
Abstract


Fernanda Laezza is an Assistant Professor of Pharmacology and Toxicology at the University of Texas Medical Branch. She was born in Italy and came to the United States in 1995 as a graduate student at Emory University to work with Raymond Dingledine. Her work as a graduate student provided the first demonstration of a prominent role of GluA2-lacking glutamate receptors in the induction of synaptic plasticity in the hippocampal circuit. Her interest in the physiology of ion channels and their relationship to excitability and synaptic transmission brought her to Washington University in St. Louis where she undertook her postdoctoral training, initially with Ann Marie Craig and later with David Ornitz and Jeanne Nerbonne. While at Washington University, she developed an interest in voltage-gated sodium channels. At University of Texas Medical Branch (UTMB), Dr. Laezza is establishing a research program focusing on the regulation of neuronal voltage-gated sodium channels by protein–protein interaction. Her research focuses on developing new approaches to studying macromolecular complexes within ion channels and leveraging this innovative source of protein–protein interaction interfaces toward discovery of new intracellular pathways that regulate neuronal excitability in vivo and therapeutic agents that target human channelopathies.
Dr. Laezza, what is the physiological role of Nav channels?
Voltage-gated Na+ (Nav) channels are highly conserved membrane proteins expressed in all excitable cells. In neurons, Nav channels play a fundamental role in membrane excitability by providing the source of intracellular sodium that gives rise to the action potential (AP), a rapidly propagating waveform that constitutes the major functional outputs of these cells. Nav channels are complex heteromeric transmembrane proteins that consist of a pore-forming α-subunit (Nav1.1–Nav1.9), and accessory β-subunits (β1–β4). We are just starting to appreciate that there is a great deal of complexity in terms of the number of intracellular interacting proteins that bind to the intracellular portion of the channels and modulate their function. This complex network of intricate protein–protein interactions (PPIs) determines the activity, surface expression, and membrane localization of the channel, and ultimately controls channel function. These PPIs are of emerging interest in the neuroscience field and in the pharmacology field for drug discovery purposes, and one of these in particular is the main focus of my research at present.
Nav channels are typically silent at the membrane resting potential—meaning that they are closed and not functional; however, they are rapidly activated in response to membrane depolarization. Through a series of conformational changes, the channel opens, inactivates, and closes, and small changes in any step of this cycle can have a dramatic effect on excitability, with significant consequences for neuronal output. For example, slight modifications in the inactivation process, which is a nonconducting state in which the channel is not formally closed but does not conduct current, can dramatically affect the shape and frequency of APs. This knowledge has therapeutic value. For example, slowing or blocking inactivation is actually the mode of drug action of some anti-epileptic drugs, such as lamotrigine or phenytoin.
In neurons, pattern expression analysis of Nav channels shows that they are clustered at a high concentration in the proximal region of the axon—the axonal initial segment (AIS)—where they have a critical role in the initiation and propagation of APs. They are also present throughout the axon in lower concentrations, which allows the AP to propagate down to the end of the axon to the presynaptic terminal, inducing the release of vesicles filled with neurotransmitter and mediating synaptic transmission to neighboring neurons.
Importantly, although Nav channels are concentrated in the initial portion of the axon, they are also expressed, though diffuse, in the soma and in dendrites, where they play critical roles in a phenomenon called back-propagation of the AP, which influences synaptic plasticity and signal integration in neurons. So, Nav channels are fundamental players in intrinsic excitability, synaptic transmission, and signal integration.
Because the AP is the major electrical output in neurons and the activity of Nav channels is needed for the generation and propagation of this signal, Nav channels are critical for many cellular aspects of neurons and brain circuitry throughout development and in adulthood. For example, if you were to block Nav channels for a few days in a tissue preparation, you would induce a global remodeling of brain circuitries as part of a homeostatic process that controls synaptic strength. The point is that modulation of Nav channels produces both acute and chronic effects.
If we look at genetic deletion of Nav channels in animal models or mutations of Nav channels in patients, these can be either lethal (in the case of genetic deletion) or can lead to epilepsy, chronic pain, migraine, and other neurological or cardiac conditions that are still in need of efficacious treatments. This emphasizes the importance of Nav channels as therapeutic targets.
An interesting and puzzling finding in my mind is the fact that Nav channels appear to be expressed even in some metastatic cancer cells. Their role in these cells remains an open question, but we know that pharmacological inhibition or silencing of these channels using genetic tools can prevent metastatic spread, suggesting that Nav channel expression contributes to malignant phenotypes. By investigating how they influence malignant cells, we might discover some unexpected roles of these channels in the brain, which may be unrelated to their ability to conduct electrical signals.
What are some of the mechanisms understood to regulate Nav channel expression and function?
In general, much more is known about their function. Nav channels were among the first channels to be studied by biophysicists, so much is known about how the channels open and close and are activated and inactivated. This information has since been confirmed using molecular techniques. One of the most fascinating discoveries was the mechanism of transition of the Nav channel from the open to the inactivation state in response to membrane depolarization. This was first modeled by Clay Armstrong and Francisco Bezanilla 1 and named a “ball and chain” mechanism based on the movement of part of the intracellular portion (the loop connecting domains III and IV) of the channel into the pore, clogging the pore and preventing sodium flux. We now know that mutations in this intracellular region of the channel can lead to epilepsy and other neurological disorders in humans.
Nav channel expression is less well understood than function, and certainly one of the barriers to breach is to develop better probes to tag or follow the expression of these channels at the single-cell level. Unfortunately, these tools are still suboptimal for Nav channels, and questions about how Nav channel proteins are trafficked to the cell membrane and targeted to specific subcellular compartments or what mechanisms control maturation of these channels remain open. Part of my current research focuses on how channel expression and subcellular targeting are regulated by PPIs, specifically by intracellular fibroblast growth factor (iFGF; FGF11–FGF14) (Fig. 1). This family of genes is very unique and fascinating and is fundamental for brain function. We and others have shown that iFGFs play a critical role in regulating Nav channel function. If, for example, you use molecular tools to delete or introduce mutations in FGF14, Nav channel clustering at the AIS, the site of generation of APs, is lost and excitability is impaired, affecting brain circuitries and behavior. This is one example of the evidence to date showing how disruption of these PPI networks can impact neuronal function and emphasizing how important it is to understand the mechanisms of these intracellular interactions to gain a more complete knowledge of how Nav channels (and other ion channels) operate in vivo.
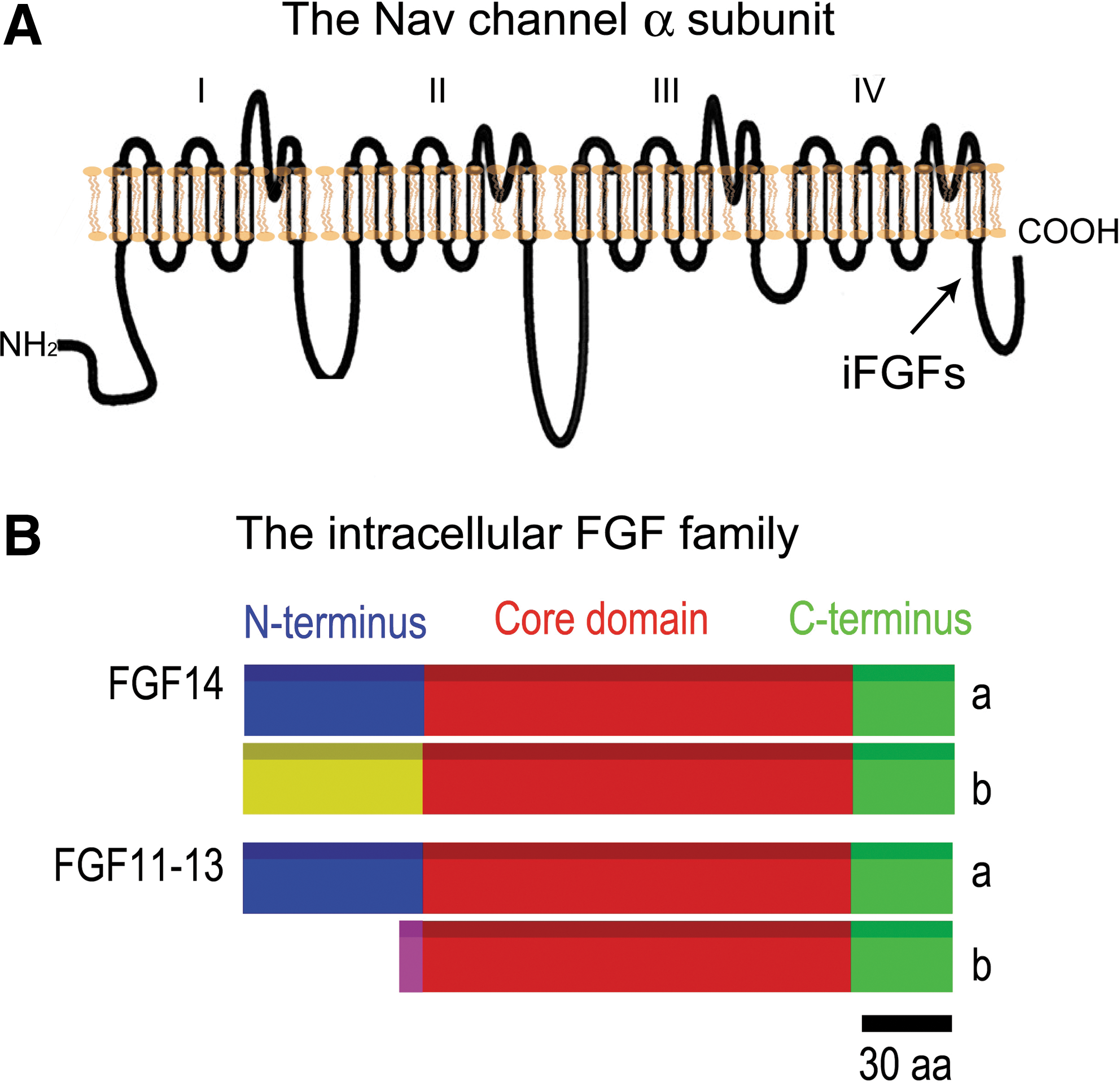
Schematic topology of a typical voltage-gated Na+ (Nav) channel.
How is your work contributing to a better understanding of these mechanisms?
I began as an electrophysiologist studying the role of glutamate receptors in the hippocampal circuitry of the brain, and then came to realize how critical PPIs are in determining the phenotypic expression of native currents and what impact PPIs have on synaptic transmission, excitability, and synaptic plasticity. Ultimately, it is through PPIs that information is decoded from chemical cascades into an electrical output, but how that occurs mechanistically is largely unknown. I started to study Nav channels by chance, focusing on iFGF and discovering with others that those molecules are major binding partners of the Nav channels and critical regulators of their function. We know that PPIs mediated by iFGFs and other proteins within the Nav channel complex have a tremendous impact on Nav channel physiology. Mutations in some of these interacting proteins can lead to mistargeting of these channels in neurons, loss in excitability, and neurological symptoms. Unfortunately, though, the knowledge of the network of proteins that composes the Nav channel in vivo is still far from being complete, and we are quite far behind in having a complete map of this network of proteins in comparison to other ion channels such as the glutamate receptor. I intend for my research to contribute to deciphering the functional significance of the iFGFs and other PPIs toward a better understanding of how the native Nav channelosome operates. Because PPIs can be very specific, an emerging area of research is to target these interactions for drug discovery purposes against brain, peripheral nervous system, or cardiac diseases. Many times we see that a particular disease is caused by a mutation in a specific channel isoform, and targeting PPIs might be an innovative approach for designing channel isoform-specific drugs, a long-standing goal in neuropharmacology.
In the long term, a complete map of these PPIs and their dynamic regulation through post-translational modifications will definitely contribute to a better understanding of how Nav channels function in vivo and possibly provide new platforms for drug development. As Nav channels are large, complex transmembrane proteins, it will be very challenging to achieve these goals, but, hopefully with the implementation of new high-throughput screening technologies, we might have an answer in the near future.
Can you please elaborate on the role of iFGFs and why this protein family is a focus of the research underway in your laboratory?
As opposed to classical FGFs—which comprise the majority of the 24 FGFs and are secreted molecules—iFGFs (FGF11, 12, 13, and 14) remain intracellular and are not secreted. iFGFs are similar to classical FGFs in sequence and structure, but appear functionally different. iFGFs are highly expressed in neurons and exhibit selective regional brain localization.
There are at least two different splice variants for each iFGF, which brings the number up to eight variants (Fig. 1), and each variant appears to have a unique expression pattern. For example, there is an FGF14 a and b, and the b variant is more highly expressed in the brain. For most of the variants, we know that they bind to Nav channels and modify the phenotypic expression of sodium currents. They bind directly to the intracellular carboxy tail of the pore-forming α subunit of the Nav channel, and each interaction results in a different phenotypic expression of the sodium current. More than 10 combinations have been tested so far. 2 –8
We are currently focusing most of our work on FGF14-1b, which is a unique nonredundant molecule required for central nervous system function. With an N-terminal domain distinct from any of the other iFGFs, FGF14 is the only iFGF directly linked to a human brain disorder, such that functional silencing of FGF14 in humans results in spinocerebellar ataxia (SCA)27 characterized by severe mental retardation, ataxia, and neurodegeneration.
Our initial work showed that FGF14 is an integral component of the AIS where it joins Nav channels. Binding of FGF14 controls channel gating and channel expression. We have learned by studying the functional activity of the naturally occurring FGF14F145S dominant negative mutation that loss of FGF14 wild-type function has an impact on neuronal excitability. If expressed in neurons, this mutation decreases sodium currents, impairs neuronal excitability, and disrupts localization of Nav channels at the AIS. Conversely, over-expression of wild-type FGF14 causes opposite phenotypes. 9 This emphasizes how important FGF14 is in the brain and the fact that Nav channels can be controlled bi-directionally by FGF14.
FGF13 is another critical iFGF, with a possible role in the peripheral nervous system. FGF12 and FGF11 have been less well studied. FGF12 knock-out mice are available, and although they do not show pronounced phenotypes in the CNS, they can aggravate FGF14 knock-out mice phenotypes, suggesting a potential functional synergy between different members of the same protein family. This hypothesis still needs to be validated.
We are now focusing on understanding the dynamic regulation of the FGF14:Nav complex using a combination of integrated technologies, including bioluminescence, confocal imaging, and patch-clamp in brain slices, with the long-term goal of translating this knowledge into animal models of disease and hopefully creating new platforms for drug discovery. We envision being able to use this information to design better therapies for channelopathies and other human diseases associated with Nav malfunction.
Your studies on iFGF have provided evidence for an important role of PPIs in regulating Nav channel function. How can this knowledge translate into new drug development?
In general, there is an increasing interest in targeting PPIs for drug discovery, an approach that has been applied in a number of cancer types. 10
A key advantage of targeting PPIs is their specificity, and this might allow us to design very selective and potent drugs—ideally Nav-isoform specific drugs. The Nav channel medications available now originate from a few families of compounds, and they are all targeted at highly conserved portions of the channel, 11 so there is not much opportunity for isoform specificity. Many medications that target Nav or other ion channels have failed due to their cross-reactivity with nontarget channels. Therefore, the idea of using PPIs as potential druggable targets is quite appealing.
For iFGF, I think the key is that each iFGF:Nav channel complex appears to have a different effect on sodium currents. This functional diversity is likely the indication of structural diversity at the level of each iFGF and Nav channel complex. We could exploit these properties and use specific iFGF:Nav channel interfaces for drug development.
What assay strategies would you use to initiate a translational research program for ion channelopathies?
We have explored different assays to screen the PPIs at Nav channel interfaces and we have been successful in using bioluminescence in cell-based assays. 12 These assays, and specifically the split luciferase complementation assay (LCA), are fast and can be rapidly translated into animal studies. This assay strategy was introduced primarily in cancer research; it was shown that you could use cells expressing the split luciferase, implant them in animals, and look at the effects on PPIs in real time with various compounds or manipulations. The LCA uses two luciferase enzyme fragments that have no activity on their own; however, when genetically linked to two suspected PPI partners, the fragments come in close proximity as the two proteins bind, reconstitute the full luciferase enzyme, and generate light upon addition of the appropriate substrate. For now, we have focused on the analysis of FGF14 and the Nav1.6 channel C-tail. 12 In the future, if the assay is adapted to the full-length channel analysis and is combined with automated patch-clamp or ion flux assays, it could be part of a very powerful screening platform for studying PPIs within ion channel macromolecular complexes (Fig. 2).
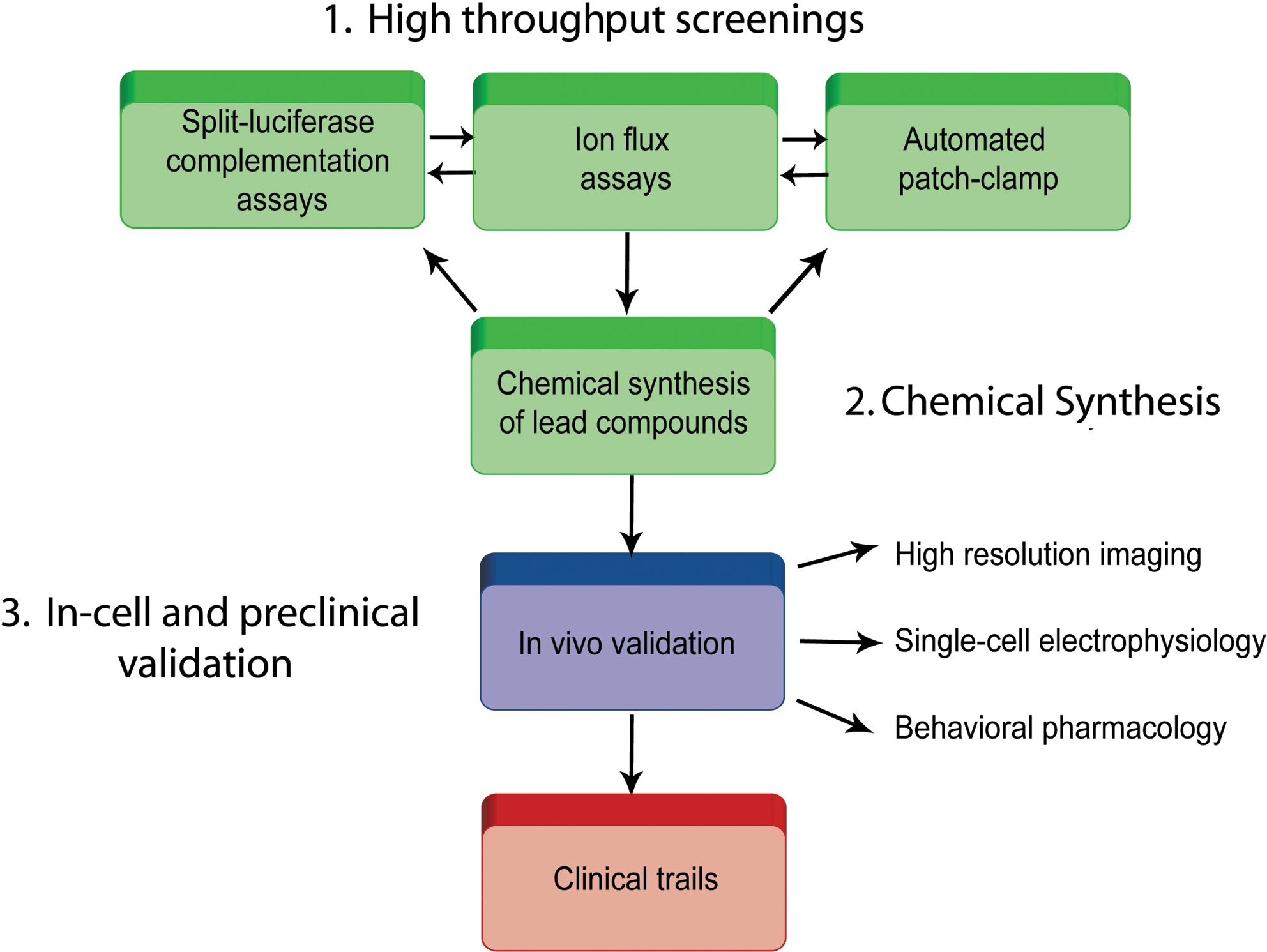
Schematic diagram of a drug discovery campaign targeting protein-protein interactions (PPI) within ion channel complexes. Relevant PPIs might be screened using a combination of protein-fragment recombination techniques, such as the split-luciferase complementation assay, combined with ion flux assays and automated patch-clamp. Following synthesis and validation of new chemical leads, compounds might be validated in-cell and in animal models as a prelude to clinical trails.
When thinking about assay strategies, we need to consider time, money, outcomes, and the target. Once a significant target is identified, then these rapid, in-cell approaches are advantageous because they allow you to skip a lot of in vitro purified protein studies of protein complexes.
What tools does your laboratory employ to study ion channel function and regulation?
We use a combination of approaches including molecular biology, biochemistry, confocal imaging, and electrophysiology. For screening compounds that regulate the FGF14:Nav channel interface we have used LCA, but we continue to use patch-clamp technology in cultured neurons and slice preparation for functional analysis of Nav channels. We also use confocal imaging for studying trafficking of these channels and understanding how PPIs affect their protein expression and distribution in neurons.
We also use recombinant systems in heterologous cells because this is a fast way to validate results before moving into native preparations. Practically speaking, when your research focuses on a gene or a group of genes, it is critical to consider the entire spectra of techniques, ranging from the most reduced to the most complex. Hopefully that will give us the power to understand gene function in its entirety.
What technologies are you aware of that can advance our understanding of ion channel biology and why?
In my mind, one of the current challenges in understanding the biology of ion channels is to resolve the complex regulation underlying PPIs and the dynamic regulation of these PPIs by intracellular and/or extracellular signaling cascades. This is going to be a challenge, but once it is done, it will revolutionize the way we conceive channel function, increase our ability to intervene in ion channel function, and bring us closer to deciphering the “alphabet” of our cells, at least at the level of electrical signals.
How can we achieve this? I do not think there is one single technology that will do it; it will require the integration of many technologies and, most importantly, our ability to analyze and interpret data will make the difference. For example, for PPIs, high-throughput bioluminescence assays, fluorescence-based assays, automated patch-clamp robotic stations, and in vivo validations, combined with appropriate bioinformatics analysis, might be the way to go.
One other unresolved issue is the fact that, believe it or not, we still do not have the structure of any of the mammalian Nav channels—either a crystal or high-resolution structure—although a nuclear magnetic resonance–based structure of the Nav1.2 carboxy-tail 13 and the first crystal structure of a Nav channel from bacteria have recently been made available. 14 I think, overall, structure resolution and large-scale integrated screening for resolving PPI networks and dynamic regulation of PPIs in vivo are the missing links at present.
Are there barriers to the study of ion channel biology that could be surmounted with an appropriate technology? Please elaborate.
I think that one of most fascinating and evolving technologies is the automated patch clamp in combination with evolving high-throughput methods for in-cell analysis of ion channels. For example, toward the goal of resolving the function of specific PPIs or post-translational modifications within ion channels, large, efficient, and fast robotic stations will provide an advantage in time that will make the difference. For resolving millions of combinations of interactions, automation will be essential.
Another barrier to surmount is how to probe ion channels in vivo. Besides pharmacological and genetic approaches, what else is available? We might want to focus on developing new antibodies or chemical probes that could be labeled and followed in real time. Some of the sophisticated optical approaches for activating channels with lasers, such as the laser-activated rhodopsin channel, are very intriguing and give us the power to dissect circuitry with a previously unimaginable precision. You can then transfer these channels to animals, and simply by shining light on the animal, you can fine tune the opening and closing of a channel.
How do you think PPIs will guide drug discovery against ion channels?
As I mentioned, PPIs have recently emerged as attractive targets for drug discovery.
The specificity of the PPI at the binding interface is regulated by a few amino acid “hot-spots.” Peptides, peptidomimetics, or small molecules designed to target these hot-spots can serve as highly selective and potent scaffolds for drug development. The large network of PPIs that comprises the ion channelosome controls neuronal function and could provide a rich source of targets for drug discovery. In the field of Nav channels, the holy grail would be to use PPIs to design isoform-specific drugs.
However, we need to consider the challenges in initiating a drug discovery campaign against a PPI for ion channels. First, it is important to consider what chemistry we want to invest in and what types of problems we might have to contend with further down the road in the drug discovery pipeline. We might develop a peptide that could mimic one of the hot-spots on one of the protein components, for example. Or we might screen against small molecules and look for a compound that can modulate the interaction. Peptides that target an intracellular PPI present several potential challenges: although they are highly selective, they may not penetrate the cells efficiently and reach the target in vivo; they are rapidly degraded; they are structurally unstable in solution; and their active conformations are not easily predictable theoretically or defined experimentally. Nanoparticles for drug delivery might offer a solution to overcome the issue of cell penetration.
On the other hand, small molecules have much better pharmacological profiles, but they are more promiscuous and less specific. When initiating a drug discovery campaign against a target, careful evaluation of all of these elements is essential before investing time and money.
Overall, we need more targeted, specific drugs against ion channels and we have to find innovative approaches to design new medications. Exploring PPIs provides an opportunity to extend the drug search to unexplored chemical space, which is estimated at 1063 molecules! 15 Current drugs cover a very small portion of this chemical space.
What is SCA27? What is the prevalence of SCA27 and how well understood is its etiology?
This is an interesting disease, and it is what first attracted me to the field of iFGF. When we were studying the cellular phenotypes of this disease, we knew that a single amino acid mutation (phenylalanine 145 to serine) in the FGF14 human gene can lead to the disease. It is an autosomal dominant disease with 100% gene penetration. Not many genes that code for Nav channel interactors, when mutated, cause phenotypes as profound as those seen in SCA27.
SCA27 is a rare disease; it has been identified across four generations in a family living in The Netherlands. It is a very severe form of complex ataxia in which patients show a degree of mental retardation, cognitive impairment, tremors, and other neurological signs.
We know that the SCA27 mutant does not bind to Nav channels, but has the ability to associate with the wild-type molecule. In neurons, the FGF14 mutant has strong effects on excitability. It reduces sodium currents, blocks Nav targeting to the AIS, and impairs excitability; this, we believe, will be sufficient to cause the disease. One model proposed is that the SCA27 mutant is acting as a dominant negative, inhibiting the wild-type copy of the gene by binding to it to form a nonfunctional heterodimer. 9 Whether or not there are additional mechanisms of toxicity of the SCA27 mutant is unknown.
One important lesson learned with SCA27 is the importance of investigating rare diseases. It was through SCA27 studies that we unraveled a critical role of FGF14 in controlling excitability and subcellular targeting of Nav channels. We need to continue to explore rare diseases as a means of discovering more unexplored mechanisms and gaining new knowledge that can be translated into other systems. An international effort to bank biopic tissues or other specimens and relevant information on patients with rare diseases presents tremendous challenges, but it would definitely be of value for the rapid translation of animal study results into medication development for humans.
How might one attempt to develop a treatment for this neurodegenerative disease? Is there sufficient understanding of the disease to begin exploring therapeutic strategies?
The easiest and obvious answer is to restore the FGF14 wild-type function and to block SCA27 mutant activity. An immediate approach might involve trying to restore binding of the SCA27 mutant to Nav, or perhaps to design a strategy to eliminate the mutant from circulation and to reinstate the wild-type gene into the body, using a genetic approach or even delivery of recombinant FGF14, which is a relatively small protein comprised of 252 amino acids. One provocative idea to block the mutant would be to create a SCA27 vaccine, similar to what has been proposed and is currently the focus in tremendous research efforts against β-amyloid in the Alzheimer's field.
—Interview by Vicki Glaser
Footnotes
Acknowledgment
We would like to thank the PhRMA Foundation for supporting our research through the Research Starter Grant.