Sela-Passwell N, Kikkeri R, Dym O, Rozenberg H, Margalit R, Arad-Yellin R, Eisenstein M, Brenner O, Shoham T, Danon T, Shanzer A, Sagi I: Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2012;18:143–147.
Abstract: Endogenous tissue inhibitors of metalloproteinases (TIMPs) have key roles in regulating physiological and pathological cellular processes. Imitating the inhibitory molecular mechanisms of TIMPs while increasing selectivity has been a challenging but desired approach for antibody-based therapy. TIMPs use hybrid protein-protein interactions to form an energetic bond with the catalytic metal ion, as well as with enzyme surface residues. We used an innovative immunization strategy that exploits aspects of molecular mimicry to produce inhibitory antibodies that show TIMP like binding mechanisms toward the activated forms of gelatinases (matrix metalloproteinases 2 and 9). Specifically, we immunized mice with a synthetic molecule that mimics the conserved structure of the metalloenzyme catalytic zinc histidine complex residing within the enzyme active site. This immunization procedure yielded selective function-blocking monoclonal antibodies directed against the catalytic zinc-protein complex and enzyme surface conformational epitopes of endogenous gelatinases. The therapeutic potential of these antibodies has been demonstrated with relevant mouse models of inflammatory bowel disease. Here we propose a general experimental strategy for generating inhibitory antibodies that effectively target the in vivo activity of dysregulated metalloproteinases by mimicking the mechanism employed by TIMPs.
Commentary:Targeting metalloproteases in a tissue-specific manner has been challenging due to difficulties associated with dialing in selectivity based on active enzyme conformations involving the catalytic metal center. Such catalytic metal-protein clefts have been deemed nonimmunogenic due to the poor epitope presentation and instability during B-cell surface display, thus precluding generation of highly specific antibodies. Here, a mimicry-based approach was adopted in which the catalytic zinc-containing center consisting of the zinc metal typically liganded by three histidines was mimicked by a zinc ion coordinated by a tridentate ligand incorporating three imidazoles (see first figure). This inorganic complex was used in a mouse immunization scheme followed by hybridoma generation and screening for antibodies directed against both the zinc tripod hapten and the metalloprotease MMP-9. Two clones, SDS3 and SDS4, were selected for detailed biophysical investigation. The antibodies bound MMP-9 at nanomolar affinity and displayed double-digit nanomolar inhibition of the catalytic activity of the enzyme. The antibodies were also found to be nonreactive again a range of metal-containing enzymes, such as carbonic anhydrase and alcohol dehydrogenase, as well as against the related MMP-1, MMP-7, and MMP-12. An X-ray crystal structure revealed that the new antibodies possess a unique wide concave-shaped cleft with protruding light- and heavy-chain complementarity-determining regions. One of the new antibodies, SDS3, was evaluated for therapeutic utility in a model for Crohn's disease and related ulcerative colitis syndromes by testing the effect of the antibody on dextran sulfate (DSS)–induced colitis in mice (see second figure). While the untreated mice presented severe colon damage 13 days after DSS insult, the SDS3-treated animals had limited inflammation and a well-preserved mucosal architecture of the colon. Treatment with nonspecific antibody failed to produce a protective effect. This study represents a key milestone not only for the field of protein engineering in general, by showing that an antibody possessing a unique antigen binding architecture can be generated using metal complex mimicry approach, but also because it demonstrates that antibodies generated through this approach can in fact be useful in treating a debilitating disease. Contributed by Anton Simeonov.
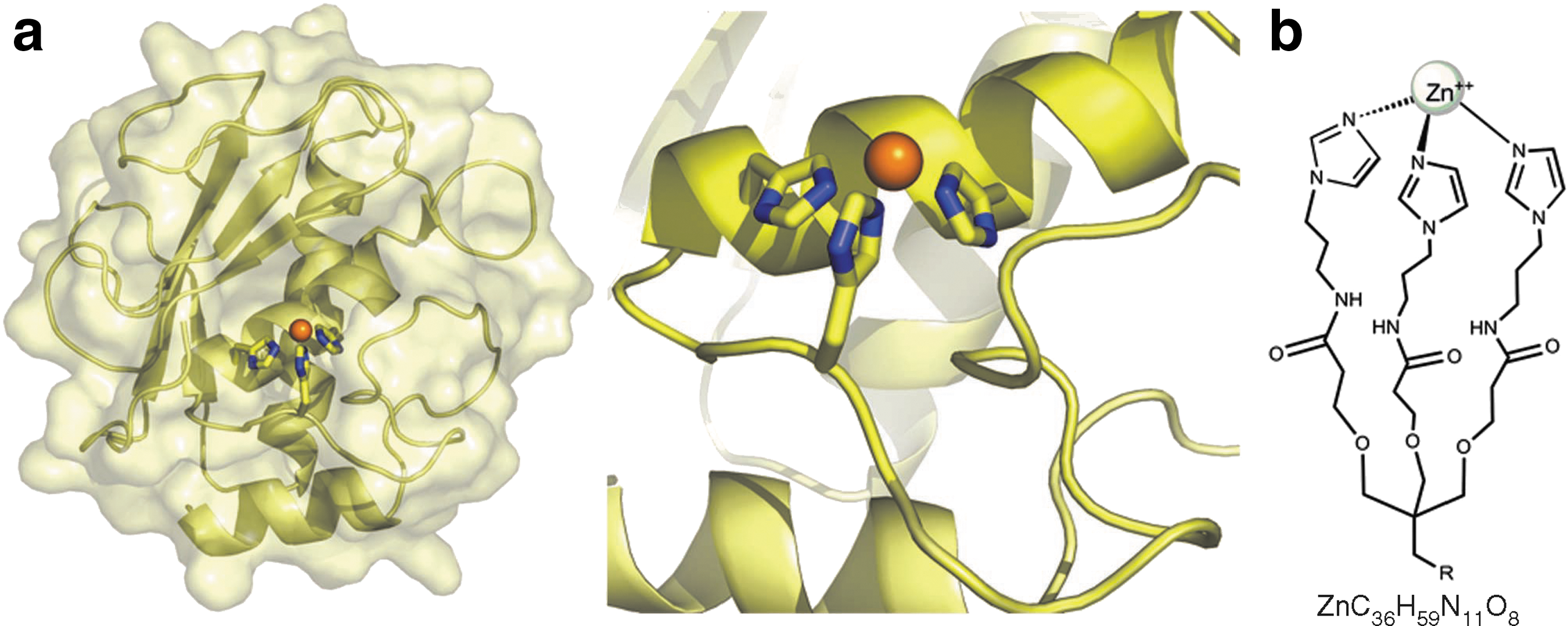
Mimicry of the zinc-protein motif of MMPs. Based on the resolved structure of the conserved HEXXHXXGXXH zinc-binding motif (where X is any amino acid) in MMPs, a symmetrical tripodal tris-imidazol–zinc complex (Zn-tripod; ZnC36H59N11O8) was designed and synthesized as a mimicry complex of the natural tetrahedral zinc-protein motif in MMPs. (a) Left, MMP-9 catalytic domain shown in a secondary structure representation with a semitransparent surface. Right, a close-up view of the catalytic metalloprotein site. The three histidine side chains and a water molecule (not shown) bind the zinc ion (orange sphere) in a tetrahedral conformation. (b) Chemical structure of Zn-tripod in which three imidazoles and a water molecule (not shown) bind the zinc ion (sphere) in a tetrahedral conformation, thus structurally and chemically mimicking the zinc–protein interactions in MMPs.
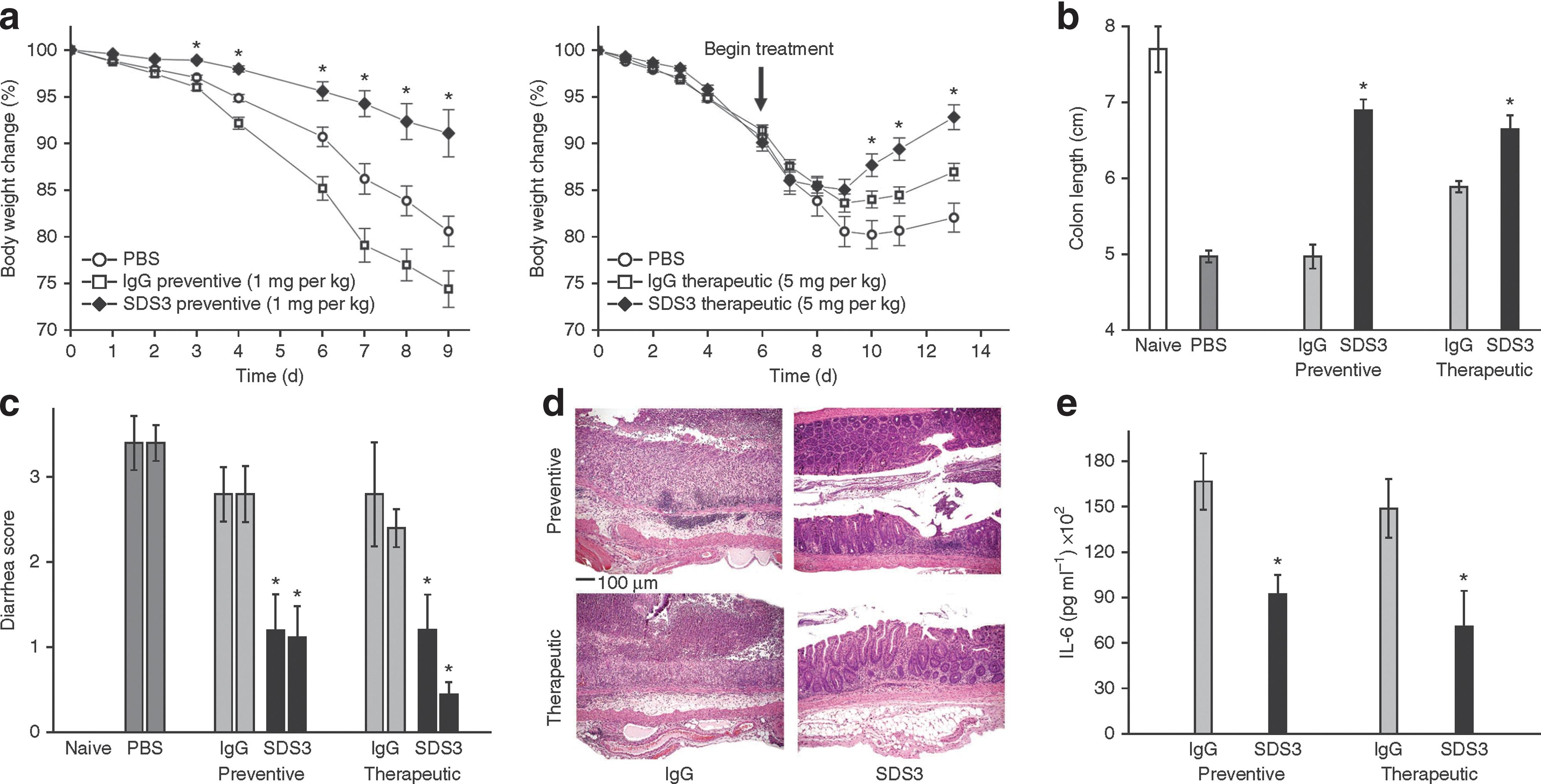
Prophylactic and therapeutic treatment with SDS3 protects C57BL/6J mice against DSS-induced colitis. (a–c) Clinical colitis severity, as monitored by body weight loss (a), colon length (b) and diarrhea on day 9 (left bar) and on day 14 (right bar) after DSS induction (c). (d) Representative photomicrographs of colons following prophylactic (top) and therapeutic (bottom) treatment with antibodies (control IgG [left] and SDS3 [right]). H&E stain; magnification: × 10. (e) Colonic IL-6 cytokine analysis 9 d after DSS induction. *P < 0.05 compared with mice treated with nonspecific mouse immunoglobulin control (IgG) and mice treated with vehicle (PBS). n = 10 mice per group in all panels except (e); n = 6 for IL-6 analysis. Results are expressed as mean ± s.e.m.
Enhancing the Anti-cancer Effect of Cytarabine with a Wee Bit of Wee1
Tibes R, Bogenberger JM, Chaudhuri L, Hagelstrom RT, Chow D, Buechel ME, Gonzales IM, Demuth T, Slack J, Mesa RA, Braggio E, Yin HH, Arora S, Azorsa DO: RNAi screening of the kinome with cytarabine in leukemias. Blood 2012;119:2863–2872.
Abstract: To identify rational therapeutic combinations with Cytarabine (Ara-C), we developed a high-throughput, small-interference RNA (siRNA) platform for myeloid leukemia cells. Of 572 kinases individually silenced in combination with Ara-C, silencing of 10 (1.7%) and 8 (1.4%) kinases strongly increased Ara-C activity in TF-1 and THP-1 cells, respectively. The strongest molecular concepts emerged around kinases involved in cell-cycle checkpoints and DNA-damage repair. In confirmatory siRNA assays, inhibition of WEE1 resulted in more potent and universal sensitization across myeloid cell lines than siRNA inhibition of PKMYT1, CHEK1 or ATR. Treatment of 8 acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), and chronic myeloid leukemia (CML) cell lines with commercial and the first-in-class clinical WEE1 kinase inhibitor MK1775 confirmed sensitization to Ara-C up to 97-fold. Ex vivo, adding MK1775 substantially reduced viability in 13 of 14 AML, CML, and myelodysplastic symdromes patient samples compared to Ara-C alone. Maximum sensitization occurred at lower to moderate concentrations of both drugs. Induction of apoptosis was increased using a combination of Ara-C and MK1775 compared with using either drug alone. WEE1 is expressed in primary AML, ALL, and CML specimens. Data from this first siRNA-kinome sensitizer screen suggests that inhibiting WEE1 in combination with Ara-C is a rational combination for the acute lymphoblastic leukemia treatment of myeloid and lymphoid leukemias.
Commentary:Drug combination therapy is a promising avenue for the treatment of cancer. Testing all possible combinations of drugs to look for synergies, however, can quickly become quite a daunting task. The approach used here involved a high-throughput RNAi screen on nonadherent cells to identify targets that synergize with a drug called Ara-C that is used in induction treatment for several blood cancers, including AML. Ara-C is a compound that contains cytosine with an arabinose sugar rather than deoxyribose. This change leads to a disruption in DNA synthesis when this cytosine mimic is incorporated into DNA and ultimately leads to cell death in rapidly dividing cells. While Ara-C can be quite effective in treating AML patients, the outcome for relapsed or refractory patients is not as good. Ara-C has been tried in combination with other drugs in the clinic, but there is still significant room for improvement. CHEK1 kinase had been shown previously to synergize with Ara-C in leukemias. Because kinases have been determined to be activated in various blood cancers, the siRNA library selected here targeted 572 kinases with four siRNA sequences per gene. The authors identified around 10 kinases that were possible sensitizers to Ara-C, including WEE1, PKMYT1, and CHEK1. These kinases tend to have elevated expression levels in myeloid and lymphoid cancers. The authors focused on inhibition or knockdown of WEE1, which increases the potency of Ara-C by up to 97-fold in AML, CML, and ALL cell lines. The small molecule clinical candidate MK1775 targets WEE1. The authors show that K562 cells did not reach their usual plateau in their response to Ara-C when co-dosed with MK1775. In primary patient cells for AML, MDS, or CML, the two drugs were shown to synergize ex vivo (see figure) and to reduce cell number by induction of apoptosis. The most potent combinations were found at the low to moderate doses of Ara-C and MK1775. The authors point out that a combination clinical trial is now under development for Ara-C and MK1775. The strategy of using RNAi to determine which targets might synergize with a given cytotoxic drug is quite promising and will hopefully continue to provide guidance into the design of new drug combinations. Contributed by Mindy I. Davis.
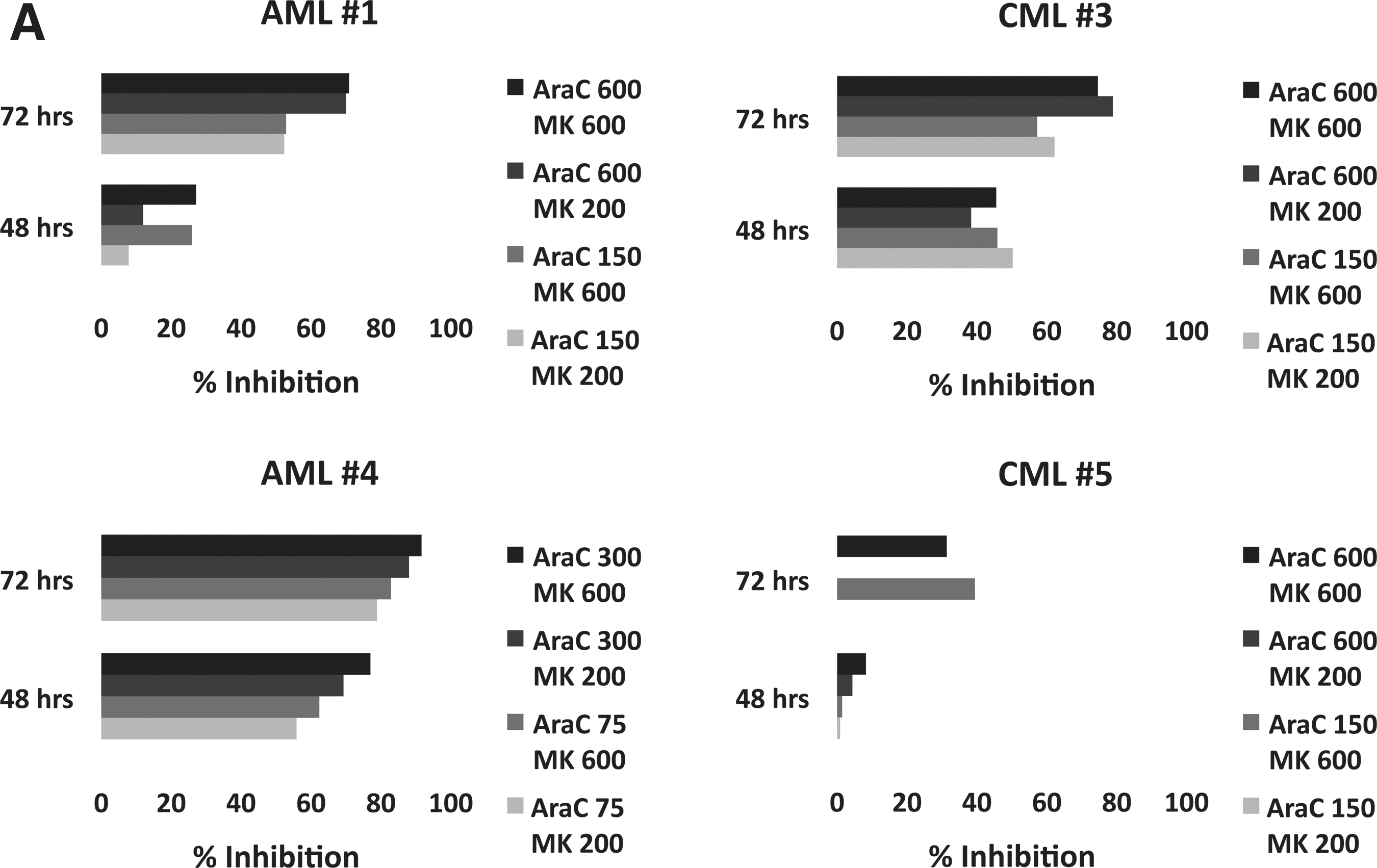
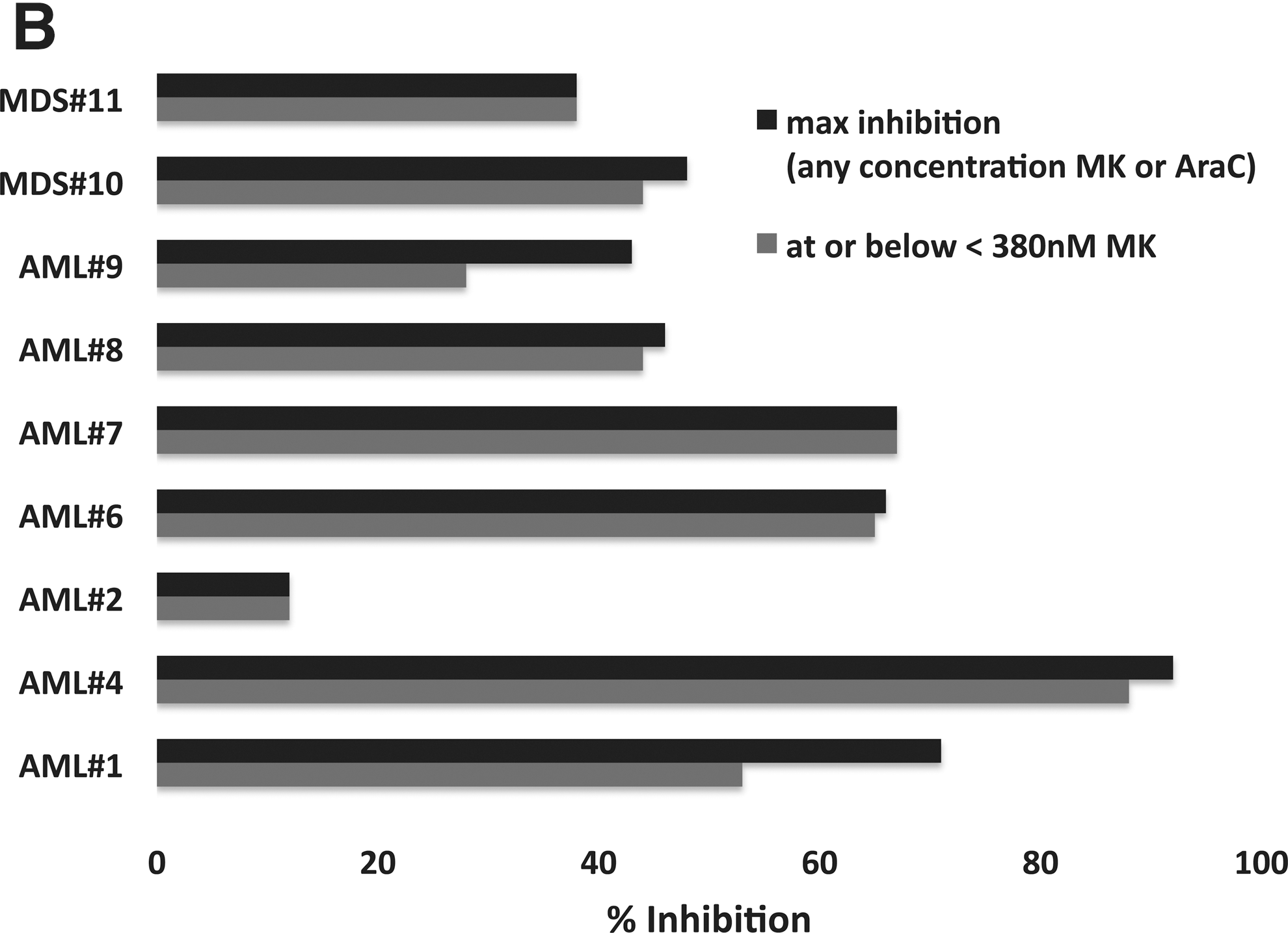
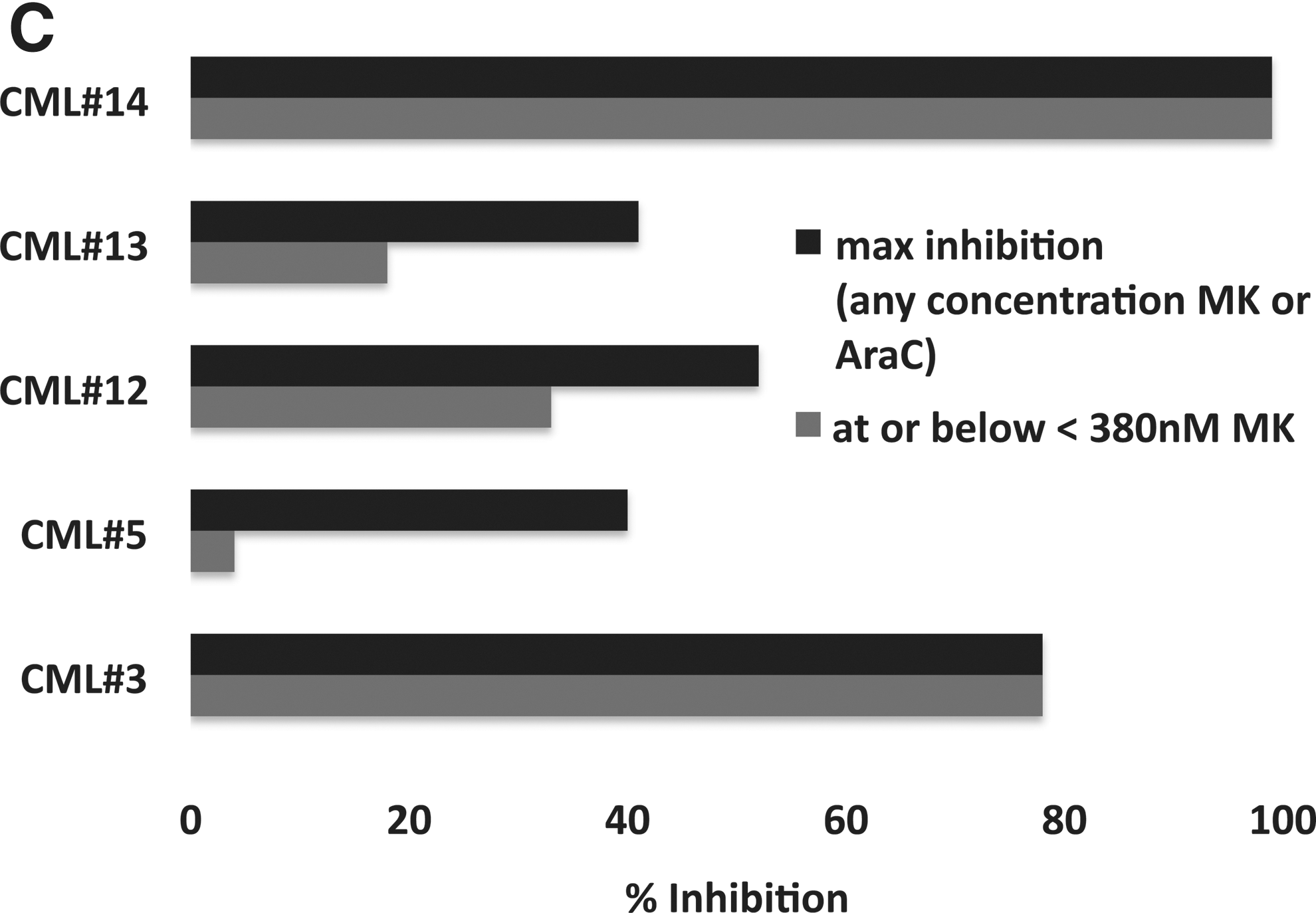
MK1775 is synergistic with Ara-C (cytarabine) ex vivo. Percent inhibition of relative cell number for the combination of Ara-C and MK1775 over Ara-C alone in primary AML, MDS and CML patient samples. (A) Initial four (of five) primary samples as described in the text. (B) AML and MDS primary samples. (C) CML primary samples. For (B) and (C), maximum percent inhibition is shown for any concentration of MK1775 or Ara-C (darkly shaded bars) and for the greatest inhibition observed at or below 380 nM.
Know your enzyme forms
Lin Y, Fan H, Frederiksen M, Zhao K, Jiang L, Wang Z, Zhou S, Guo W, Gao J, Li S, Harrington E, Meier P, Scheufler C, Xu Y-C, Atadja P, Lu C, Li E, Gu XJ: Detecting S-adenosyl-L-methionine-induced conformational change of a histone methyltransferase using a homogeneous time-resolved fluorescence-based binding assay. Anal Biochem 2012;412:171–177.
Abstract: A homogeneous time-resolved fluorescence (HTRF)-based binding assay has been established to measure the binding of the histone methyltransferase (HMT) G9a to its inhibitor CJP702 (a biotin analog of the known peptide–pocket inhibitor, BIX-01294). This assay was used to characterize G9a inhibitors. As expected, the peptide–pocket inhibitors decreased the G9a–CJP702 binding signal in a concentration dependent manner. In contrast, the S-adenosyl-L-methionine (SAM)–pocket compounds, SAM and sinefungin, significantly increased the G9a–CJP702 binding signal, whereas S-adenosyl-L-homocysteine (SAH) showed minimal effect. Enzyme kinetic studies showed that CJP702 is an uncompetitive inhibitor (vs. SAM) that has a strong preference for the E:SAM form of the enzyme. Other data presented suggest that the SAM/sinefungin-induced increase in the HTRF signal is secondary to an increased E:SAM or E:sinefungin concentration. Thus, the G9a–CJP702 binding assay not only can be used to characterize the peptide–pocket inhibitors but also can detect the subtle conformational differences induced by the binding of different SAM–pocket compounds. To our knowledge, this is the first demonstration of using an uncompetitive inhibitor as a probe to monitor the conformational change induced by compound binding with an HTRF assay.
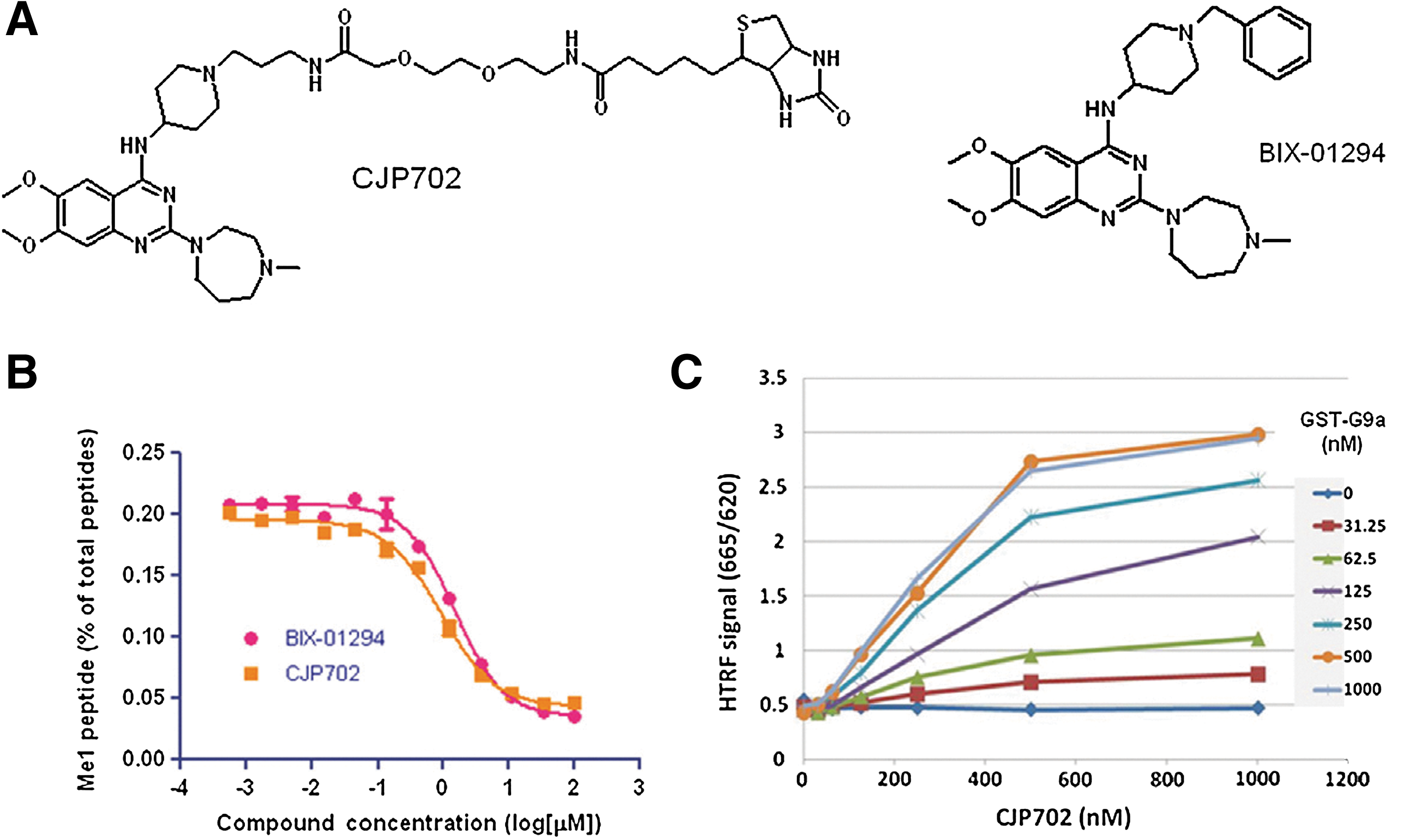
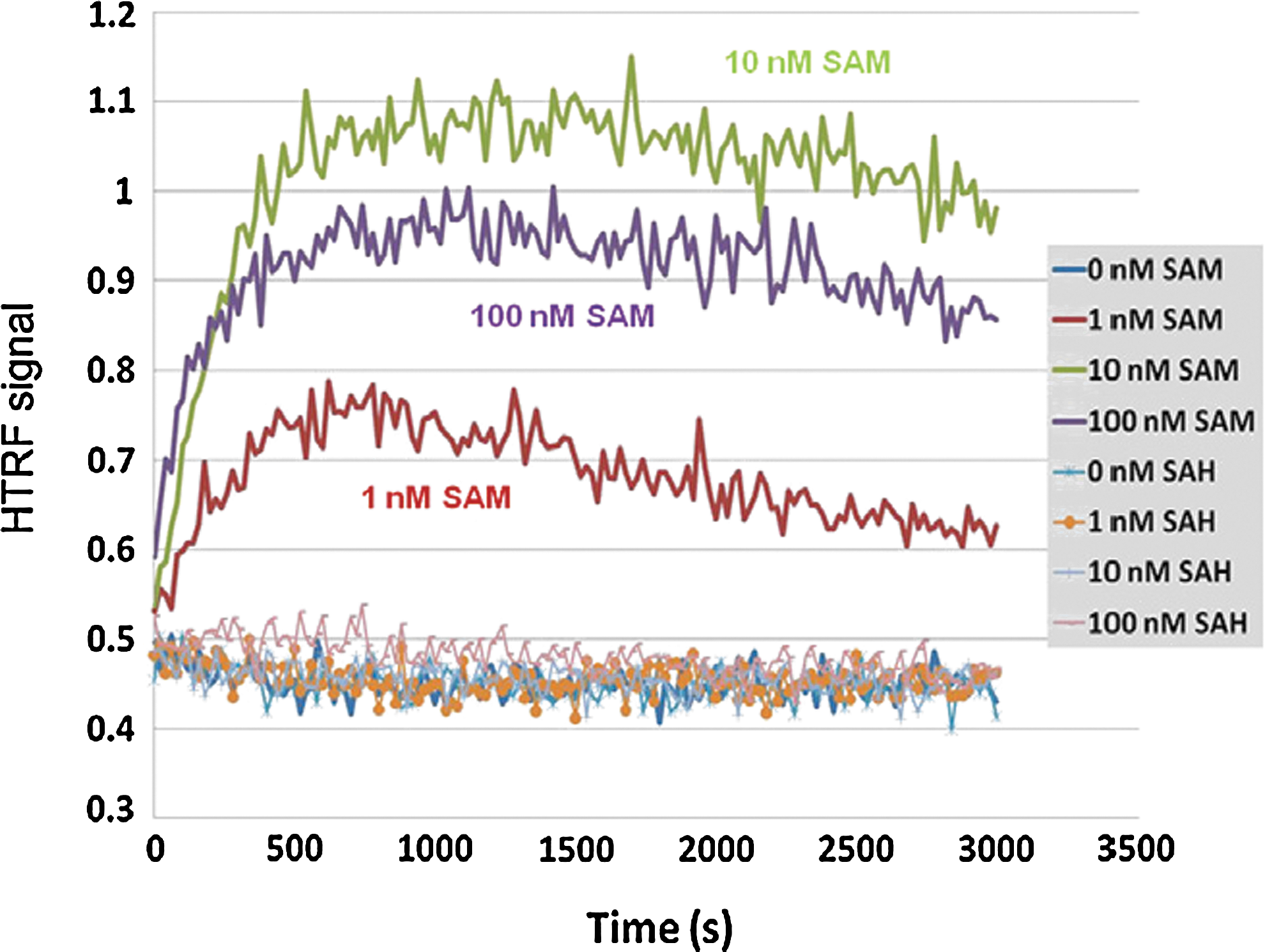
Commentary:Choosing the right enzyme construct is an important step in assay development. Whether to use full-length or truncated variants and which posttranslational modifications to include are a few of the key questions when developing the assay. Furthermore, the order of the enzyme reaction should be understood when setting substrate and cofactor concentrations. An interesting finding for the histone methyltransferase (HMT) G9a is described in this article in which a substrate peptide binding assay was found to be sensitive to conformational changes mediated by the S-adenosyl-L-methionine (SAM) pocket. The HMT family of enzymes all use a SAM cofactor to transfer a methyl group to either lysine or arginine of proteins. This study used a number of tool compounds, which included peptide competitive compounds. BIX-01294 (see first figure) is one of the first peptide-binding site inhibitors, and to construct a competitive-binding assay the authors first synthesized CJP702, a biotin analog of BIX-01294. An HTRF binding assay was enabled using a GST-tagged G9a and streptavidin-APC. Compounds known to be competitive with peptide were used to validate the assay and these compounds showed the same rank order in potency as the enzyme assay, although the IC50 values were weaker in the binding assay compared with the enzyme assay. SAM competitive compounds were then tested, but these were found to increase the HTRF signal. Reducing the concentration of GST-G9a and CJP702 from 500 nM to 60 nM and 250 nM, respectively, showed a very low basal signal, which was likely due to nonspecific interactions because this signal could not be reduced by peptide inhibitors. However, addition of SAM or sinefungin (and analog of the enzymatic product SAH) caused a large increase in the HTRF signal. CJP702 was derived from BIX-01294, which is an uncompetitive inhibitor with respect to SAM. Therefore, the reason for increased signal upon addition of SAM to the binding assay was thought to be likely due to CJP702 also acting as an uncompetitive inhibitor, and detailed kinetic analysis confirmed this mode of inhibition for CJP702. Interestingly, SAH did not promote an increase in HTRF signal, suggesting that SAH binds to a different conformation. With this mind, the HTRF signal reports on conformational changes due to SAM-pocket binding. The reconfigured peptide binding assay was then used to measure SAM-pocket binding compounds in a kinetic mode (see second figure). The signal increases are due to conformational changes, which involve several steps, including binding of SAM to free enzyme, resulting in a conformational change in the enzyme allowing CJP702 binding. The rate-determining step is unknown but the t1/2 from kinetic analysis sets the lower limit on the forward rate constants at ∼2.5 min. Uncompetitive inhibitors can be common in certain two-substrate–requiring enzyme classes such as dehydrogenases, and this study suggests that HMTs may also have this property. Contributed by Doug Auld.
Binding of GST–G9a to CJP702 in the HTRF-based assay. (A) Chemical structure of BIX-01294 and CJP702. (B) Inhibition of G9a's enzymatic activity by BIX-01294 and CJP702. (C) Binding of GST–G9a to CJP702 detected in the HTRF assay.
Time course of SAM- and SAH-induced changes (or lack of them) in the G9a–CJP702 binding signal. Here, 50 nM GST–G9a and 200 nM CJP702 were preincubated with 1 nM anti-GST–Eu and 7 μg/ml SA–APC for 2 h. After that, 1, 10, or 100 nM SAM or SAH was added to the binding mixture, and the HTRF signals were monitored every 20 s for 50 min.
Can Kinase Inhibitors Resensitize Antibiotic-Resistant Bacteria?
Shakya T, Stogios PJ, Waglechner N, Evdokimova E, Ejim L, Blanchard JE, McArthur AG, Savchenko A, Wright GD: A small molecule discrimination map of the antibiotic resistance kinome. Chem Biol 2011;18:1591–1601.
Abstract: Kinase-mediated resistance to antibiotics is a significant clinical challenge. These enzymes share a common protein fold characteristic of Ser/Thr/Tyr protein kinases. We screened 14 antibiotic resistance kinases against 80 chemically diverse protein kinase inhibitors to map resistance kinase chemical space. The screens identified molecules with both broad and narrow inhibition profiles, proving that protein kinase inhibitors offer privileged chemical matter with the potential to block antibiotic resistance. One example is the flavonol quercetin, which inhibited a number of resistance kinases in vitro and in vivo. This activity was rationalized by determination of the crystal structure of the aminoglycoside kinase APH(2″)-IVa in complex with quercetin and its antibiotic substrate kanamycin. Our data demonstrate that protein kinase inhibitors offer chemical scaffolds that can block antibiotic resistance, providing leads for co-drug design.
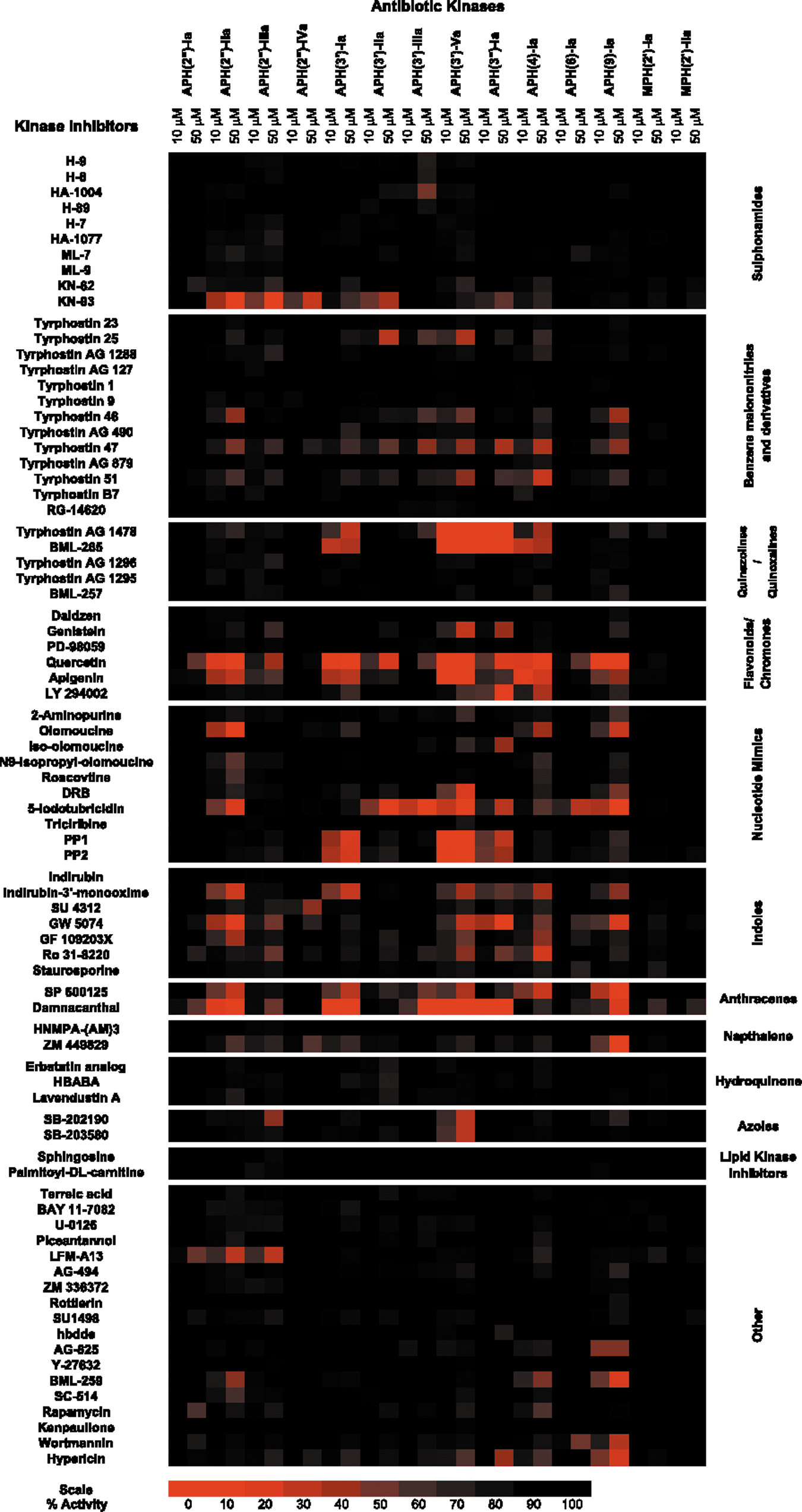
Commentary:Antibiotic resistance is a growing problem that requires novel solutions. Despite the significant energy and resources that have been focused on novel antibiotic scaffolds and inhibiting novel targets and pathways, new drugs are still desperately needed to overcome the increasing resistance to available drugs. Bacteria can gain resistance to antibiotics in several ways, and researchers are now trying to combat these resistance mechanisms head on. For example, the β-lactam–containing antibiotics can be inactivated by β-lactamases. Combinations of a β-lactam antibiotic with a β-lactamase inhibitor have been shown to irreversibly block β-lactamase activity and restore the antibiotic activity. Here, the authors sought to find inhibitors of kinases involved in phosphorylating, and thereby inactivating, the aminoglycoside- and macrolide-containing antibiotics. Fourteen representative antibiotic resistance-conferring kinases were screened against a commercial library of 80 protein kinase inhibitors to generate a small molecule antibiotic kinase interaction map. The authors identified compounds, such as the flavonol quercetin, that inhibited many of the kinases tested, as well as more selective compounds, such as the MAP kinase inhibitor SB-203580 (see first figure). The kinases that confer antibiotic resistance to the macrolide antibiotics were largely unaffected by the compounds in the protein kinase library. Aminoglycoside kinases provide a major route to inactivation of the aminoglycoside family of antibiotics, such as kanamycin. In May 2011, the first structure of a eukaryotic kinase inhibitor with a bacterial aminoglycoside kinase was reported (D.H. Fong et al., PLoS One 2011;6:e19589). Here, a crystal structure of quercetin, the antibiotic substrate kanamycin, and an aminoglycoside kinase are reported (see second figure). Quercetin binds to the hinge region of the kinase and is competitive with ATP as expected. The lack of specificity seen for quercetin binding to the kinases tested here was attributed to the conservation in the sidechains and backbone atoms for the residues involved in binding. The Ki was determined to be 25 μM and the ARF (antibiotic rescue factor; a normalized measure of the increase in cell death from the antibiotic combination with the inhibitor versus the antibiotic alone) was determined to be 0.45 for APH(2″)-Ia. While several of the inhibitors tested here, including quercetin, were able to greatly reduce the antibiotic resistance, none of the inhibitors were able to fully overcome it. Perhaps the potency of the hits could be improved by medicinal chemistry optimization. The co-crystal structure of the antibiotic and kinase inhibitor with the kinase also advances the opportunities for structure-based drug design for this nascent field of aminoglycoside inhibitors. Quercetin is a very promiscuous inhibitor among human protein kinases, as can be gleaned from a search of PubChem, and as such it may not be a viable drug. However, it does provide additional support for targeting a kinase that imparts resistance as a viable strategy for overcoming antibiotic resistance. While this article takes advantage of the similarities in the nucleotide binding sites between human and bacterial kinases, it may be interesting to instead target their differences to develop a selective inhibitor for the bacterial kinase while leaving the human kinases unaffected. Contributed by Mindy I. Davis.
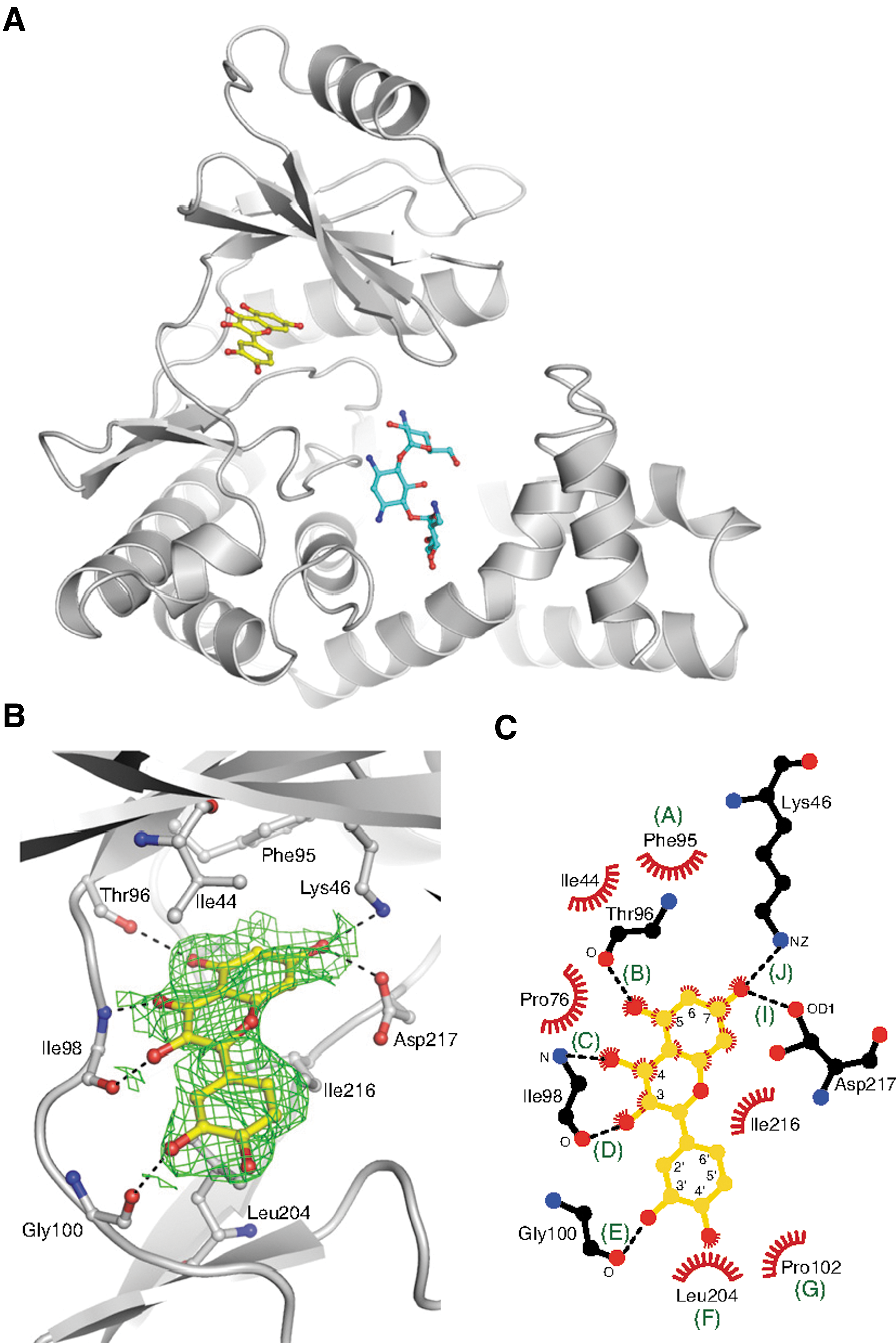
Primary screen of AK (antibiotic kinase) against protein kinase inhibitors. A 2D array of the 14 AKs against the 80 kinase inhibitors tested from the Screen-Well kinase inhibitor library. The average of the residual activities, at both 10 μM and 50 μM, were plotted and assigned a color based on the potency of the inhibitor. Red denotes complete inhibition, and black is no effect. The compounds have been ordered based on relatedness by chemical substructure.
Crystal structure of the APH(2″)-IVa-kanamycin-quercetin complex. (A) Structure of the APH(2″)-IVa-kanamycin-quercetin complex, showing one of the two complexes in the asymmetric unit. (B) Quercetin molecules bound to chain A of APH(2″)-IVa, showing simulated annealing omit Fo-Fc density at 1.0σ (green). Side chains forming interactions with the inhibitor are labeled and hydrogen bonds are indicated with dashed red lines. (C) Mapping of interactions between quercetin and APH(2″)-IVa. Interaction sites in which the distance between the flavonoid and APH enzyme are within 4 Å are labeled with letters A through J, with H-bonds indicated by dashed black lines, and van der Waals interactions indicated by red fans.
DNA Origami
Douglas SM, Bachelet I, Church GM: A logic-gated nanorobot for targeted transport of molecular payloads. Science 2012;335:831–834.
Abstract: We describe an autonomous DNA nanorobot capable of transporting molecular payloads to cells, sensing cell surface inputs for conditional, triggered activation, and reconfiguring its structure for payload delivery. The device can be loaded with a variety of materials in a highly organized fashion and is controlled by an aptamer-encoded logic gate, enabling it to respond to a wide array of cues. We implemented several different logical AND gates and demonstrate their efficacy in selective regulation of nanorobot function. As a proof of principle, nanorobots loaded with combinations of antibody fragments were used in two different types of cell-signaling stimulation in tissue culture. Our prototype could inspire new designs with different selectivities and biologically active payloads for cell-targeting tasks.
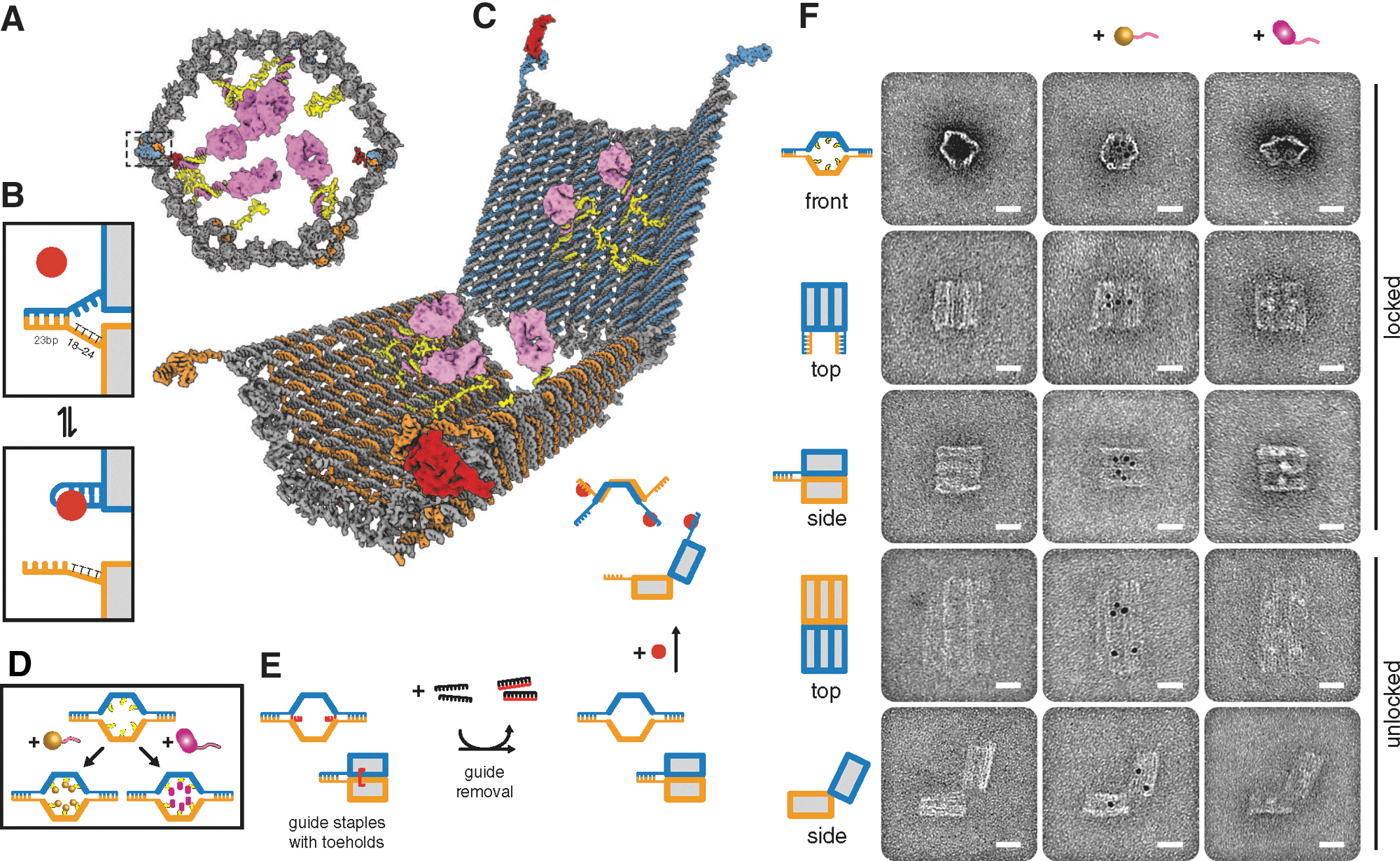
Commentary:Approaches to therapeutic development that go beyond the use of low-molecular compounds have been fruitful, as demonstrated by the success of antibodies to specifically target proteins involved with disease (see also “Conjugation Side Effects” commentary, this issue, p. 122). The field of nanotechnology is also beginning to show promise, with either designed particles or molecular size “nano-machines” being developed to target disease. This article uses DNA as a polymer to fold structures with specific functions. A computer program (“cadnano”) was used to suggest DNA oligonucleotides that are likely to fold into a desired structure. In the study a hexagonal barrel that folds to enclose a payload was made with dimensions of 35 nm × 35 nm × 45 nm. The structure can be unlocked in an “entropic spring” mechanism when the correct antigen binds to DNA aptamer-based locks holding the structure in the closed state (see figure). The synthesized structure was analyzed by atomic force microscopy and transmission electron microscopy (TEM) (see figure). Payloads were placed inside the nanorobot through a 15-base single-stranded DNA linker. It was found that staples, 8-base overhangs placed at the end of the cage, were needed to obtain 97.5% of the nanorobot in the closed form. Several assays were used to validate the delivery of payloads to the specific sites by the nanorobot. For example, six different nanorobots were constructed to contain fluorescently labeled antibody Fab fragments to human leukocyte antigen subtypes, and these were mixed with different cell types containing the appropriate “keys.” Analysis by flow cytometry showed that only cells containing the correct key combinations were able to open the cage and release the labeled Fab fragments. The nanorobot could be engineered so that either a single key or two different keys were needed to open the cage. Thinking of the various combinations of aptamer-encoded locks and keys as a series of 0s and 1s provides a “logic gated” function to the robots. The work also describes payloads that modulated signaling pathways of specific cell types. Nanodevices as described here could be used as new tools to study specific signaling pathways in cells and hold promise for selective delivery of drug payloads to specific cells and compartments. Contributed by Doug Auld.
Design and TEM analysis of aptamer-gated DNA nanorobot. (A) Schematic front orthographic view of closed nanorobot loaded with a protein payload. Two DNA aptamer locks fasten the front of the device on the left (boxed) and right. (B) Aptamer lock mechanism, consisting of a DNA aptamer (blue) and a partially complementary strand (orange). The lock can be stabilized in a dissociated state by its antigen key (red). Unless otherwise noted, the lock duplex length is 24 bp, with an 18- to 24-base thymine spacer in the nonaptamer strand. (C) Perspective view of nanorobot opened by protein displacement of aptamer locks. The two domains (blue and orange) are constrained in the rear by scaffold hinges. (D) Payloads such as gold nanoparticles (gold) and antibody Fab′ fragments (magenta) can be loaded inside the nanorobot. (E) Front and side views show guide staples (red) bearing 8-base toeholds aid assembly of nanorobot to 97.5% yield in closed state as assessed by manual counting. After folding, guide staples are removed by addition of fully complementary oligos (black). Nanorobots can be subsequently activated by interaction with antigen keys (red). (F) TEM images of robots in closed and open conformations. Left column, unloaded; center column, robots loaded with 5-nm gold nanoparticles; right column, robots loaded with Fab′ fragments. Scale bars, 20 nm.
Resolving Pharmacological Responses
Miller OJ, Harrak AE, Mangeat T, Baret J-C, Frenz L, Debs BE, Mayot E, Samuels ML, Rooney EK, Dieu P, Galvan M, Link DR, Griffiths AD: High-resolution dose–response screening using droplet-based microfluidics. Proc Natl Acad Sci USA 2012;109:378–383.
Abstract: A critical early step in drug discovery is the screening of a chemical library. Typically, promising compounds are identified in a primary screen and then more fully characterized in a dose–response analysis with 7–10 data points per compound. Here, we describe a robust microfluidic approach that increases the number of data points to approximately 10,000 per compound. The system exploits Taylor–Aris dispersion to create concentration gradients, which are then segmented into picoliter microreactors by droplet-based microfluidics. The large number of data points results in IC50 values that are highly precise (±2.40% at 95% confidence) and highly reproducible (CV = 2.45%, n = 16). In addition, the high resolution of the data reveals complex dose–response relationships unambiguously. We used this system to screen a chemical library of 704 compounds against protein tyrosine phosphatase 1B, a diabetes, obesity, and cancer target. We identified a number of novel inhibitors, the most potent being sodium cefsulodine, which has an IC50 of 27 ± 0.83 μM.
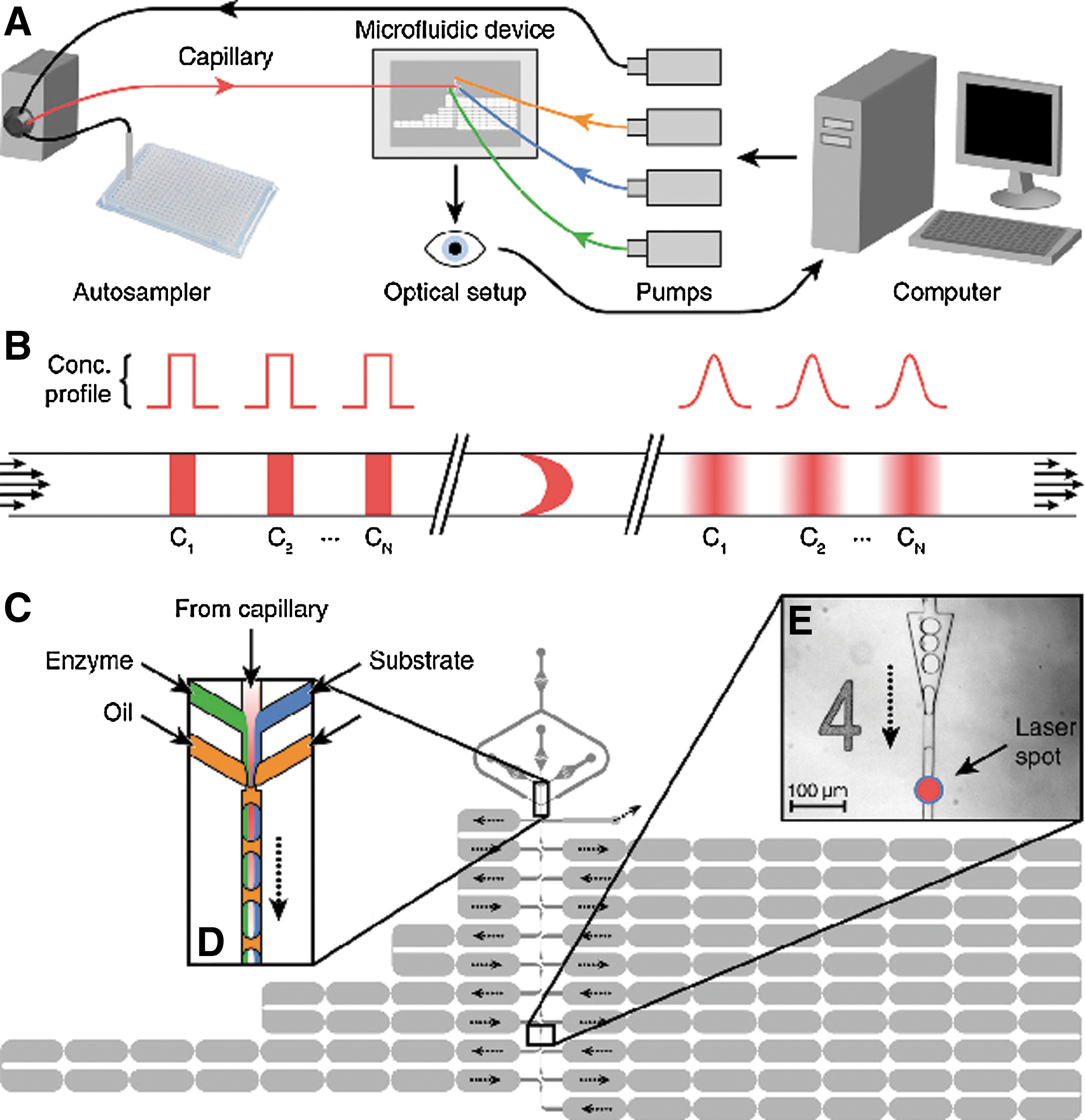
Commentary:Understanding pharmacology stems from accurately measuring concentration–effect relationships. Generally, concentration response curves (CRCs) can be divided into three regions. These include a region of biological deficiency occurring at low doses at which no effect is observed (characterized by the “NOEL” point, the “no-effect-limit”), a region of biological efficiency at which a biological effect is observed, and a region of toxicity at which deleterious effects are observed at higher doses. Exactly how all three of these regions are modulated by a particular substance can lead to many types of complex pharmacology, including bell-shaped CRCs, partial agonism/antagonism, and steep or shallow Hill slopes—all of which can occur over several logs in concentration. For compounds, several strategies are used to capture and measure the activity in bioassays. For large chemical libraries (>1 million compounds) high-throughput screening (HTS) is used in which libraries are screened at typically one concentration and “hits” are then followed-up with 8- to 16-point CRCs.Another HTS approach (qHTS) uses a flexible plate archive allowing screening of medium-size libraries (up to ∼350,000 compounds has been accomplished; see Michael et al., Assay Drug Dev Technol 2008;6:637–658) at up to seven concentrations which can capture complex pharmacological relationships during the screen. This article provides a system that uses droplet-based microfluidics to generate highly precise CRCs containing ∼10,000 data points. In the biochemical assays shown, these CRCs could be collected at rate of 1 every ∼2.6 min. These high density data come from mixing the assay components with a compound concentration gradient that is formed inside the microfluidics (see figure). An initial bolus of compound is injected that undergoes Taylor-Aris dispersion, which converts the rectangular concentration profile into a smooth Gaussian profile. To calculate the concentration in the gradient, an encoder fluorophore (a near-infrared dye, DY-682) is added to the compound sample. The detected concentration range can cover approximately 3 orders of magnitude, although the authors note that this could be increased to 4 or 5 orders of magnitude by using an improved fluorescence detector. The enzyme assay is dispersed into fluorinated oil, which forms small aqueous drops that are mixed with portions of the compound concentration gradient (see figure). This procedure requires approximately 18 times less reagent than a traditional 8-point CRC. Therefore, this technology provides for highly precise measurements of CRCs with low reagent consumption. The article shows collection of high resolution CRCs for β-galactosidase and PTP1B and the use of this technology to screen small libraries in a dose–response mode. The microfluidic system should be applicable to any fluorescent enzyme assay. In the article, a complex CRC was characterized for suramin against PTP1B in which inhibition and activation of enzyme activity occurred over a narrow range. The use of a concentration gradient as described here eliminates some issues with compound presentation such as carry-over on pintools. Adapting this to rapid response cell-based assays could enable the use of primary cells due to the low volumes involved. Increasing the throughput of the system would allow addressing larger libraries with very low reagent consumption, which would expand on the paradigm of qHTS that aims to reduce false positives and negatives by measuring the pharmacology of compounds at the level of the primary screen. Contributed by Doug Auld.
The microfluidic screening system. (A) Overview of the system. (B) Schematic showing how sequential injections of different compounds from the autosampler (C1 to CN) are transformed into smooth pulses by Taylor–Aris dispersion in the capillary. Each pulse gradually rises and then falls in concentration as it arrives at the subsequent microfluidic device. (C) Design of the microfluidic device (plan view) showing the two depths of channel: 25 μm (dark gray) and 75 μm (pale gray). Dotted black arrows show the route the droplets take through the device. (D) Schematic of the droplet production region of the device. Smoothed compound pulses from the capillary are combined with the enzyme (green) and the substrate (blue) and are then segmented into droplets by two streams of fluorinated oil (yellow). Each droplet contains a different concentration of the compound but constant concentrations of enzyme and substrate. (E) Light micrograph of one of the 10 analysis points with the position of the laser spot indicated just after the triangular droplet-respacing feature.
Oligobody for Immuno-PCR
Kazane SA, Sok D, Cho EH, Uson ML, Kuhn P, Schultz PG, Smider VV: Site-specific DNA-antibody conjugates for specific and sensitive immuno-PCR. Proc Natl Acad Sci USA 2012;109:3731–3736.
Abstract: Antibody conjugates are widely used as diagnostics and imaging reagents. However, many such conjugates suffer losses in sensitivity and specificity due to nonspecific labeling techniques. We have developed methodology to site-specifically conjugate oligonucleotides to antibodies containing a genetically encoded unnatural amino acid with orthogonal chemical reactivity. These oligobody molecules were used in immuno-PCR assays to detect Her2+ cells with greater sensitivity and specificity than nonspecifically coupled fragments, and can detect extremely rare Her2+ cells in a complex cellular environment. Such designed antibody-oligonucleotide conjugates should provide sensitive and specific reagents for diagnostics, as well as enable other unique applications based on oligobody building blocks.
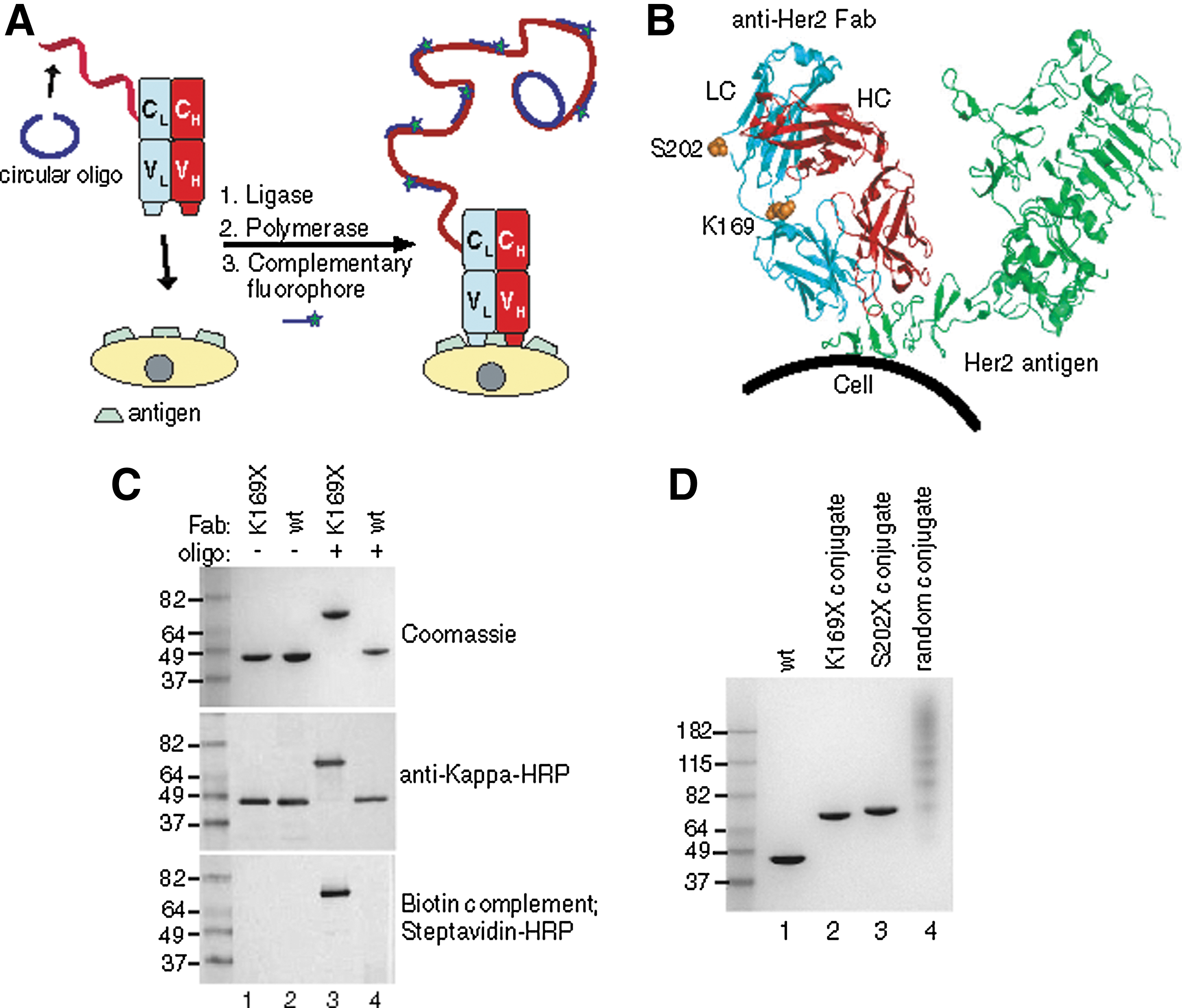
Commentary:Recent advances in the polymerase chain reaction (PCR) method for nucleic acid amplification, as well as in the general field of sample preparation, have made it possible to detect a single copy of target nucleic acid in a range of sample backgrounds. At the same time, however, the detection of rare cell types has remained a daunting task. Advances in this field and the related detection of extremely low-concentration protein biomarkers are going to make possible the detection of circulating tumor cells (CTCs) and ultralow-abundance disease-associated protein variants, thus greatly empowering clinical diagnostics. A recent breakthrough in this area includes the development of immuno-PCR type methods in which a protein analyte of interest (free or cell surface–residing antigen) is recognized through a specific monoclonal antibody, which in turn carries a short single-stranded oligonucleotide. Upon analyte capture (typically achieved in a sandwich-ELISA format with the aid of a capture antibody) and removal of unbound antibody, the complex is detected through a nucleic acid amplification step using the short oligonucleotide as a target. A persistent challenge for practicing such a method remains the difficulty of obtaining a uniform population of site-specifically labeled antibody: this requirement is in place in order to reduce the fraction of antibody molecules with the oligonucleotide tag attached to sites proximal to the antigen recognition hypervariable and adjacent loops. The traditional techniques of reacting activated oligonucleotide fragments with the amines present on the antibody lysine residueshave invariably produced heterogeneous reagents that preclude improvement of the limit of detection. In the present article, Kazane and colleagues generate an antibody that carries a genetically encoded unnatural amino acid with a unique chemical reactivity, thus ensuring a single defined conjugation site. As a model system, the authors used the anti-Her2 receptor antibody trastuzumab and engineered an acetyl-phenylalanine at sites distal to the antigen binding pocket (see first figure). To attach the nucleic acid fragment, a thiol-modified oligonucleotide was coupled to the antibody through the use of an aminooxy-hexyl maleimide bifunctional linker; the identity and precise stoichiometry of the single-site conjugate was verified through electrophoretic analyses (see first figure) and mass spectrometry. Interestingly, in detailed studies of the new conjugate, the authors prepared a comparison reagent using a traditional conjugation technique, through coupling of lysine residues. In parallel tests, the conjugate prepared through the traditional technique was shown not only to be heterogeneous in constitution but also to carry a much higher background, presumably due to the distortion of the antigen recognition pocket by one or more oligonucleotide tags. Finally, the new conjugate was tested in a cell detection model of extremely rare Her2-positive CTCs. In an experiment in which just 11 Her2-positive cells were spiked into approximately 1.4 million white blood cells (WBCs), all 11 cells were detected (see second figure). This represents a powerful example of protein engineering advances being directly used to significantly enhance a clinical diagnostic application. Contributed by Anton Simeonov.
Oligobody construction for immuno-PCR. (A) Scheme depicting site-specific immuno-PCR. A complementary single-stranded oligonucleotide (68 nt) (blue) is annealed to the oligobody (red). T4 ligase is added to form a circular oligonucleotide, which is now the template for RCA. The antibody-oligonucleotide conjugate is bound to Her2 on cells and phi29 polymerase is added to isothermally amplify the DNA, creating multiple copies of the 20 nt sequence. Small complementary oligonucleotides derivatized with Alexa Fluor 488 (20 nt) are then added to obtain a fluorescent signal. (B) Residues (K169 or S202) in anti-Her2 Fab mutated to pAcF for site-specific conjugation are shown in sphere form in orange, in the light chain (LC) in blue and the heavy chain (HC) in red. The Her2 antigen (green) is distant from all mutations. (C) Oligonucleotide conjugation to Fab. Either anti-Her2 pAcF (K169X, lanes 1, 3) or wild-type Fab (lanes 2, 4) was incubated without (lanes 1, 2) or with (lanes 3, 4) 3 mM aminoxy-modified ssDNA (100 mM methoxy aniline, 37°C, 16 h). Reactions were analyzed by SDS-PAGE (top), or transferred to nitrocellulose and incubated with anti-kappa-HRP (middle) or a biotinylated antisense oligonucleotide, then detected with streptavidin- HRP (bottom). The anti-kappa-HRP and streptavidin-HRP blots were developed colorimetrically with the metal enhanced DAB kit (Pierce). (D) Purified site-specific oligonucleotide conjugates and nonspecifically labeled oligonucleotide conjugate. Lanes 2 and 3 correspond to oligonucleotide site-specifically coupled to anti-Her2 pAcF mutant Fab. Lane 4 has multiple oligonucleotides (1–6) coupled to various lysines in anti-Her2 wild-type Fab.
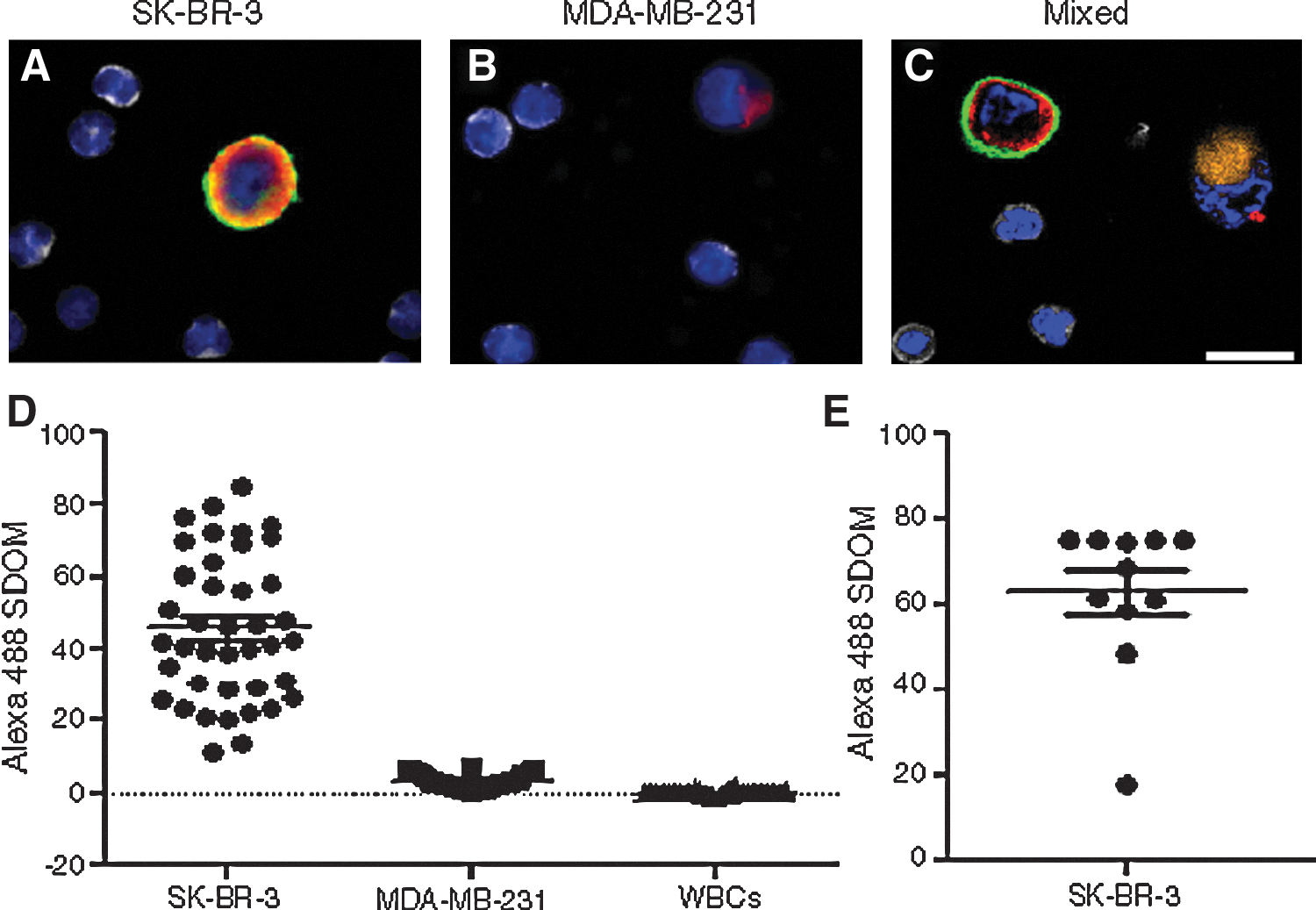
Immuno-PCR in a complex mixture derived from human blood. Cells are identified by nuclear staining (blue). The anti-CK antibodies (anti-CK19, 1:100, Dako and anti-panCK, 1:100, Sigma) (red) stained both the (A) SK-BR-3 cells and (B) MDA-MB-231 cells, whereas the Her2 immuno-PCR with K169pAcF oligobody (green) only stained (A) SK-BR-3 cells. Anti-CD45 (1:125, AbD Serotec) (gray) only stained WBCs. (C) MDA-MB-231 cells were stained with CellTracker Red (Invitrogen) (orange) and both SK-BR-3 and MDA-MB-231 cells were spiked into WBCs. SK-BR-3 cells were stained with Her2 immuno-PCR (green) and CK antibodies (red), whereas MDA-MB-231 cells were only stained with CK antibodies (red). (D) Approximately 1,000 cells were spiked into approximately three million WBCs and scanned on a fluorescent microscope (1:3,000). The Alexa Fluor 488 standard deviation of the mean (SDOM) of a representative amount of cells were graphed. (E) Eleven SK-BR-3 cells were spiked into 1.4 million WBCs, scanned and all cells were identified by immuno-PCR. Scale bar, 25 μm.
Conjugation Side Effects
Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR: Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 2011;30:184–189.
Abstract: The reactive thiol in cysteine is used for coupling maleimide linkers in the generation of antibody conjugates. To assess the impact of the conjugation site, we engineered cysteines into a therapeutic HER2/neu antibody at three sites differing in solvent accessibility and local charge. The highly solvent-accessible site rapidly lost conjugated thiol-reactive linkers in plasma owing to maleimide exchange with reactive thiols in albumin, free cysteine or glutathione. In contrast, a partially accessible site with a positively charged environment promoted hydrolysis of the succinimide ring in the linker, thereby preventing this exchange reaction. The site with partial solvent-accessibility and neutral charge displayed both properties. In a mouse mammary tumor model, the stability and therapeutic activity of the antibody conjugate were affected positively by succinimide ring hydrolysis and negatively by maleimide exchange with thiol-reactive constituents in plasma. Thus, the chemical and structural dynamics of the conjugation site can influence antibody conjugate performance by modulating the stability of the antibody-linker interface.
Commentary:The work by Shen and colleagues represents a first-in-kind systematic study of the in vivo stability and efficacy of antibody–drug conjugates (ADCs) as a function of the type and length of linker used, and the site of drug conjugation. ADCs have risen to a new level of prominence recently, with at least one approved product (Brentuximab vedotin, also known as Adcetris) and multiple candidate agents in various stages of development. ADCs consist of an antibody or its respective reduced-size fragment, designed to address itself to a specific cancer cell, and a typically cytotoxic drug covalently attached to it. In the case of Adcetris, an antibody targeting the cell surface antigen CD30 is conjugated to several molecules of the antimitotic agent monomethyl auristatin E (MMAE). The present study compared several key aspects of the ADC as a new therapeutic agent. First, the method for attaching the drug moiety was investigated by comparing the traditionally used methods for conjugation through reactive thiols generated by selective reduction of interchain disulfide cysteine linkages, or through side-chain amines, with a strategy (dubbed THIOMABs) recently developed by the authors in which specific reactive cysteines available for drug conjugation are prepared via protein engineering. Second, different antibody variants were selected for testing based on differences in their local structural environments: three reactive-cysteine variants of trastuzumab were chosen based on whether the conjugation site existed within a particular charged amino acid environment and whether the site was solvent accessible or buried (see first figure). MMAE was attached to the three trastuzumab clones through a maleimido-caproyl-valine-citruline-p-amino-benzyloxy carbonyl (MC-vc-PAB) linker. All three conjugates were shown by LC-MS analysis to contain 1.7–1.9 molecules of the drug, representing a fairly narrow distribution. In a cell assay using the HER2-amplified breast cancer SK-BR-3 cell line, all conjugates displayed comparable activity (see first figure). When the conjugates were tested in vivo, they displayed significant differences in efficacy, presumably due to differences in their pharmacological profiles. Because the clearance rates for the three conjugates were found to be similar, the differences could only be attributed to differential linker stabilities. After detailed investigations into the plasma stability and mechanisms of degradation of the conjugates, the authors concluded that the rate of conjugate clearance in vivo is dependent on the relative rates of catabolism (degradation) and deconjugation of the thiol-reactive linkers in the plasma (see second figure). Cysteine sites with high solvent accessibility are hypothesized to promote linker-maleimide exchange from the antibody to albumin, cysteine, or glutathione in the plasma. In turn, conjugation sites that exist within a positively charged environment are proposed to undergo succinimide ring hydrolysis in the linker, thereby preventing further linker-maleimide exchange and enabling stability. This model of relative conjugate stability was further tested by the generation and studies of AlexaFluor antibody conjugates and by limited studies of the conjugates in mice and cynomolgous monkeys. The lessons learned from this detailed investigation, in particular the environment dependency of succinimide hydrolysis, should inform those working to generate antibody conjugates for therapeutic and diagnostic uses. Contributed by Anton Simeonov.
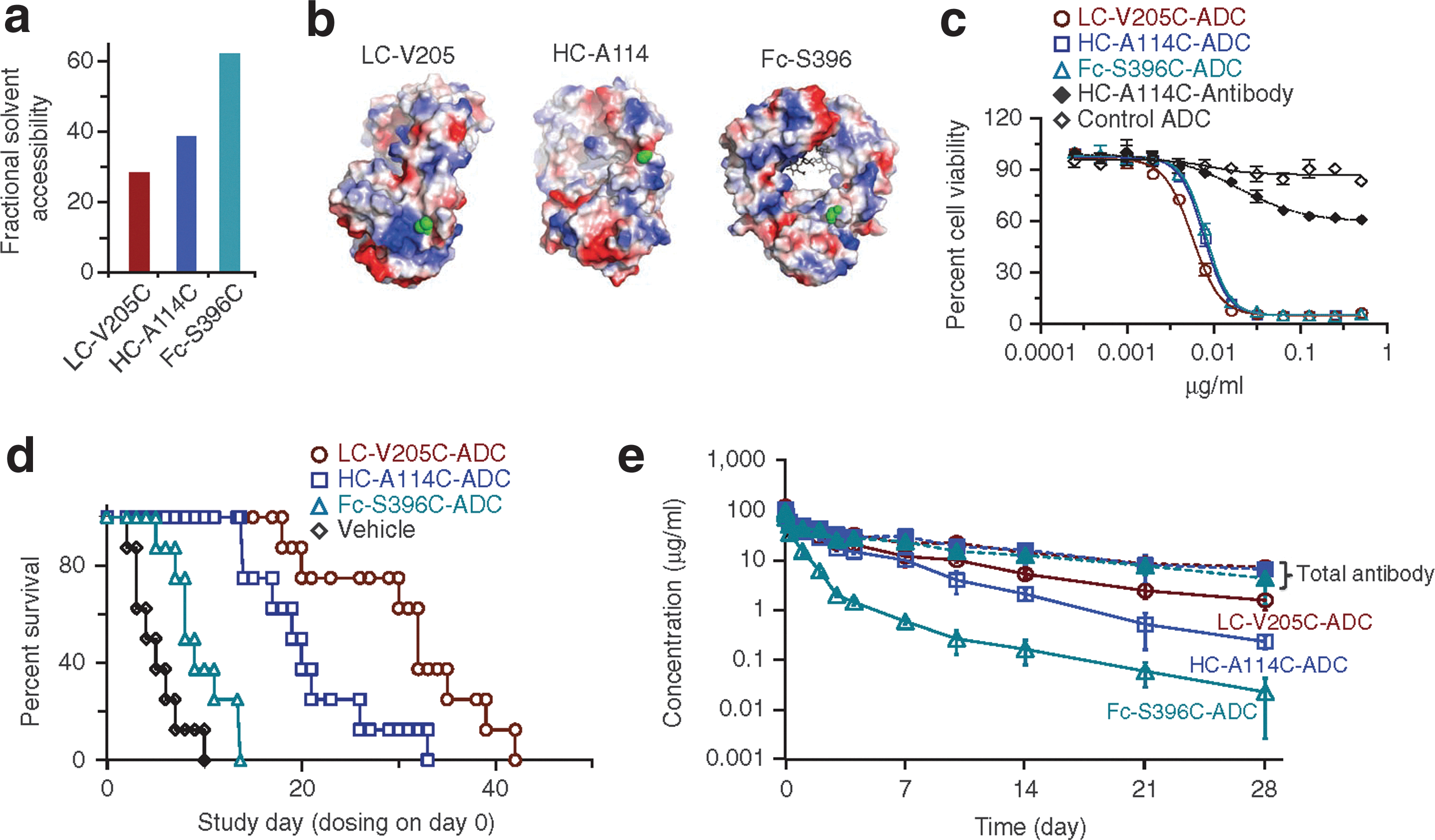
The site of conjugation modulates the therapeutic activity of antibody conjugates. (a,b) Rationale for conjugation site selection. The calculated fractional solvent accessibility values for the selected unconjugated thio-trastuzumab variants (LC-V205C, HC-A114C and Fc-S396C) (a). Electrostatic charge distribution (calculated within PyMOL and colored to make +65 kT/e blue and −65 kT/e red) for surfaces of unconjugated antibody-Fab and Fc reveal that the LC-V205C conjugation site is immediately adjacent to a region of net positive electrostatic charge, and that the HC-A114C and Fc-S396C sites are in neutral electrostatic charge environments. The selected conjugation sites are depicted in green (b). (c) Thio-trastuzumab-MC-vc-MMAE ADCs show similar in vitro potencies. LC-V205C-ADC, HC-A114C-ADC and Fc-S396C-ADC, unconjugated HC-A114C-thio-trastuzumab and negative control ADC (thio-anti-CD22-HC-A114C-MC-vc-MMAE conjugate, which binds only B cells) are examined for their ability to inhibit proliferation in HER2-expressing SK-BR-3 cells in vitro. (d) LC-V205C, HC-A114C and Fc-S396C ADCs differ in their in vivo efficacies. Kaplan-Meier plots for delayed time to progression (number of days taken for a tumor to double in size from the day of randomization or for an animal death) in response to ADC treatment. Vehicle is the formulation buffer control without ADC. The difference in time to tumor progression distributions between groups was determined using a nonparametric log-rank test, with a P ≤ 0.05 being considered significant. Fc-S396C-ADC versus LC-V205C-ADC or HC-A114C-ADC: P < 0.0001; and LC-V205C-ADC versus HC-A114C-ADC: P = 0.025. (e) Equally dosed LC-V205C, HC-A114C and Fc-S396C ADCs differ in their pharmacokinetic properties in mice. At the indicated time points, blood was drawn for determination of total antibody (conjugated and unconjugated, solid symbols) and ADC (conjugated only, open symbols) levels using ELISA. The LC-V205C conjugate exhibited greater stability in the mouse plasma than the HC-A114C and Fc-S396C conjugates.
Proposed mechanism for the influence of the conjugation site on linker stability and therapeutic activity of antibody conjugates. (a,b) Consistent mass decreases are associated with each linker-drug loss observed from HC-A114C-MC-vc-MMAE (a, ∼1,200 Da) or HC-A114C-MC-Alexa488 (b, ∼580 Da) conjugates in the plasma. Antibody conjugates were incubated in human plasma at 37°C for 96 h and the samples were analyzed by affinity capture LC-MS to determine the corresponding molecular weights of the ADC or AFC species upon drug/fluorophore loss in the plasma. (c) The maleimide exchange from the antibody conjugate and hydrolysis of succinimide ring in the linker are key steps that influence conjugate stability and therapeutic activity.
Crystallization Screening by ThermoFluor
Santos SP, Bandeiras TM, Pinto AF, Teixeira M, Carrondo MA, Romão CV: Thermofluor-based optimization strategy for the stabilization and crystallization of Campylobacter jejuni desulforubrerythrin. Protein Expr Purif 2012;81:193–200.
Abstract: Desulforubrerythrin from Campylobacter jejuni has recently been biochemical and spectroscopically characterized. It is a member of the rubrerythrin family, and it is composed of three structural domains: the N-terminal desulforedoxin domain with a non-heme iron center, followed by a four-helix bundle domain harboring a binuclear iron center and finally a C-terminal rubredoxin domain. To date, this is the first example of a protein presenting this kind of structural domain organization, and therefore the determination of its crystal structure may unveil unexpected structural features. Several attempts were made in order to obtain protein crystals, but always without success. As part of our strategy the thermofluor method was used to increase protein stability and its propensity to crystallize. This approach has been recently used to optimize protein buffer formulation, thus yielding more stable and homogenous protein samples. Thermofluor has also been used to identify cofactors/ligands or small molecules that may help stabilize native protein states. A successful thermofluor approach was used to select a pH buffer condition that allowed the crystallization of Campylobacter jejuni desulforubrerythrin, by screening both buffer pH and salt concentration. A buffer formulation was obtained which increased the protein melting temperature by 7°C relatively to the initial purification buffer. Desulforubrerythrin was seen to be stabilized by lower pH and high salt concentration, and was dialyzed into the new selected buffer, 100 mM MES pH 6.2, 500 mM NaCl. This stability study was complemented with a second thermofluor assay in which different additives were screened. A crystallization screening was carried out and protein crystals were rapidly obtained in one condition. Protein crystal optimization was done using the same additive screening. Interestingly, a correlation between the stability studies and crystallization experiments using the additive screening could be established. The work presented here shows an elegant example where thermofluor was shown to be a key biophysical method that allowed the identification of an improved buffer formulation and the applicability of this technique to increase the propensity of a protein to crystallize is discussed.
Dupeux F, Röwer M, Seroul G, Blot D, Márquez JA: A thermal stability assay can help to estimate the crystallization likelihood of biological samples. Acta Crystallogr D Biol Crystallogr 2011;D67:915–919.
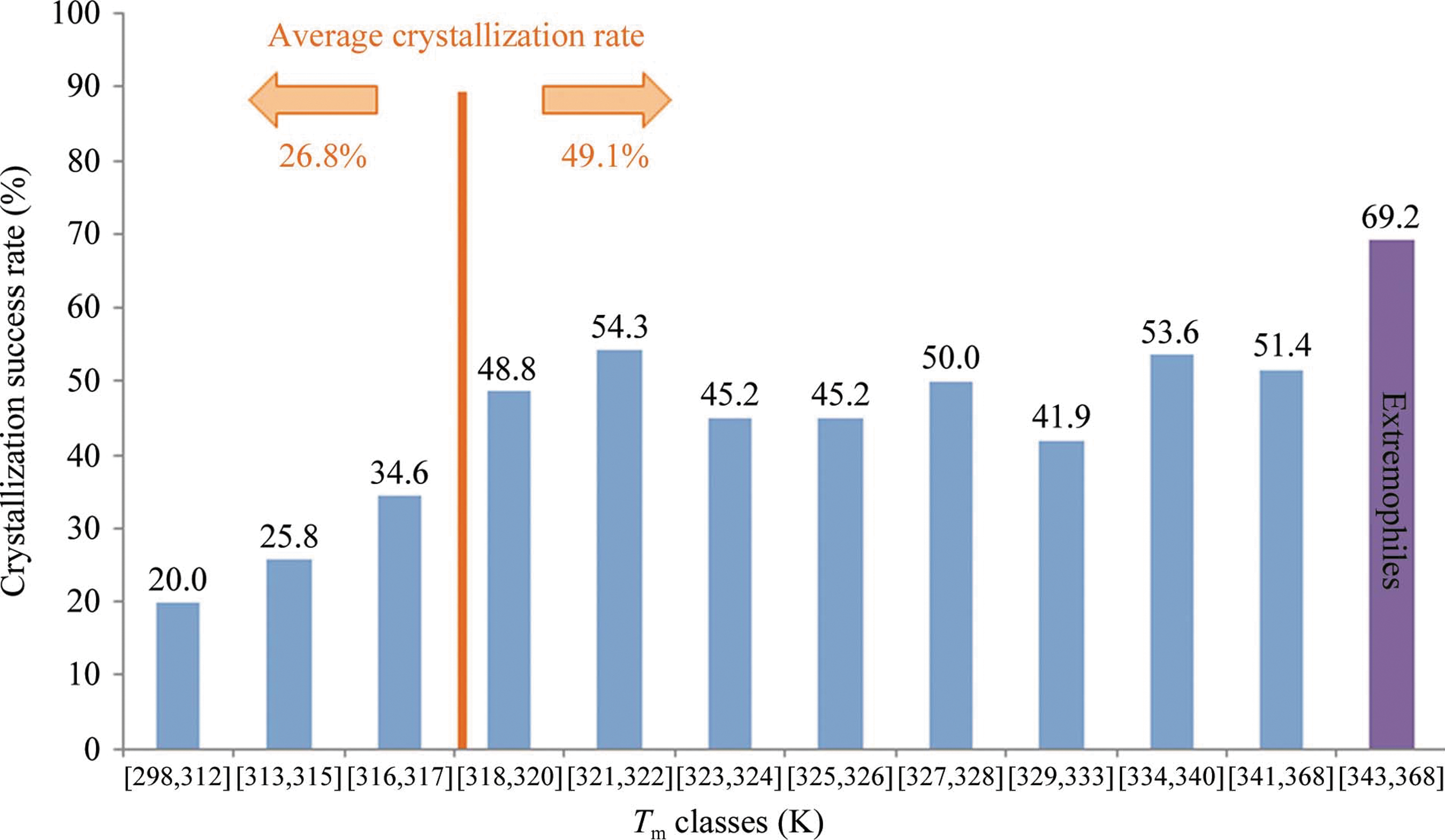
Abstract: The identification of crystallization conditions for biological molecules largely relies on a trial-and-error process in which a number of parameters are explored in large screening experiments. Currently, construct design and sample formulation are recognized as critical variables in this process and often a number of protein variants are assayed for crystallization either sequentially or in parallel, which adds complexity to the screening process. Significant effort is dedicated to sample characterization and quality-control experiments in order to identify at an early stage and prioritize those samples which would be more likely to crystallize. However, large-scale studies relating crystallization success to sample properties are generally lacking. In this study, the thermal stability of 657 samples was estimated using a simplified Thermofluor assay. These samples were also subjected to automated vapour-diffusion crystallization screening under a constant protocol. Analysis of the data shows that samples with an apparent melting temperature (Tm) of 318K or higher crystallized in 49% of cases, while the crystallization success rate decreased rapidly for samples with lower Tm. Only 23% of samples with a Tm below 316K produced crystals. Based on this analysis, a simple method for estimation of the crystallization likelihood of biological samples is proposed. This method is easy, rapid and consumes very small amounts of sample. The results of this assay can be used to determine optimal incubation temperatures for crystallization experiments or to prioritize certain constructs. More generally, this work provides an objective test that can contribute to making decisions in both focused and structural genomics crystallography projects.
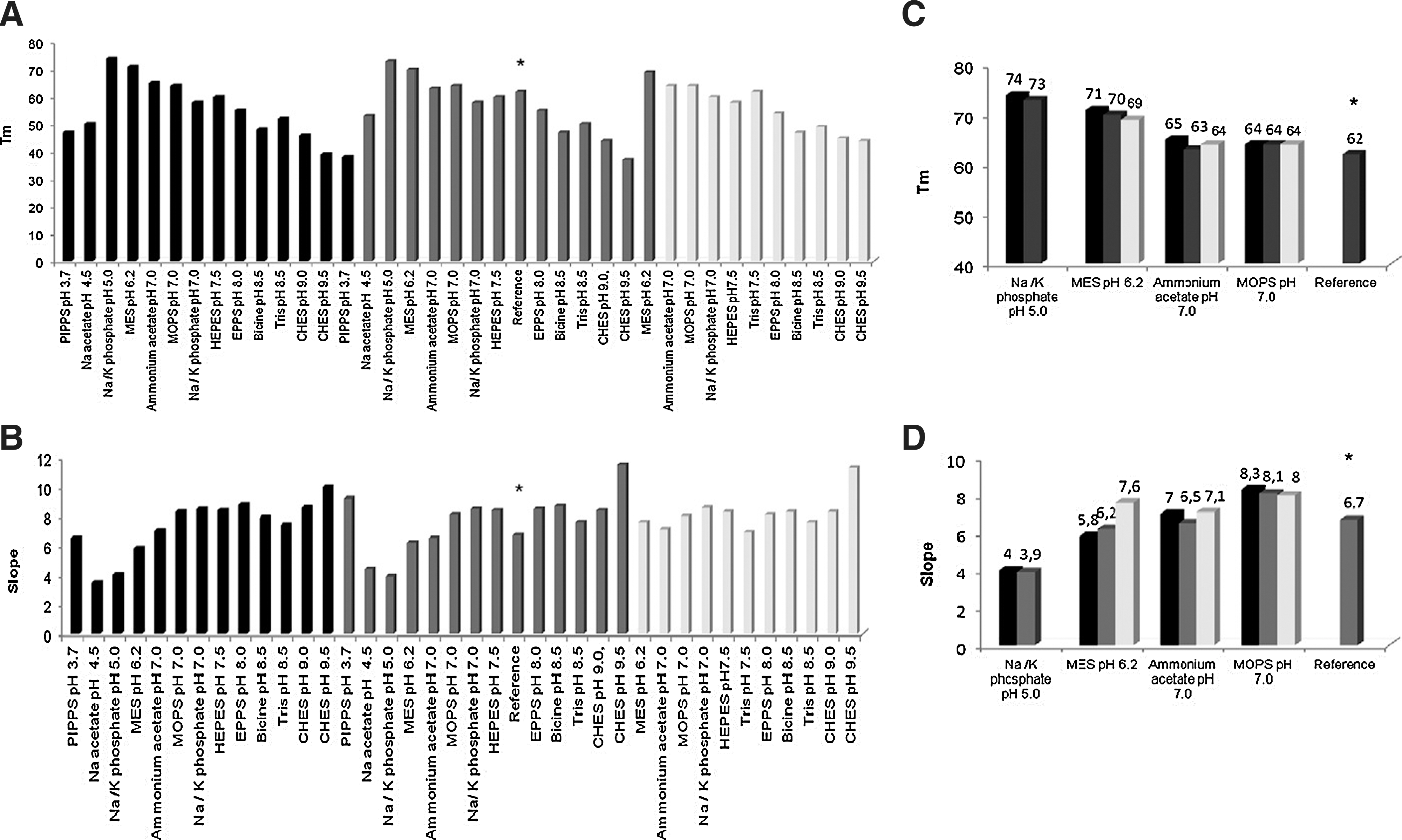
Commentary:ThermoFluor, also known as differential scanning fluorimetry (DSF), is a technique that measures protein stability (quantified by the protein melting temperature Tm) through the fluorescence change in hydrophobicity-dependent dyes. Since its first microplate-based description in 2001 (Pantoliano et al., J Biomol Screen 2001;6:429–440), within the broad context of protein stability applications, DSF has been implemented in small molecule screening (see “Melting Proteins” in Assay Drug Dev Technol 2004;2:579–580). DSF has been commonly applied for the identification of function(s) of unknown protein targets or for additional biophysical characterization of inhibitors to aid the development of structure–activity relationship. DSF is also applicable in screening optimal buffer formulation in order to improve the success rate of protein crystallization, but few reports in this area exist. Herein, two articles are highlighted in which DSF was utilized as a platform to search for optimal crystallization conditions. In the article by Santos et al., the authors performed a series of DSF-based screens with the aim to obtain protein crystals for desulforubrerythrin (DRbr), a protein isolated from Campylobacter jejuni and proposed to be involved in hydrogen peroxide reduction. Functions of DRbr and its family members are yet to be established, with DRbr possessing a unique three-domain (an N-terminal Dx domain, a four-helix bundle domain, and a C-terminal rubredoxin domain) structural organization, rendering its structural determination important in further characterization of this family of proteins. In the initial buffer additives screens, the authors found that the protein yielded both a high Tm and a high transition slope in 100 mM MES pH 6.2, when supplemented with 500 mM NaCl (see first figure). A single sharp protein melt transition is a criterion for the determination of optimal buffer conditions as such transition signifies a homogeneous protein preparation, and for multi-domain proteins, a highly cooperative unfolding pathway (Phillips et al., Curr Prot Mol Biol 2011;10.28.1–10.28.15). For DRbr, a pH of 6.2 lies in the vicinity of the pI of one of the protein's domains that is prone to degradation, the Dx domain, and this pH may further stabilize the protein by neutralizing the domain's charge and reducing its repulsion with other domains. In the subsequent crystallization screens with the addition of different additives that favored the protein's stability, the authors further identified TCEP as the most promising additive improving the quality of protein crystals. Conditions that were found to stabilize protein in the DSF screens correlated well with the quality of protein crystals, further validating the criterion (a single sharp Tm transition, i.e., a high Tm and a high slope at Tm) used during buffer condition assessment and the usefulness of DSF for crystallization screening in general.
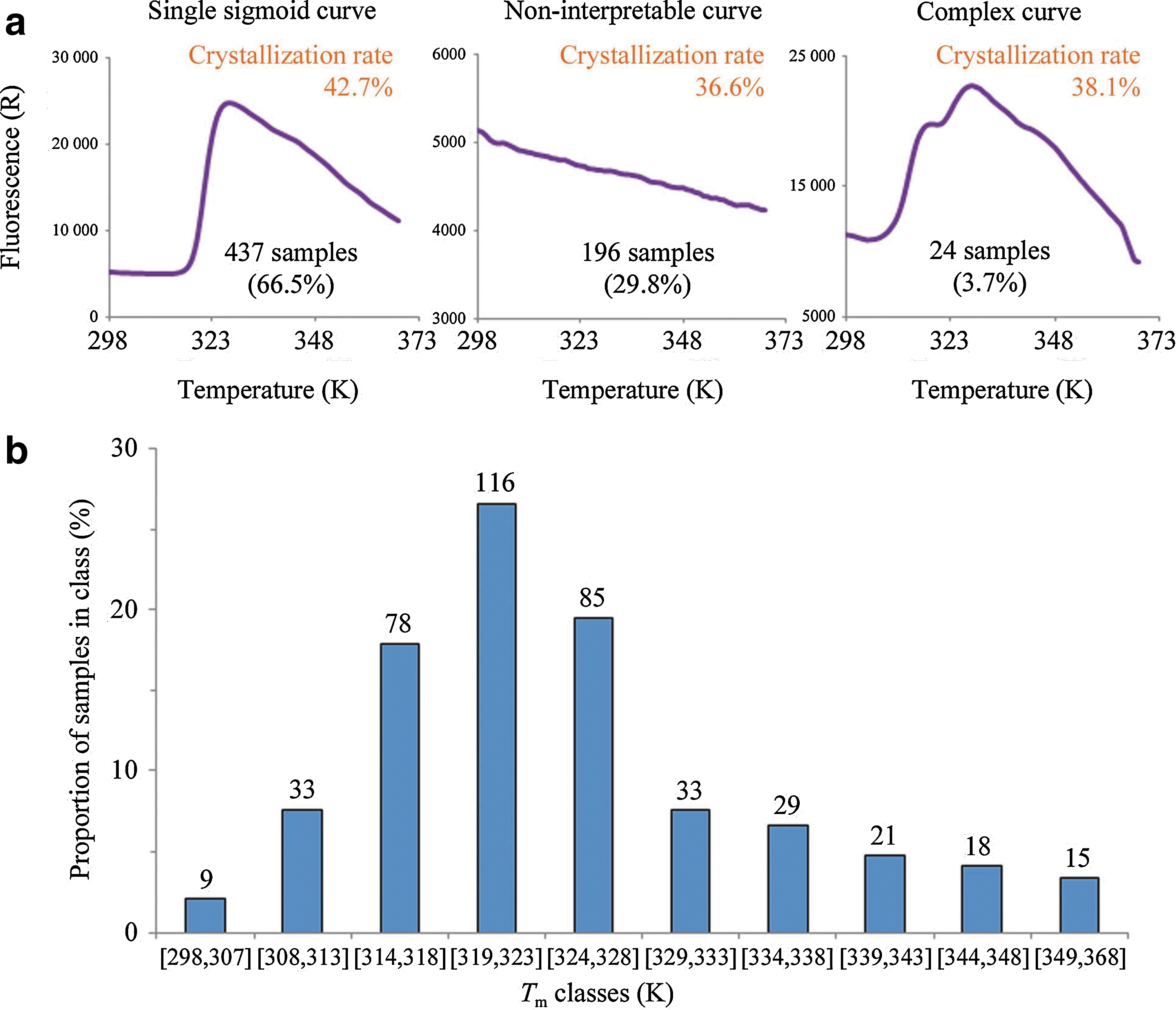
In a complementary article by Dupeux et al., the authors conducted a larger scale thermal stability and crystallization screen, using 657 samples that included proteins from more than 40 species. In a DSF-based screen, the authors identified three groups of proteins, based on the number of melt transitions (0, 1, and 2) (see second figure, panel a). Similar to the criterion applied in the article by Santos et al., Dupeux and coworkers observed that 66.5% of the samples exhibited a single sharp melt transition, and among these samples, the population of their Tm followed an unsymmetrical bell-shaped distribution pattern (see second figure, panel b). Importantly, the authors could establish a relationship between the crystallization success rate and the protein's Tm: samples with their Tm at least 25 degrees higher than the crystallization incubation temperature had a maximized likelihood to crystallize, whereas samples with lower Tms exhibited a sharp decrease in their crystallization success rate (see third figure). This could be explained by the basic concept of protein stability where small perturbation around the protein's Tm could have a much stronger effect on protein unfolding pathway than at temperature regions far away from its Tm. In order to further validate this relationship, using two proteins with lower Tm (313K), the authors reduced the crystallization incubation temperature from the original 293K to 278K so that the incubation temperature would be more than 25 degrees lower than the proteins' Tm. Protein crystals were successfully produced at the adjusted condition while no crystals were observed at the original condition (see Figure 3 in the article by Dupeux et al.). The identification of crystallization success rate as a function of protein Tm could serve as a general guideline in future efforts towards diverse crystallography studies. DSF has been illustrated in both articles as a useful tool in selecting optimal buffer formulation to maximize crystallization success rate. Both studies utilized readily accessible PCR instruments, and the technique called for a relatively lower amount of protein (1–10 μM) owing to the miniaturizable nature of the technique. These are specific examples showcasing the ability of DSF to bridge the gap between protein purification and structural determination. Contributed by Wendy Lea.
Midpoint temperatures of the protein-unfolding transition (Tm) (A, C) and its slope (B, D) for Campylobacter jejuni DRbr. (A, B) in the presence of the buffers that gave a clear thermal transition, and (C, D) in the presence of the four best buffers: sodium/potassium phosphate pH 5.0, MES pH 6.2, ammonium acetate pH 7.0, MOPS pH 7.0. In this study the concentration of salt (NaCl) was zero (black), 150 mM (gray) and 500 mM (light-gray). The control experiment represented as reference (*) was prepared with the final purification buffer, 20 mM Tris-HCl pH 7.2, 150 mM NaCl.
ThermoFluor assay and Tm histogram. (a) According to the results of the ThermoFluor experiment samples can be classified into three groups: those producing a sharp sigmoidal denaturation curve (left), those with no clear temperature transition (middle) and those with complex denaturation curves (right). The crystallization success rates for each of these groups are indicated. (b) Histogram of Tm for the 437 samples producing single sigmoidal curves. The numbers over the bars indicate the number of samples in the class.
Tm and crystallization success rate: crystallization success rates as a function of Tm for samples assayed for crystallization at 293K (excluding samples originating from extremophilic organisms). The crystallization success rate for 12 proteins originating from extremophilic organisms (with Tm between 343K and 368K) is also shown. The numbers over the bars indicate the crystallization success rates for each class. The crystallization success rate decreases rapidly for samples with Tm<318K.