31 antibodies, viability, DNA content, and relative cell size). The signaling behavior of cell subsets spanning a defined hematopoietic hierarchy was monitored with 18 simultaneous markers of functional signaling states perturbed by a set of ex vivo stimuli and inhibitors. The data set allowed for an algorithmically driven assembly of related cell types defined by surface antigen expression, providing a superimposable map of cell signaling responses in combination with drug inhibition. Visualized in this manner, the analysis revealed previously unappreciated instances of both precise signaling responses that were bounded within conventionally defined cell subsets and more continuous phosphorylation responses that crossed cell population boundaries in unexpected manners yet tracked closely with cellular phenotype. Collectively, such single-cell analyses provide system-wide views of immune signaling in healthy human hematopoiesis, against which drug action and disease can be compared for mechanistic studies and pharmacologic intervention.
Commentary:Multiplexing using fluorescent probes is limited by the spectral overlap of the emission spectrum. This restricts the number of measured parameters and for most laboratories, multiplexing more than four fluorophores can be challenging. Atomic mass spectrometry (DVS Sciences), which measures the mass of individual atoms as opposed to the mass of the intact molecule, provides high resolution and sensitivity and offers the ability to detect multiple parameters by employing elements of the periodic table as tags. In the method shown here, antibodies are labeled with a common tag containing a polymer that can chelate ∼30 metal ions. To boost the signal, three such polymers can be linked to an antibody so that each antibody carries nearly 100 metal ions. Choosing metals not found in cells contained within a single series of the periodic table such as lanthanide metals allows for use of a single chelator for the labeling step and sensitive detection of antigen. Following labeling, the cells are treated with the test conditions and then analyzed in a flow cytometer in which the detection is based on atomic mass spectrometry. Cells are passed through an inductively coupled argon plasma beam operating at a temperature approximately equal to that found on the surface of the sun (∼5500 K). This temperature effectively vaporizes the cells and disintegrates their components into a cloud of fully ionized atoms. The ionized cloud of elemental ions is then passed through a time-of-flight atomic mass spectrometer, and the signals for each of the metals previously contained within single cells are quantified (see
figure
). This article demonstrates a tour-de-force of this technology for the analysis of human hematologic cell types. The differentiation states of hematopoietic cells can be determined by “cluster of differentiation” (CD) markers. To create an “immunophenotype” panel, antibodies that monitored 13 core CD surface markers and 18 subset-specific cell-CD surface markers were used. In addition, a second panel was constructed using antibodies containing the same 13 core markers as well as 18 intracellular markers reflective of intracellular signaling events (e.g., through phosphoprotein detection). In addition three labels for total DNA, cell length, and cell viability were also used, so each panel measured 34 parameters. Comparing the data for various phosphoproteins from atomic mass cytometry with conventional fluorescence cyotometry showed that both qualitatively and quantitatively similar patterns were obtained with each method. The data can be exported as typical .fcs files but the wealth of data generated is overwhelming. To address this issue, data analysis was performed using SPADE (spanning-tree progression analysis of density-normalized events; see: Qiu et al., Nat Biotechnol 2011;29:886–891). SPADE analysis provides unsupervised clustering of the data and the results can be visualized with two-dimensional tree plots. For example, in the immunophenotype panel, each node in the tree plot represents a cell cluster having a similar phenotype in the 13 dimensional space defined by the core surface markers. Well-defined cell types such as T cells or monocytes can provide internal standards for the nodes that enabled capturing unexpected transitional cell types and defining lineage by determining the relationships between each cell cluster. The functional state of each cell type can be similarly treated and visualized. Significant signaling events within each subtype were examined. In addition to providing a system-level view of hematopoiesis this study examines pharmacological modulation with well-known kinase inhibitors such as dasatinib, which points to polypharmacology, which may explain the efficacy of this compound in certain B-cell malignancies. This approach demonstrates a method for the analysis of pathways using the resolving power of atomic mass spectrometry. Assay development of multiplexed formats using fluorophores is difficult due to the large differences in fluorescent brightness between probes and the spectral overlap. In mass cytometry the isotope signals vary within a twofold range and the isotopic masses are readily separated upon detection, which allows for a much greater number of parameters to be simultaneously measured and more facile assay optimization. The technology demonstrates a unique approach to high content methods that enables pathway analysis from primary cell types. Contributed by Doug Auld.
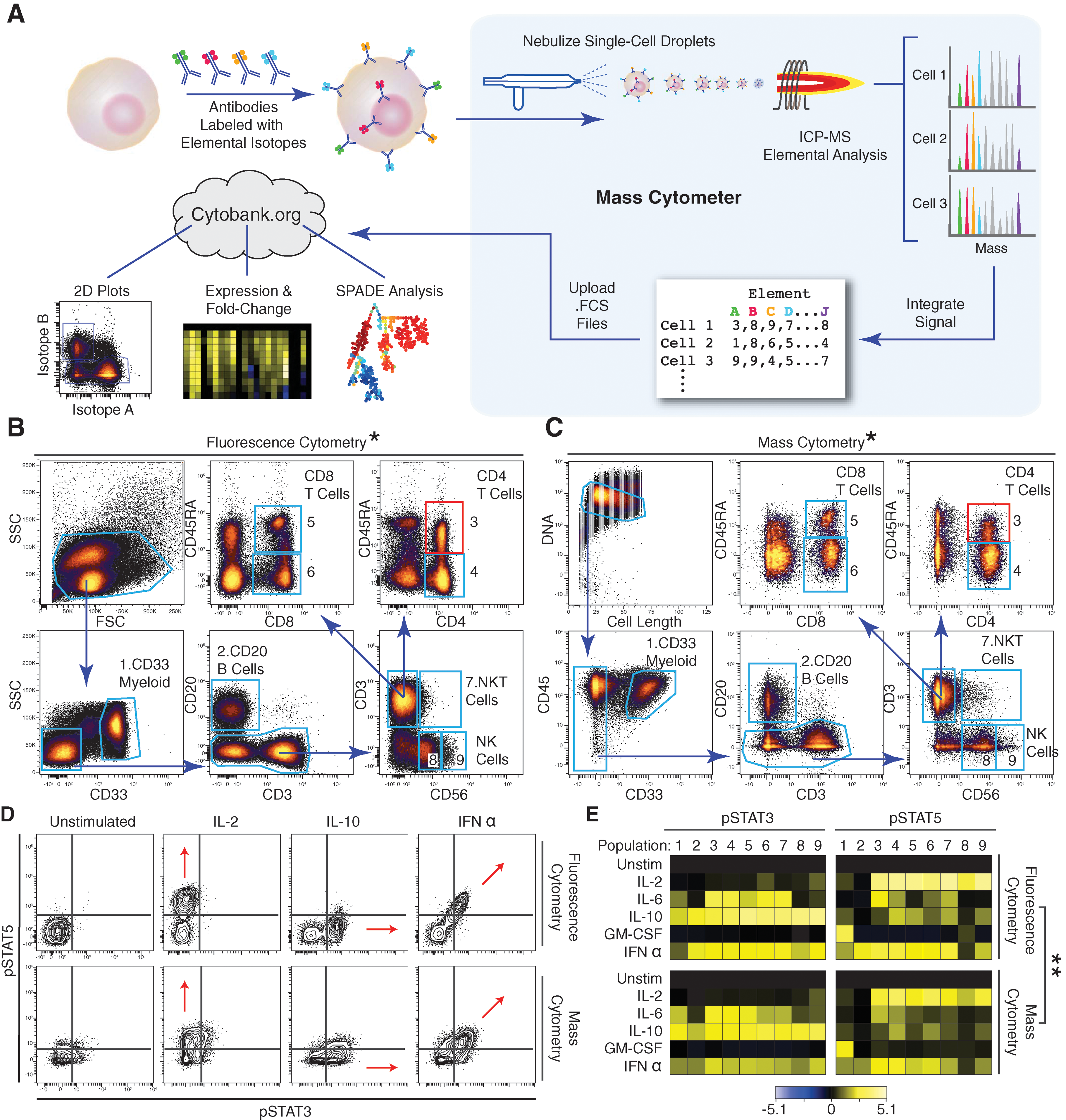
Mass cytometry profiling of immune cell response patterns. (A) Workflow summary of mass cytometry analysis. Cells are stained with epitope-specific antibodies conjugated to transition element isotope reporters, each with a different mass. Cells are nebulized into single-cell droplets, and an elemental mass spectrum is acquired for each. The integrated elemental reporter signals for each cell can then be analyzed by using traditional flow cytometry methods as well as more advanced approaches such as heat maps of induced phosphorylation and tree plots. (B and C) Representative antibody surface-staining results and cell population definitions (“gating”) for (B) fluorescence and (C) mass cytometry analysis of fixed PBMCs from the same donor. Replicate analysis of a second donor is provided in Fig. S1A and S1B in the article's Supporting Online Material. (D) Induction of STAT3 and 5 phosphorylation by various ex vivo stimuli in naive CD4+CD45RA+ T cells [(B) and (C), red boxes] as measured by (top) fluorescence and (bottom) mass cytometry. Red arrows indicate the expected shift along the STAT phosphorylation axes. (E) Heatmap summary of induced STAT phosphorylation in immune populations from the PBMC donor defined in (B) and (C) [column headers refer to blue polygons in (B) and (C)]. Responses to the indicated stimuli in each row were measured by (top) fluorescence and (bottom) mass cytometry. Color scale indicates the difference in log2 mean intensity of the stimulated condition compared with the unstimulated control. Signaling responses of a second donor are provided in Fig. S1D in the article's Supporting Online Material. *Pearson correlation between frequencies measured by fluorescence or mass cytometry, including both donors (r = 0.99, p < 0.000001, two-tailed t test) (Table S1 and Fig. S1C in the article's Supporting Online Material). **Pearson correlation between signaling induction measured by fluorescence or mass cytometry, including both donors (pSTAT3: r = 0.92; p < 0.000001, two-tailed t test [Fig. S1E in the article's Supporting Online Material]; pSTAT5: r = 0.89, p < 0.000001, two-tailed t test [Figs. S1E and S1F in the article's Supporting Online Material]). PBMC, peripheral blood mononuclear cell.
Can We Prevent Cancer Cells from Developing Feet?
Quintavalle M, Elia L, Price JH, Heynen-Genel S, Courtneidge SA. A cell-based high-content screening assay reveals activators and inhibitors of cancer cell invasion. Sci Signal 2011;4:ra49.
Abstract: Acquisition of invasive cell behavior underlies tumor progression and metastasis. To further define the molecular mechanisms underlying invasive behavior, we developed a high-throughput screening strategy to quantitate invadopodia, which are actin-rich membrane protrusions of cancer cells that contribute to tissue invasion and matrix remodeling. We tested the LOPAC 1280 collection of pharmacologically active agents in a high-content, image-based assay and identified compounds that inhibited invadopodium formation without overt toxicity, as well as compounds that increased invadopodia number. The chemotherapeutic agent paclitaxel increased both the number of invadopodia and the invasive behavior of various human cancer cell lines, effects that have potential clinical implications for its use before surgical removal of a primary tumor (neoadjuvant therapy) or in patients with chemoresistant tumors. Several compounds that inhibited invasion have been characterized as cyclin-dependent kinase (Cdk) inhibitors, and loss-of-function experiments determined that Cdk5 was the relevant target. We further determined that Cdk5 promoted both invadopodium formation and cancer cell invasion by phosphorylating and thus decreasing the abundance of the actin regulatory protein caldesmon.
Commentary:When cancer cells leave the primary tumor site and spread to other tissues and organs (i.e., metastasize), the patient's mortality rate increases. One of the mechanisms by which cancer cells can migrate to remote locations is by developing invadopodia. Invadopodia have been observed in many cancer cell lines, including those from breast cancer, squamous cell carcinoma of the head and neck, melanoma, and glioblastoma. As the name suggests, invadopodia are foot-like protrusions that correlate with a cell's invasiveness. These actin-rich membrane protrusions enable the cell to extend into and degrade the extracellular matrix (ECM). Inhibition of the formation of invadopodia may be a way to prevent cancer cell metastasis. The authors developed a high-content screening (HCS) assay that can monitor for regulators of the presence of invadopodia. Instead of human cancer cells, NIH 3T3 cells transformed with activated SRCwere used because they have many invadopodia that are often found in rings or rosettes, which enables the automatic high-throughput detection of their presence. The
figure
shows the results from a screen of the 1280 compounds in the LOPAC (library of pharmacologically active compounds) set screened in duplicate at 10 μM. After removing cytotoxic compounds and applying thresholds to the data, seven compounds were obtained. In various cancer cell lines, it was found that the chemotherapeutic paclitaxel, which targets microtubules in the cytoskeleton, increased both the number of invadopodia and their invasiveness at a dose achievable in the clinic (2 μM); i.e., it was an activator. Paclitaxel is known to bind and stabilize microtubules, and microtubules are present in mature invadopodia. The authors point out that this may have clinical implications for how paclitaxel should be used during cancer treatment, and indeed paclitaxel treatment increased metastasis in animals with paclitaxel-resistant leukemia cells. Several inhibitors of invadopodia were identified. The target of several inhibitors was CDK5 (cyclin-dependent kinase 5), which phosphophorylates caldesmon, an actin regulatory protein, leading to its degradation. CDK5 was shown to promote invadopodia and cancer cell invasiveness and conversely inhibition of CDK5 correlated with an inhibition of invadopodia formation (EC50=0.2 μM for purvalanol A and GCP74514A). After mining the LOPAC set for other known CDK inhibitors, the images were re-evaluated and another two CDK inhibitors were determined to inhibit invadopodia formation. These had been previously discarded due to irreproducibility later attributed to edge effects. An additional CDK inhibitor was remade and was then found to inhibit invadopodia. Before undertaking a larger high-throughput screening campaign, the assay robustness should be optimized to try to minimize compounds, such as the three CDK inhibitors initially missed as hits here, from scoring as false negatives. The development of this assay provides for a new strategy to identify anti-metastatic drugs, which may be of interest as single agents or for use in combination therapies. This new assay also provides a way to assess whether a compound may actually encourage the formation of invadopodia and lead to tumor invasiveness, and therefore whether its use may be contraindicated. Contributed by Mindy I. Davis.
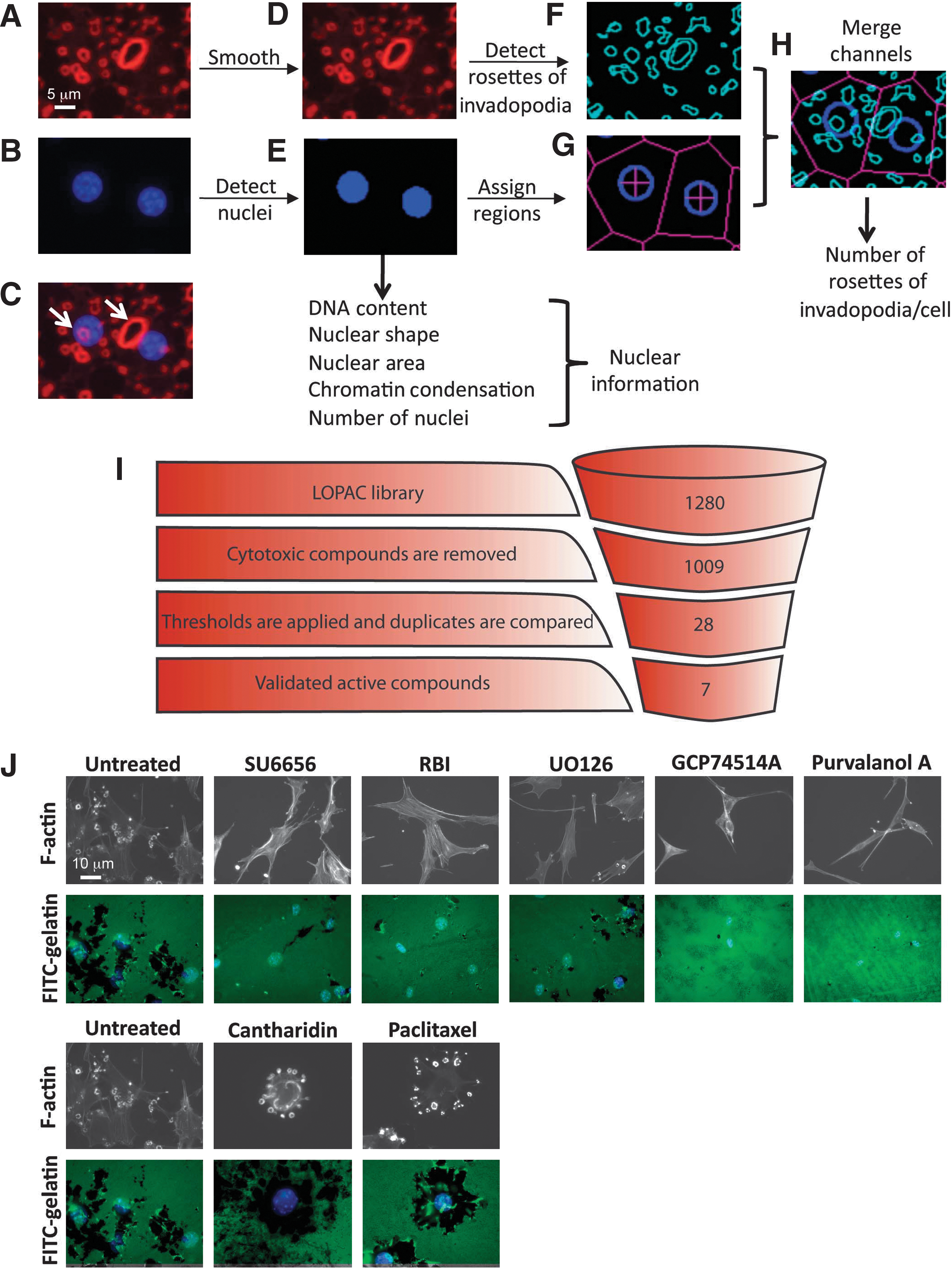
HCS image analysis. (A–H) Image analysis for the screen of invadopodium formation. Cells stained for actin (red) (A) and with DAPI to stain nuclei (blue) (B). The overlay of both channels is displayed in (C). The image background for the red channel was subtracted and a 3-by-3 median filter was applied to smooth the image and remove fine actin fibers (D). Nuclei detection (Cytoshop) was applied (E) and nuclear parameters were extracted from the segmented DAPI image. From the nuclei masks (E), cellular regions were assigned to each nucleus using tessellation in Cytoshop (G). The red channel was segmented using Cytoshop's fluorescence aggregate detection to find rosettes (F). Finally, the detected nuclei, assigned cellular regions, and detected rosettes were merged (H) and the number of detected rosettes was calculated on a cell-by-cell basis. Representative rosettes of invadopodia are highlighted by white arrows in (C). (I) Screening results: 1280 compounds were screened in duplicate. Cytotoxic compounds were removed from the active compound list. Thresholds for inhibition and activation were applied. Visual confirmation led to a final list of seven active compounds. (J) Validation of active compounds. Src-3T3 cells were plated on fluorescently labeled gelatin-coated coverslips. Representative images of gelatin degradation (lower row) and F-actin staining (upper row) are shown.