Farra R, Sheppard NF Jr, McCabe L, Neer RM, Anderson JM, Santini JT Jr, Cima MJ, Langer R. First-in-human testing of a wirelessly controlled drug delivery microchip. Sci Translational Med 2012;4:122ra21.
Abstract: The first clinical trial of an implantable microchip-based drug delivery device is discussed. Human parathyroid hormone fragment (1–34) [hPTH(1–34)] was delivered from the device in vivo. hPTH(1–34) is the only approved anabolic osteoporosis treatment, but requires daily injections, making patient compliance an obstacle to effective treatment. Furthermore, a net increase in bone mineral density requires intermittent or pulsatile hPTH(1–34) delivery, a challenge for implantable drug delivery products. The microchip-based devices, containing discrete doses of lyophilized hPTH(1–34), were implanted in eight osteoporotic postmenopausal women for 4 months and wirelessly programmed to release doses from the device once daily for up to 20 days. A computer-based programmer, operating in the Medical Implant Communications Service band, established a bidirectional wireless communication link with the implant to program the dosing schedule and receive implant status confirming proper operation. Each woman subsequently received hPTH(1–34) injections in escalating doses. The pharmacokinetics, safety, tolerability, and bioequivalence of hPTH(1–34) were assessed. Device dosing produced similar pharmacokinetics to multiple injections and had lower coefficients of variation. Bone marker evaluation indicated that daily release from the device increased bone formation. There were no toxic or adverse events due to the device or drug, and patients stated that the implant did not affect quality of life.
Commentary:Implanted medical devices have been around for a while and their uses range from simple mechanical support (joint and bone replacements) to delivery of a drug through an external pump, and include direct control of key organs, such as the pulse of the heart through a pacemaker. While the existing devices are sometimes fairly complex, the next-generation implantables are expected to serve even more complex functions as in the example of a fully implanted drug delivery unit that is responsive to the changing needs for the drug being delivered. Unlike a drug delivery pump, which typically remains attached to the body and has only a feeder line implanted into the patient with all the device controls and access ports being available on the outside, a fully implanted device would be expected to remain autonomous for extended periods of time. Under such conditions, issues such as drug stability, reliability of drug release, acceptance of the device by the host, and stability of communication to control the drug delivery regimen, will all have to be resolved. The present work provides key evidence that producing such small, fully implanted, remotely controlled drug delivery devices is possible. Robert Langer's team used human parathyroid hormone fragment 1–34 (hPTH(1–34)) as the proof-of-concept drug in a clinical trial of an implanted microchip-based drug delivery device controlled through wireless communication (first figure). hPTH(1–34) is the active ingredient of teriparatide, a drug used to treat osteoporosis in patients with elevated risk of bone fractures. The delivery of teriparatide needs to be controlled very carefully because its continuous administration can lead to the unwanted effect of promoting bone loss. The implanted delivery device therefore consisted of two microchips containing 10 small drug delivery reservoirs each; these were designed to deliver individual drug doses of 40 μg upon electrothermal ablation of a thin metallic membrane that covered each reservoir. Seven patients were subjected to the trial, which involved implantation of the device in their abdominal area through an outpatient surgery. After waiting for almost 2 months to create conditions of a fully tissue-encapsulated device, the authors initiated daily dosing of the drug for 20 days; the devices were removed after approximately 3.5 months. PK monitoring of the drug level indicated overall stable and adequate levels of hPTH(1–34), with good rates of absorption and distribution. Evaluation of the histology of the tissue surrounding the devices (referred to as capsule tissue) indicated normal wound-healing responses with very few inflammatory cells, and no malignancies or bacterial infections were noted (second figure). Moreover, the bone markers indicated adequate dosing and in-range responses to the drug, while the test subjects did not experience any out-of-the-ordinary discomfort and managed the post-implantation and -explantation pain with 2-day administration of over-the-counter pain medications. It is expected that these very encouraging results will be followed by the expanded testing of a device that ensures delivery of a drug for at least a 1-year period. Contributed by Anton Simeonov.
The microchip-based drug delivery device and overview of study design. (A, B) Microchip-based hPTH(1–34) drug delivery device (54 mm × 31 mm × 11 mm, l × w × h) (A) containing two microchips with 10 reservoirs each (13.0 mm × 5.4 mm × 0.5 mm, l × w × h) (B). (C) Schematic cross section of microchip assembly showing drug releasing from one reservoir. (D) Timeline of study events.
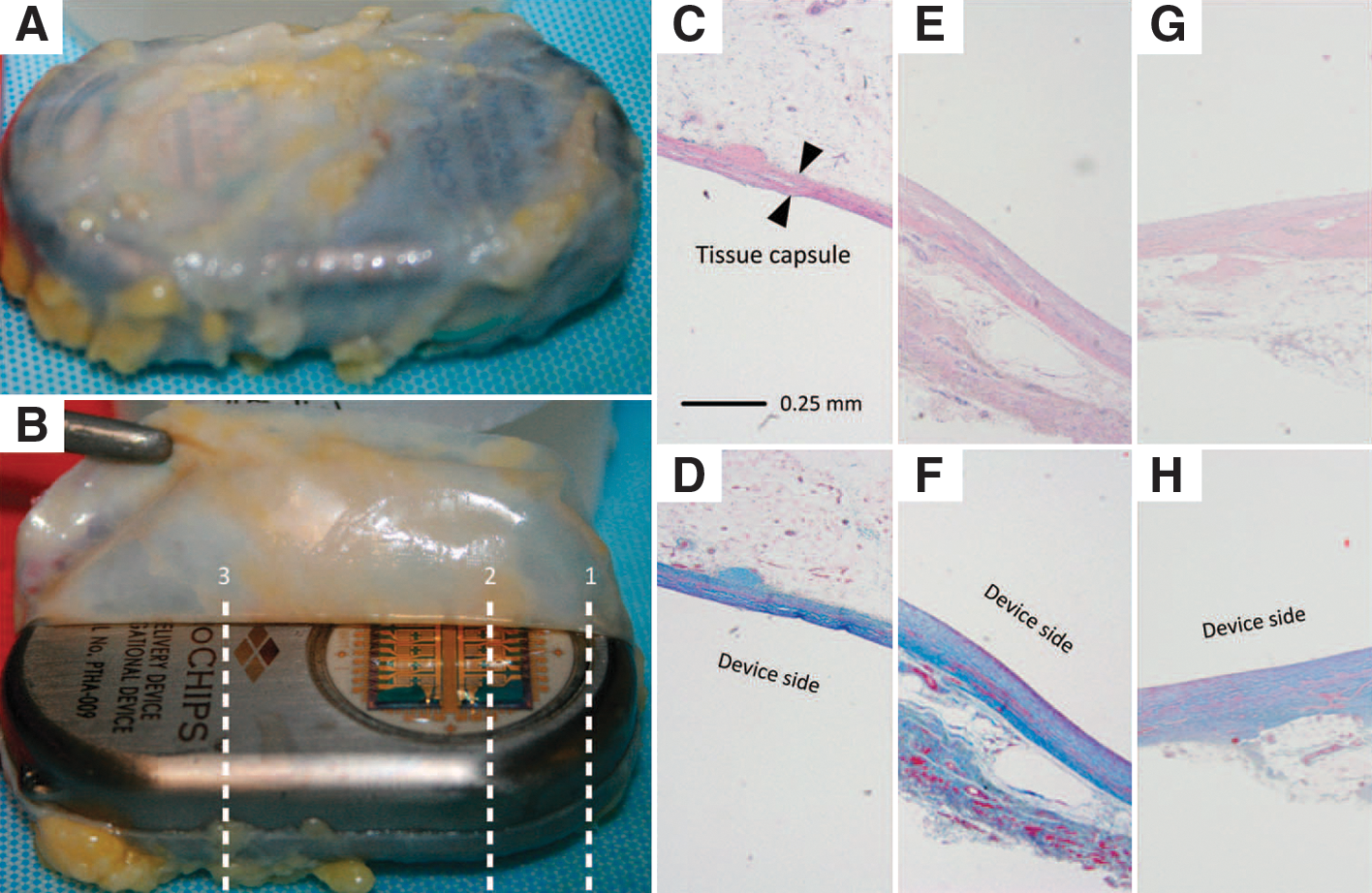
Tissue histology results from a representative patient, MC-0012. (A, B) Two representative macroscopic images of the tissue capsule surrounding the device after explantation. (C–H) Micrographs of the tissue capsule from each patient consisted of three total images from both the dorsal (antenna, toward skin) and the ventral (microchip, toward muscle) sides. Top row, H&E stain; bottom row, Masson's trichrome stain. (C, D) Cross-section 1 at the microchip and titanium interface (dorsal). (E, F) Cross-section 2 over the microchip (ventral). (G, H) Cross-section 3 over the titanium case (ventral).
Subverted by Bacteria
Grover RK, Cheng J, Peng Y, Jones TM, Ruiz DI, Ulevitch RJ, Glass JI, Dennis EA, Salomon DR, Lerner RA. The costimulatory immunogen LPS induces the B-Cell clones that infiltrate transplanted human kidneys. Proc Natl Acad Sci USA 2012;109:6036–6041.
Abstract: The mechanism of chronic rejection of transplanted human kidneys is unknown. An understanding of this process is important because, chronic rejection ultimately leads to loss of the kidney allograft in most transplants. One feature of chronic rejection is the infiltration of ectopic B-cell clusters that are clonal into the transplanted kidney. We now show that the antibodies produced by these B-cells react strongly with the core carbohydrate region of LPS. Since LPS is a costimulatory immunogen that can react with both the B-cell receptor (BCR) and the Toll-like receptor 4 (TLR4), these results suggest a mechanism for the selective pressure that leads to clonality of these B-cell clusters and opens the possibility that infection and the attendant exposure to LPS plays a role in the chronic rejection of human kidney transplants. If confirmed by clinical studies, these results suggest that treating patients with signs of chronic rejection with antibiotics may improve kidney allograft survival.
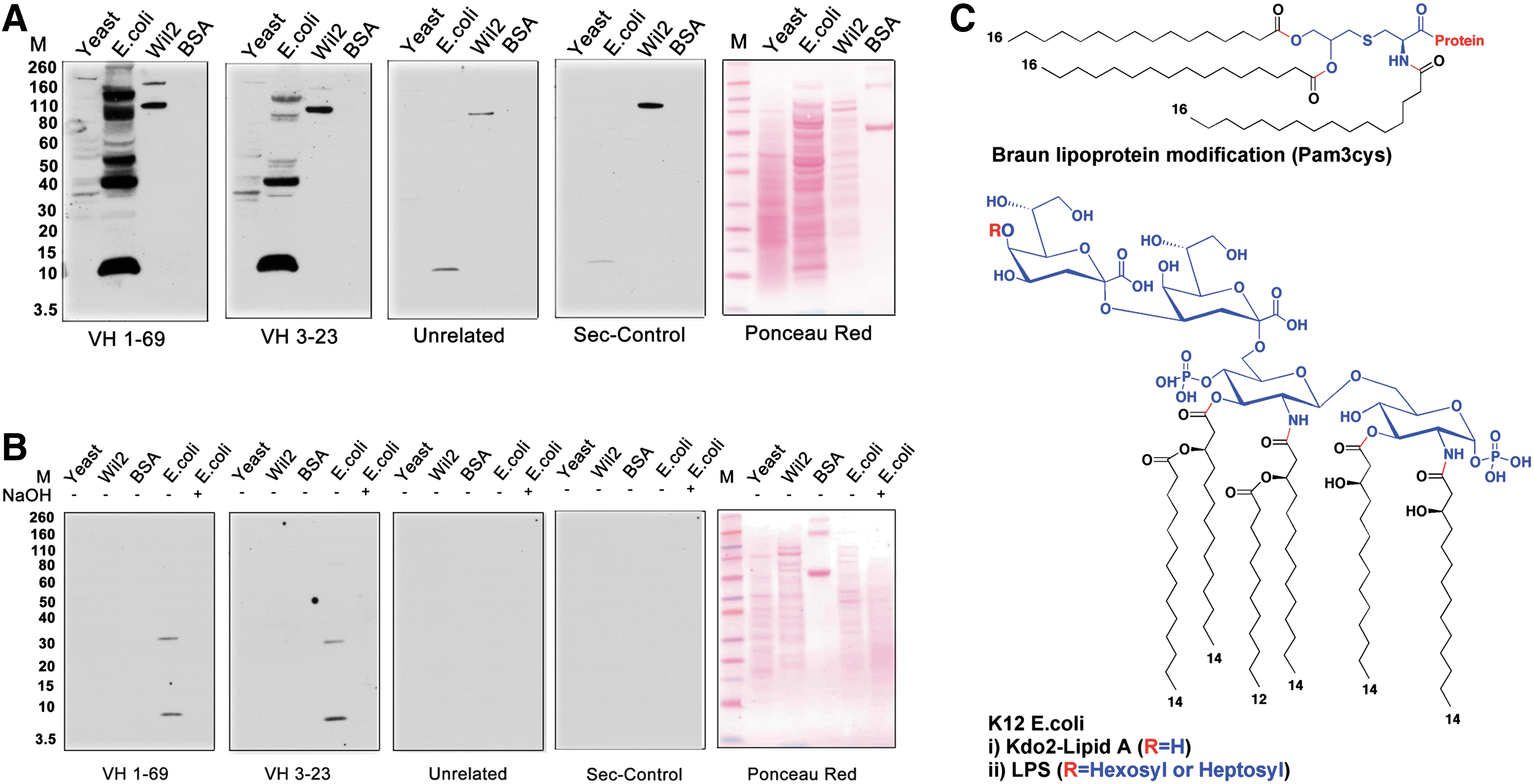
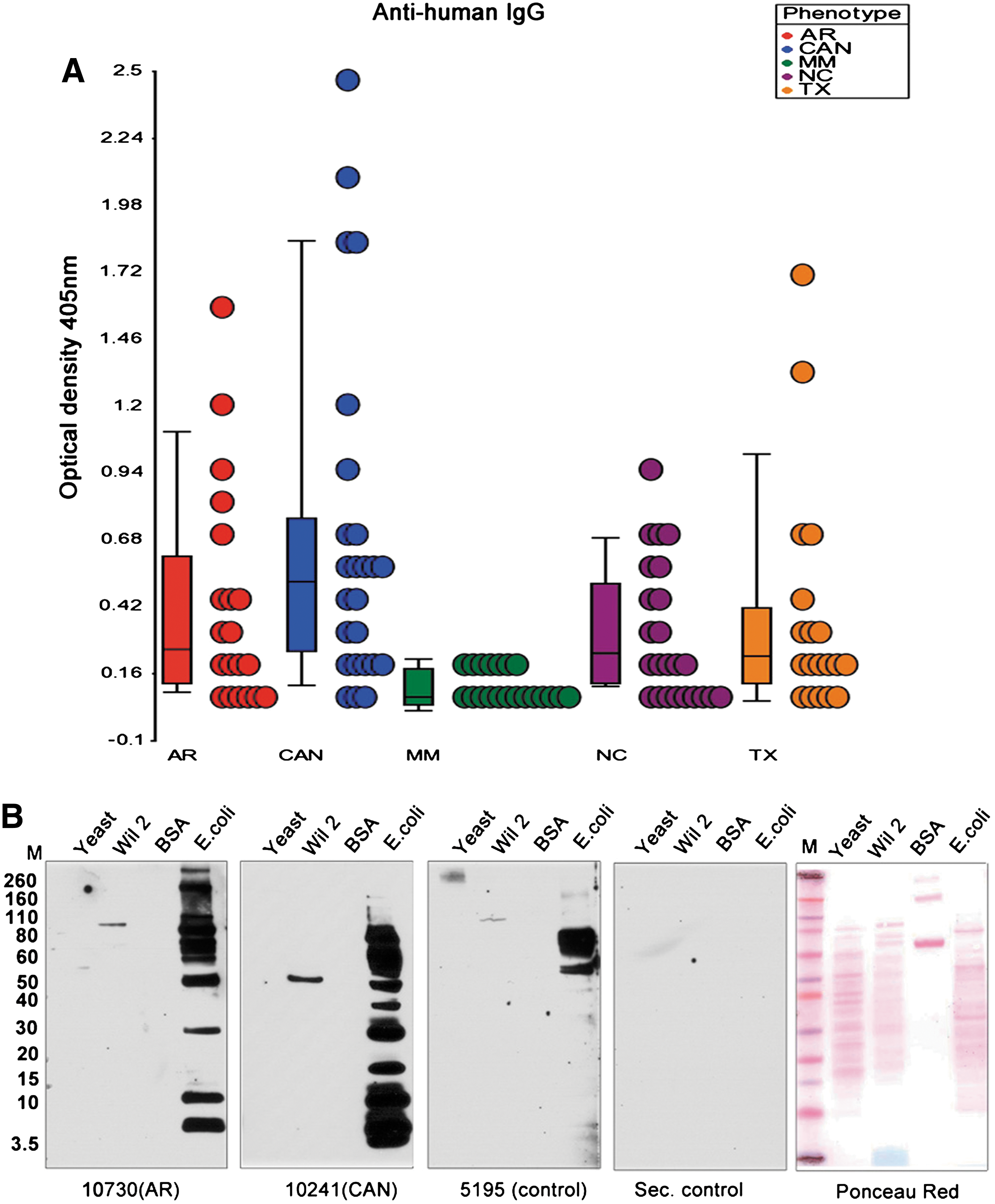
Commentary:The work by Richard Lerner, Richard Ulevitch, and colleagues sheds light on a tantalizing problem associated with kidney transplantation, namely, the unpredictable yet prevalent cases of chronic rejection. Multiple studies have suggested that a major contributor to the rejection is the expression by the host of a large set of genes associated with the innate immune system, as well as the formation of host's B-cell clusters in the transplanted organ. In a true tour-de-force investigation, the team set out to determine the driving force behind the establishment of the B-cell clusters. Recent work by Lerner's group has shown that the clusters are clonal (Proc Natl Acad Sci USA 2011;108:5560–5565), indicating that these are derived through persistent selection by a limited type antigenic pressure. Earlier work by the team had ruled out an alloantigen as the source of this selective pressure. Further, borrowing from the field of gut microbiome, in which it had been demonstrated that persistent infection by H. pylori can lead to sensitization and ultimate malignant transformation of immunocompetent cells into lymphomas, the authors searched for the stimulatory antigen among potential pathogenic bacteria and yeast that could be infecting the transplanted kidney in a recurring manner through a urinary reflux mechanism. Isolated clonal B-cell clusters were used to prepare antibody libraries, and those were tested for cross-reactivity against total cell extracts from Escherichia coli, yeast, and human lymphocytes, and the outcome measured by Western blots (first figure). Cross-reactivity was observed only with the E. coli extracts and only when using libraries derived from transplant patients. To uncover the exact antigen giving rise to the clonal B cells, the authors performed various treatments of the extracts and/or the blots, ultimately demonstrating that the epitope was localized within a 2.3-kDa molecule whose integrity was compromised by treatment with 0.05 M NaOH, indicating that the epitope contained an ester or another type of alkali-sensitive moiety (first figure, panel B). Further analyses led to the conclusion that the causative antigen was a type of lipopolysaccharide (LPS) from E. coli, with the clonal antibodies recognizing primarily the oligosaccharide portion of LPS. To further assess the relevance of the finding, the team analyzed the sera of patients with kidney transplants, by comparing 20 patients with chronic rejection complications against 20 patients whose transplants appeared to be retained without signs of rejection, and further comparing these patient groups with normal individuals and multiple myeloma patients, and found that LPS titers were increased to levels 2.5–15 times higher in the chronic-rejection patient group (second figure). Importantly, it was also shown that the antibodies produced by the B-cell clusters were capable of neutralizing LPS, indicating full functionality and pointing to a direct biological role for the antibodies as actual agents that the host is continuously producing to fight off the recurring assault from pathogenic bacteria. The impact of the present study is at least twofold. First, it demonstrates that even though antibiotics are administered to transplant patients to keep them infection-free, in at least the case of kidney transplants the somewhat unique anatomic environment provides opportunities for recurrent re-infection through the urogenital tract, which in turn leads to the establishment of clonal B-cell populations and ultimate organ rejection though B-cell proliferation, as well as the concomitant production of proinflammatory cytokines and chemokines upon the activation of Toll-like receptor by the LPS antigen. This argues for the search of additional means of controlling bacterial assaults on the transplanted kidneys through either a modified regimen of antibiotic treatments or through other approaches of limiting the LPS effects. Second, the present kidney-focused study can serve as a model for structuring similar investigations to uncover the drivers for rejection of other organs in the human body. Contributed by Anton Simeonov.
(A) Western blot analysis of the reactivity of kidney-derived ScFv antibodies VH169 and VH323 with yeast, K12 strain of E. coli, Wil2 cell extracts, and BSA. Control antibodies were an unrelated ScFv antibody against the IL1-RA protein and the secondary antiflag antibody (Sigma) alone. E. coli and yeast cells were grown in appropriate media and after centrifugation the cells were lysed according to the manufacturer's protocol using lysis buffer from Sigma Aldrich. Nucleic acids were degraded by treatment with DNAase and RNAase. A protease inhibitor cocktail (Roche) was added to prevent proteolytic degradation. The extracts were separated on SDS-PAGE gels and transferred to nitrocellulose membranes for Western blot analysis. (B) Western blot analysis of antibody reactivity after treatment of the extracts with 0.05 N NaOH (lane 5, all panels). The E. coli extract was treated with 0.05 NaOH for 2 h and neutralized to pH 7.0 with HCl. The extracts were separated on SDS-PAGE gels as described in Fig. 1A of the article. (C) Molecular structures of (top) Braun N-terminal (pam3cys) modification of bacterial lipoproteins and (bottom) LPS and kdo2-Lipid A from k12 E. coli.
(A) Analysis of anti-LPS titers in patients and normal controls. Plasma samples were obtained from control normal individuals (NC), patients with chronic allograph rejection (CAN), acute rejection (AR), well-functioning transplants (TX), and multiple myeloma (MM). Twenty samples from each cohort were analyzed by ELISA using the affinity-purified LPS-Lip12Pam3Cys preparation as an antigen. (B) Western blot analysis of Yeast, Wil2, K12 E. coli cell extracts and BSA with the sera from the different cohorts. The antigens were prepared as in first figure, panel A. The extracts were separated on SDS-PAGE gels and transferred to nitrocellulose membranes for Western blot analysis of the plasma. Binding was detected using a secondary antihuman IgG antibody. The control antibodies were normal plasma and antihuman IgG (Southern Biotech.) alone (Sec. control).
Will Less SOX2 Lead to Brain Cancer Getting Cold Feet?
Lee C, Fotovati A, Triscott J, Chen J, Venugopal C, Singhal A, Dunham C, Kerr JM, Verreault M, Yip S, Wakimoto H, Jones C, Jayanthan A, Narendran A, Singh SK, Dunn SE. Polo-like kinase 1 (PLK1) inhibition kills glioblastoma multiforme brain tumour cells in part through loss of SOX2 and delays tumour progression in mice. Stem Cells 2012;30:1064–1075.
Abstract: Glioblastoma multiforme (GBM) ranks amongst the deadliest types of cancer and given this, new therapies are urgently needed. To identify molecular targets, we queried a microarray profiling 467 human GBMs and discovered that polo-like kinase 1 (PLK1) was highly expressed in these tumours and that it clustered with the proliferative subtype. Patients with PLK1-high tumours were more likely to die from their disease suggesting that current therapies are inactive against such tumours. This prompted us to examine its expression in brain tumour initiating cells (BTICs) given their association with treatment failure. BTICs isolated from patients expressed 110–470 times more PLK1 than normal human astrocytes. Moreover, BTICs rely on PLK1 for survival because the PLK1 inhibitor BI2536 inhibited their growth in tumoursphere cultures. PLK1 inhibition suppressed growth, caused G2/M arrest, induced apoptosis and reduced the expression of SOX2, a marker of neural stem cells, in SF188 cells. Consistent with SOX2 inhibition, the loss of PLK1 activity caused the cells to differentiate based on elevated levels of GFAP and changes in cellular morphology. We then knocked-down SOX2 with siRNA and showed that it too inhibited cell growth and induced cell death. Likewise, in U251 cells, PLK1 inhibition suppressed cell growth, down-regulated SOX2 and induced cell death. Furthermore, BI2536 delayed tumour growth of U251 cells in an orthotopic brain tumour model, demonstrating that the drug is active against GBM. In conclusion, PLK1 level is elevated in GBM and its inhibition restricts the growth of brain cancer cells.
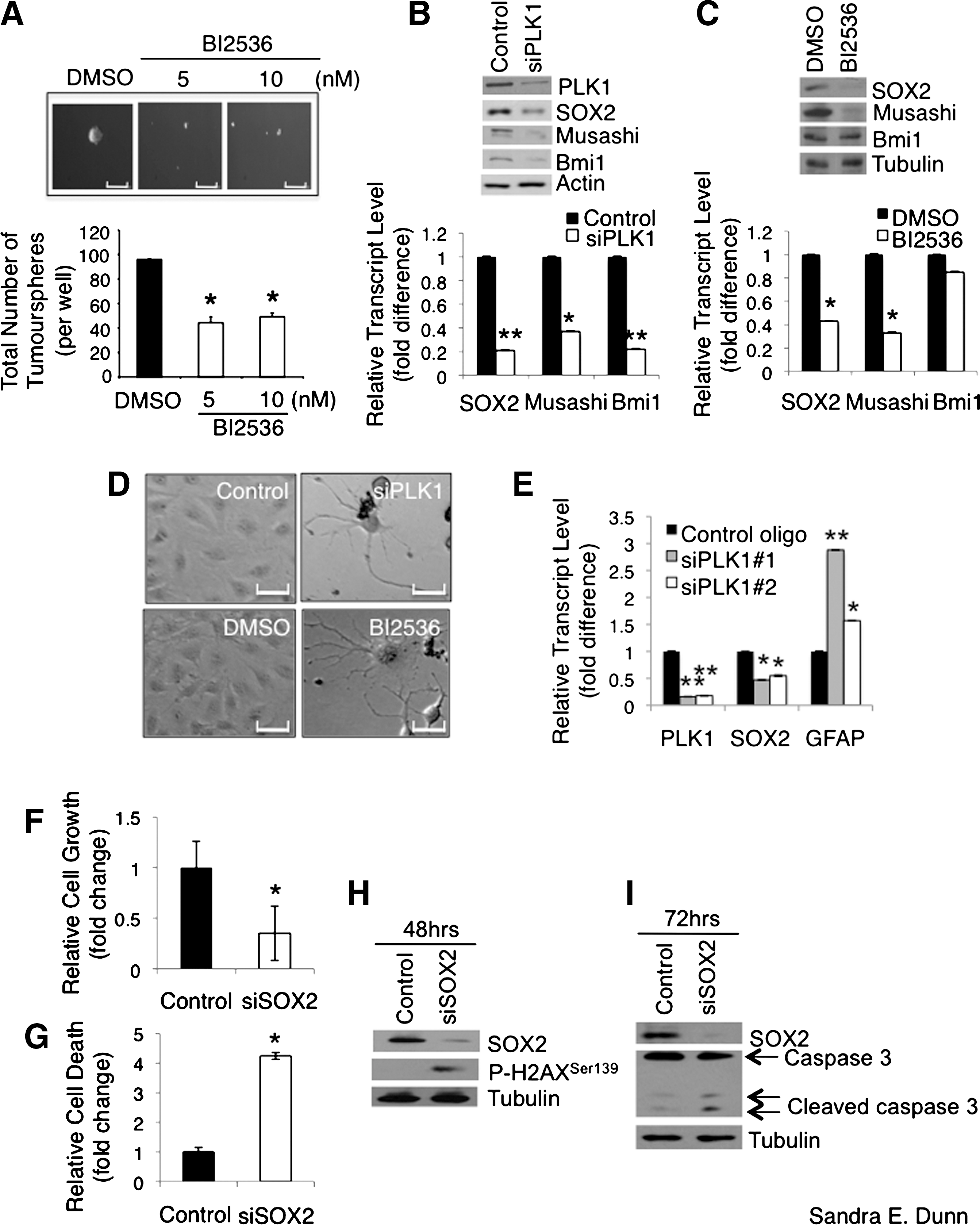
Commentary:The concept of cancer stem cells and the need to target these cells for an effective treatment of cancer has emerged over the past several years. Cancer stem cells are tumor-initiating cells that are in an undifferentiated state and are capable of self-renewal. The authors identify PLK1 (polo-like kinase 1) as a target for the treatment of glioblastoma multiforme based on both siRNA and small molecule inhibition. Glioblastoma is a brain cancer that despite much research still carries a bad prognosis. This is particularly true for the proliferative subtype of the tumors. Brain tumor initiating cells (BTICs) from patients had several hundred-fold more PLK1 as compared to the levels in normal astrocytes. The presence of PLK1 also correlated with disease severity. BTICs have been recalcitrant to chemotherapy and radiation due to their ability to limit apoptosis, repair DNA damage, and efflux drugs. Treatments that leave the BTICs behind may allow the tumor to relapse. Interestingly, the ATP-competitive PLK1 inhibitor BI2536 led to the differentiation of pediatric SF188 cells (see figure). This was accompanied by a reduction in the expression of the neural stem cell marker SOX2, which is important for self-renewal of stem cells. Cell growth was arrested at G2/M and apoptosis was induced. These phenomena were also observed in adult U251 cells and with PLK1 knockdown rather than BI2536 treatment. At low doses (5–10 nM), this drug also blocked the formation of three-dimensional tumorspheres. The ability to dose at low levels would be expected to favor the ability to obtain a therapeutic window. Mice injected intracranially with U251 cells were allowed to develop tumors and were subsequently treated with BI2536 once a week for 4 weeks. The conclusion of the study was that the drug was well tolerated at 50 mg/kg and that there was a significant delay in tumor progression with a corresponding increase in survival. BI2536 is currently in phase I and II clinical trials for other solid tumors. BI2536 is very selective but not as selective as the exquisitely selective imatinib (S[300 nM] = 0.0337 and 0.0233, respectively) (Davis et al., Nat Biotechnol 2011;29:1046–1051). The ability of BI2536 to address some of the stemness characteristics of BTICs may render these cells more susceptible to chemotherapy and radiation. In a separate study, pretreatment with BI2536 was shown to sensitize the medulloblastoma cell line Daoy, which overexpresses PLK1, to ionizing radiation in addition to reducing colony formation and cell growth, inducing apoptosis and decreasing levels of SOX2 (Harris et al., BMC Cancer 2012;12:80). The potential to use a combination therapy of traditional chemotherapeutics and/or radiation with BI2536 for the treatment of glioblastoma multiforme and medulloblastoma looks promising. Contributed by Mindy I. Davis.
PLK1 inhibition down-regulates the expression of SOX2, which is required for the growth and survival of GBM cells. (A) SF188 cells (1 × 104 cells per well in 6-well plates) were plated in neurobasal growth medium containing 5 or 10 nM BI2536 for 6 days. The total number of tumourspheres (>50 μm) in each well was counted and photomicrographs of the spheres were taken (scale bar = 500 μm). The treatments were performed in duplicates on three separate occasions. (B) The transcript and protein expression of neural stem cell markers SOX2, musashi, and Bmi1 were measured by RT-PCR and immunoblotting 36 and 48 hours, respectively, after PLK1 siRNA treatment in SF188 cells. (C) The transcript and protein expression of SOX2, musashi, and Bmi1 were measured by RT-PCR and immunoblotting 36 and 48 hours, respectively, after BI2536 treatment in SF188 cells. (D) SF188 cells were treated with 5 nM of PLK1 siRNA or BI2536 for 6 days and photomicrographs were taken on the cells that remained after the treatment. Representative photomicrographs of the cells that underwent dramatic cellular morphological alterations are shown. Scale bar = 280 μm. (E) Total RNA from the cells treated with 5 nM PLK1 siRNA #1 and #2 for 36 hours was extracted and subjected to RT-PCR to quantify the transcripts of PLK1, SOX2, and GFAP. (F) SF188 cells were treated with 100 nM SOX2 siRNA for 72 hrs. The cells were stained with Hoechst and quantified. The number of viable cells was enumerated and the relative fold difference in cell growth is shown in the bar graph. (G) The number of nonviable cells after 100 nM, 72-hour siSOX2 treatment was enumerated based on enhanced Hoechst staining due to chromatin condensation and the relative fold difference in cell death is shown in the bar chart. (H) Proteins extracted from the cells treated with 100 nM control or SOX2 siRNA for 48 hours were subjected to immunoblotting to examine the phosphorylation of H2AX at Ser139, which is a marker of early apoptosis. (I) SF188 cells were treated with 100 nM control or SOX2 siRNA for 72 hours and immunoblotting was performed on the total protein lysates. Increased caspase 3 cleavage was observed in the siSOX2-treated cells compared to the control cells.
A SPA for PMTs
Ibáñez G, Shum D, Blum G, Bhinder B, Radu C, Antczak C, Luo M, Djaballah H. A high throughput scintillation proximity imaging assay for protein methyltransferases. Comb Chem High Throughput Screen 2012;15:359–371.
Abstract: Protein methyltransferases (PMTs) orchestrate epigenetic modifications through post-translational methylation of various protein substrates including histones. Since dysregulation of this process is widely implicated in many cancers, it is of pertinent interest to screen inhibitors of PMTs, as they offer novel target-based opportunities to discover small molecules with potential chemotherapeutic use. We have thus developed an enzymatic screening strategy, which can be adapted to scintillation proximity imaging assay (SPIA) format, to identify these inhibitors. We took advantage of S-adenosyl-L-[3H-methyl]-methionine availability and monitored the enzymatically catalyzed [3H]-methyl addition on lysine residues of biotinylated peptide substrates. The radiolabeled peptides were subsequently captured by streptavidin coated SPA imaging PS beads. We applied this strategy to four PMTs: SET7/9, SET8, SETD2, and EuHMTase1, and optimized assay conditions to achieve Z′ values ranging from 0.48 to 0.91. The robust performance of this SPIA for the four PMTs was validated in a pilot screen of approximately 7,000 compounds. We identified 80 cumulative hits across the four targets. NF279, a suramin analogue, was found to specifically inhibit SET7/9 and SETD2 with IC50 values of 1.9 and 1.1 μM, respectively. Another identified compound, Merbromin, a topical antiseptic, was classified as a pan-active inhibitor of the four PMTs. These findings demonstrate that our proposed SPIA strategy is generic for multiple PMTs and can be successfully implemented to identify novel and specific inhibitors of PMTs. The specific PMT inhibitors may constitute a new class of anti-proliferative agents for potential therapeutic use.
Commentary: Scintillation proximity assays (SPAs) have been applied to adapt separation-based assays—for example, those in which substrate and products need to be separated—to assays that are suitable for high-throughput screening (HTS) using a simple “mix-and-read” format. In SPAs, a radiolabeled component is used that gives rise to a signal only when bound to beads that are impregnated with scintillate. SPAs have been widely applied to measure G-protein receptor–ligand binding, phosphorylation of peptide or protein substrates by kinases, as well as the quantification of a number of enzymatic products (for a review see Glickman et al., Assay Drug Dev Technol 2008;6:433–455.) Non–separation-based heterogeneous assays that are not radioactive, such as AlphaScreen, in which donor and acceptor beads are brought into sufficient proximity to generate a chemiluminescent signal, have become increasingly popular. However, SPA remains an attractive assay format for HTS, particularly if the assay can be configured in a generic format in which a large target class can be addressed. This article describes a generic assay for protein methyl transferases (PMTs) using SPAs. The PMT class of enzymes uses S-adenosyl-methionine (SAM) to transfer a methyl group to either lysine or arginine groups of proteins. The present article employs biotinylated peptides that are methylated with a [3H-Me]-SAM cofactor and streptavidin-coated SPA beads (see figure). When histone H3 is used as a common substrate, the SPA format provides a generic read-out for PMTs. Assays for four PMTs (SET7/9, SET8, SETD2, and EuHMTase1) were developed using this approach. The assay parameters were set using a three-step procedure. First, enzyme concentration, incubation time, dimethylsulfoxide tolerance, and stability were set in a standard filter-binding assay using a biotinylated-H3 substrate and [3H-Me]-SAM. Next, these conditions were applied to a heterogeneous assay format using streptavidin-coated sepharose beads from which the free label was removed by washing. Enzyme activity was then measured with standard scintillation counting. In the last step, the sepharose beads were replaced with SPA beads emitting a red chemiluminescence signal suitable for imaging on the LEADSeeker™ platform (imaging-based, SPIA). The filter-based assay also served as a counter-screen for SPIA-related interference by compounds; for example, biotin mimetics that appear as inhibitors in SPIA due to competition with the streptavidin beads. Following optimization of the assay parameters, pilot library testing showed good performance in all four PMT assays. The use of a single detection technology allowed for rapid identification of inhibitors that are specific for a PMT. Although other assays have been described using specific antibodies to the methylated peptide product or detection of product S-adenosyl-L-homocysteine (SAH) by mass spectrometry, the SPA format described in this work provides a robust mix-and-read generic assay format for PMTs. Contributed by Doug Auld.
Overview of the SPA-based HTS approach. (A) The 3H-methyl residue of [3H-Me]-SAM is enzymatically transferred to biotinylated PMT substrates. (B) The methylated reaction can proceed through two pathways: inactivated, in which the compound inhibits the reaction or active, in which the compound leads to no apparent effect. The peptide is then immobilized onto streptavidin-conjugated SPA beads. The proximity between β-particles and bead-coated scintillation fluid generates strong scintillation signal, which is suppressed in the inactive pathway.
New Luciferins
McCutcheon DC, Paley MA, Steinhardt RC, Prescher JA. Expedient synthesis of electronically modified luciferins for bioluminescence imaging. J Am Chem Soc 2012;134:7604–7607.
Abstract: Bioluminescence imaging with luciferase enzymes requires access to light-emitting, small-molecule luciferins. Here, we describe a rapid method to synthesize D-luciferin, the substrate for firefly luciferase (Fluc), along with a novel set of electronically modified analogues. Our procedure utilizes a relatively rare, but synthetically useful dithiazolium reagent to generate heteroaromatic scaffolds in a divergent fashion. Two of the luciferin analogues produced with this approach emit light with Fluc in vitro and in live cells. Collectively, our work increases the number of substrates that can be used for bioluminescence imaging and provides a general strategy for synthesizing new collections of luciferins.
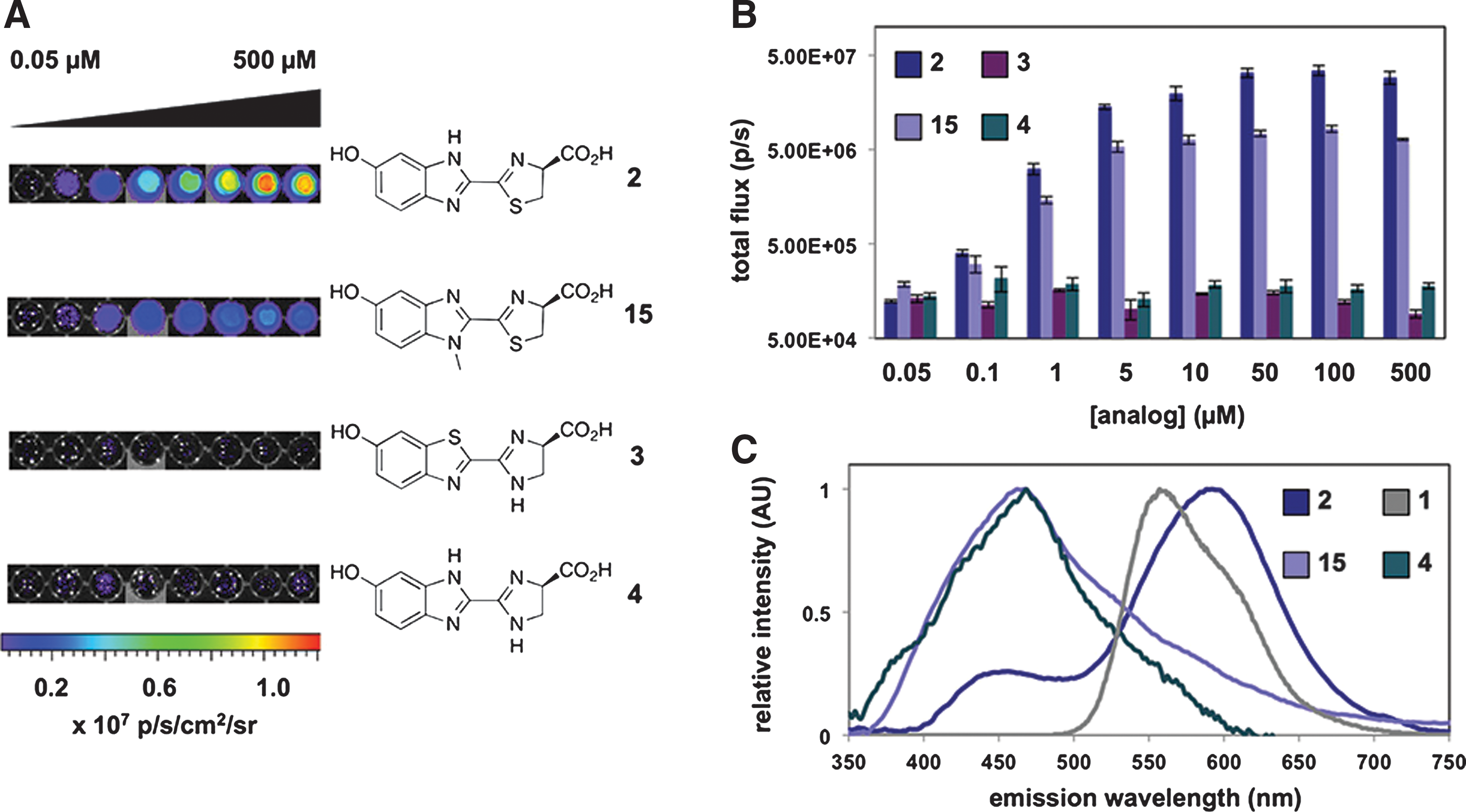
Commentary: Luciferase assays are widely used in biochemical and cell-based assays. New luciferases isolated from various bioluminescent species continue to be developed to provide highly sensitivity bioluminescence assays. Firefly luciferase (Fluc) is the most commonly used reporter in cell-based reporter-gene assays (RGAs). The bioluminescence of Fluc ranges from 500 to 700 nm with a broad emission peak at 550–570 nm. Although red-shifted variants of Fluc have been produced by mutating the D-luciferin binding site, these variant enzymes may show dimmer bioluminescence and many are not widely available. Another approach to developing luciferase assays with different emission maximums involves synthesizing new luciferins. This article describes a facile method to construct new luciferins using Appel's salt (4,5-dichloro-1,2,3-dithiazolium chloride). The authors were able to synthesize luciferins with benzimidazole and imidazoline rings. Benzimidazoles are known competitive inhibitors of Fluc, and previous work has shown that such heterocycles can be excited to emit light. After obtaining these nitrogenous luciferins, these compounds were used as substrates for Fluc in the presence of ATP and CoASH. No bioluminescence could be detected for analog 3 (see figure), and only a very weak signal was obtained for analog 4. However, analogs 2 and 15 showed detectable signals, although they were approximately 100-fold weaker than the one obtained with D-luciferin. The benzimidazole analog 2 showed a red-shifted emission maximum of 578 nm. Interestingly, analog 15 showed a greatly blue-shifted emission peak compared to other luciferins with a λmax = 460 nm. This emission is similar to what is found with coelenterazine if used as a substrate for the luciferase isolated from Renilla reniformis. These analogs were also shown to function in a cell-based FLuc assay using HEK293 cells. Studies such as these should extend the range of available substrates for Fluc providing researchers new formulations for use in both in vitro and in vivo studies. Contributed by Doug Auld.
Light production from luciferin analogues. (A) Bioluminescence images from analogues 2–4 and 15 (0.05–500 μM) incubated with Fluc or no enzyme. (B) Quantification of the images from (A). (C) Bioluminescence emission spectra for luciferins 1, 2, 4, and 15.
Antibody Quantification by Microscale Thermophoresis
Lippok S, Seidel SA, Duhr S, Uhland K, Holthoff HP, Jenne D, Braun D. Direct detection of antibody concentration and affinity in human serum using microscale thermophoresis. Anal Chem 2012;84:3523–3530.
Abstract: The direct quantification of both the binding affinity and absolute concentration of disease-related biomarkers in biological fluids is particularly beneficial for differential diagnosis and therapy monitoring. Here, we extend microscale thermophoresis to target immunological questions. Optically generated thermal gradients were used to deplete fluorescently marked antigens in 2- and 10-fold-diluted human serum. We devised and validated an autocompetitive strategy to independently fit the concentration and dissociation constant of autoimmune antibodies against the cardiac β1-adrenergic receptor related to dilated cardiomyopathy. As an artificial antigen, the peptide COR1 was designed to mimic the second extracellular receptor loop. Thermophoresis resolved antibody concentrations from 2 to 200 nM and measured the dissociation constant as 75 nM. The approach quantifies antibody binding in its native serum environment within microliter volumes and without any surface attachments. The simplicity of the mix and probe protocol minimizes systematic errors, making thermophoresis a promising detection method for personalized medicine.
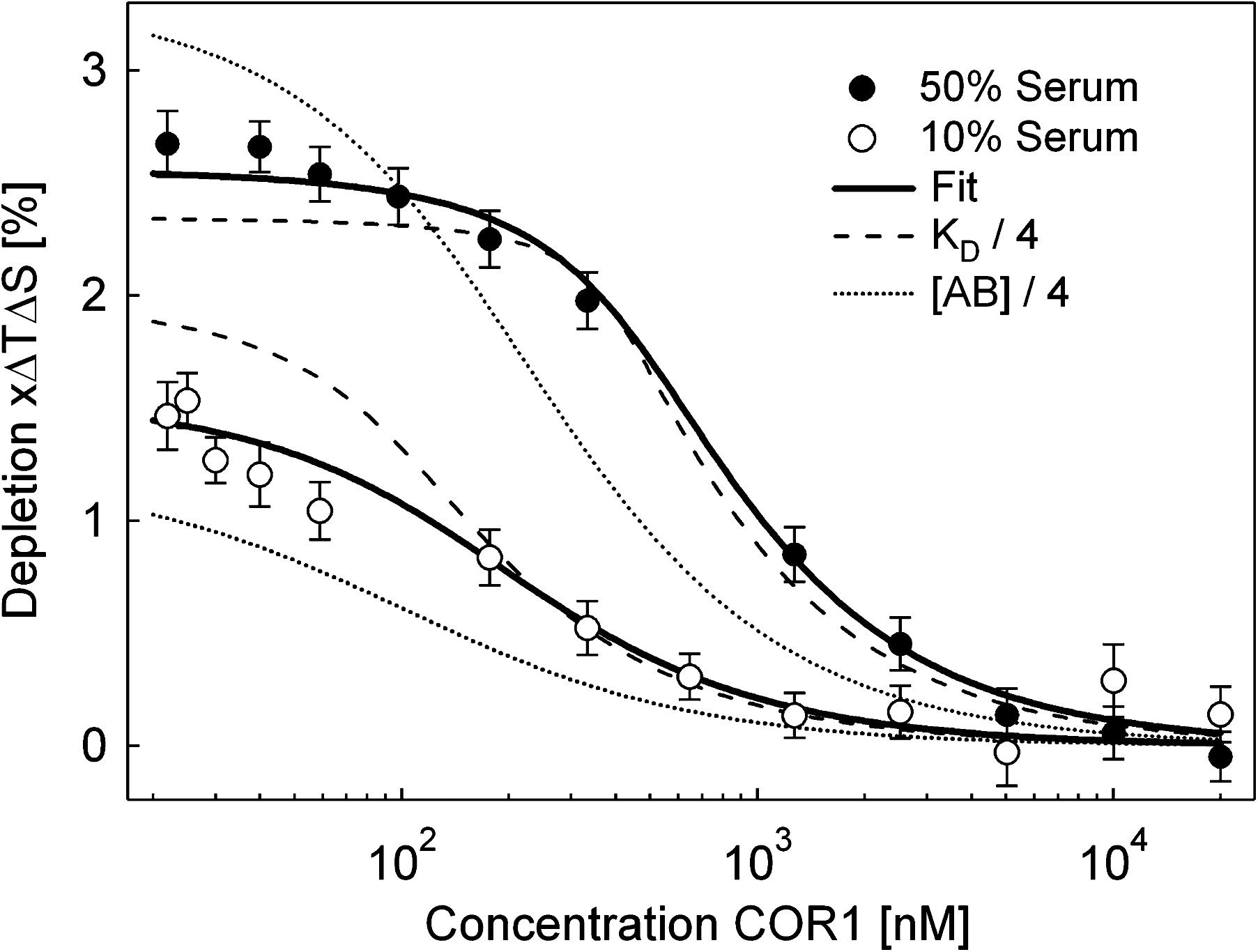
Commentary:Despite the popularity and versatility of the enzyme-linked immunosorbent assay (ELISA) for the detection of antibodies, this separation-based format can be difficult to develop, control, and standardize due to immobilization and washing steps. Solution-based methods such as radioimmunoassay and fluorescence resonance energy transfer assay require either radioactive labeling or multiple labels for signal generation. Thus, a simpler assay that can be essentially immobilization-free and require fewer or no labeling partners would be desirable. The article by Lippok et al. presents such an alternative platform for antibody quantification that possesses these improvements. Specifically, the authors developed microscale thermophoresis (MST)-based spiking, autocompetition, and dilution methods, all of which probe molecular interactions directly, based on the law of mass action. Thermophoresis, also known as the Ludwig-Soret effect, refers to the phenomenon in which particles move along a temperature gradient. Since its introduction to the bioanalytical sector in 2010, the platform has been successfully validated and applied in a wide range of interaction categories (such as protein-protein, protein-small molecule and protein-ion; for detailed experimental setup and bioanalytical examples (see Jerabek-Willemsen et al., Assay Drug Dev Technol 2011;9;:342–353). Detailed theoretical description has been discussed for molecule drift in liquids (Duhr et al., Proc Natl Acad Sci USA 2006;103:19678–19682). Herein, the authors further established two key theoretical components that served as the foundation of their subsequently devised antibody autocompetitive assays. By ensuring the application of only a small temperature gradient (ΔT≈5K–10K), the steady-state concentration of drifted molecules can be reasonably linearized from its original exponential distribution. As a result, the relative fluorescence depletion signal can be viewed as a linear function of the apparent Soret coefficient, a parameter that also takes the dye temperature dependence into consideration in addition to the thermophoretic depletion. Subsequently, the authors demonstrated that the fluorescence depletion and fraction of bound molecules (x) obeys a linear relationship. Thus, by following the thermophoretic depletion (i.e., x) obtained at varying labeled binder concentration (cB0), the ligand concentration (cL0) and dissociation constant (KD) could be determined independently through the mass action law (see first figure). To validate their theory, the authors applied dilated cardiomyophathy (DCM)-relevant antibodies and their cognate peptide COR1 as a model system. Using a previously established spiking protocol, the authors showed that the polyclonal antibody bound to a red dye (D2)-labeled COR1 in 50% serum in a specific manner with a KD of 66±25 nM. By mixing different concentrations of the antibody with a constant level of labeled COR1, a titration series of the unlabeled COR1 was introduced to each antibody + labeled COR1 mixture and an autocompetition assay was performed. Antibody concentration was derived from the fitted data (see second figure), and, regardless of the binding model that was used to fit the data (simple binding, label-dependent binding, and polyclonal labeling model), a significant correlation was shown to exist between the derived value and the input value (see second figure inset). A KD of 74 ±11 nM was independently derived, lying well within the experimental error of that obtained in the spiking protocol. Furthermore, using a dilution approach, the authors conducted similar autocompetition assays in both 50% and 10% serum. The antibody concentration and the dissociation constant were found to deviate from the ones reported from the spiking protocol by 13% and 25%, respectively. The authors further evaluated the performance of the autocompetition and dilution approach in terms of its separation resolution by showing the significantly different profiles when either antibody concentration or dissociation constant was reduced by 75% (see third figure). The current DCM relevant antibody-COR1 model system allowed a 2 nM detection limit, 35-fold below the dissociation constant of the binding partners. Improved assay sensitivity could be potentially achieved through the utilization of dimeric probes. Future directions include moving the detection platform into single molecule regime and high-throughput screening campaigns. For the latter, this is particularly interesting and useful not only for the characterization of antibody but also for other types of ligands, such as small molecules as potential drug therapeutics. Due to the principle of the MST-based assays, the platform is expected to have several competitive advantages compared to other biophysical tools, such as label free capability, no concern for diffusion limited mass transfer, quick turnaround time, small footprint, and potential for automation. Contributed by Wendy Lea.
Concentration and affinity inferred independently. The fraction x of binder B (COR1) bound to ligand L (antibody) is plotted against the binder concentration cB0 according to Equation 12 in the article. The fraction x is a linear function of thermophoretic depletion (Equation 10 in the article). Thus, a rescaled plot of the measured fluorescence ratio ΔF/F will show the same behavior. (a) A change of the ligand concentration shifts the thermophoretic depletion along the binder concentration axis under a fixed dissociation constant, KD = 200 nM. (b) A change in the dissociation constant modulates the amplitude of thermophoretic depletion under constant ligand concentration cL0 = 200 nM. The depletion is changed differently by both parameters and thus can be fitted independently.
Autocompetition assay. In 50% human serum, the concentration of total COR1 is increased by adding unlabeled COR1 to 20 nM labeled COR1. Thermophoresis only records the depletion of the labeled species. Autocompetition reduces the thermophoretic depletion as less labeled COR1 is bound to the antibody. Four binding curves are recorded for antibody concentrations of 2, 20, 80, and 200 nM. We recover the concentrations from the fit with 0.9 ± 1.5, 26 ± 3.4, 108 ± 20, and 176 ± 21 nM (SD), well within the pipetting errors. Independently, the dissociation constant was inferred to be 74 ± 11 nM. Inset: More complex binding models assuming label-dependent affinity or polyclonal binding did not change the fitted concentrations noticeably, confirming the simple binding assumption.
Concentration and affinity assay. Two autocompetition assays are performed in 50% human serum and the 5-fold dilution to 10% serum. This protocol allows the determination of both the concentration and affinity of antibodies. We find an antibody concentration of 187 ± 26 nM with a dissociation constant of KD = 73 ± 18 nM. The theoretical predictions for a 4-fold lower antibody concentration (dotted line) and a 4-fold smaller dissociation constant (dashed line) differ significantly from the measurements.
Reducing Endocytosis Assay
Cole NB, Donaldson JG. Releasable SNAP-tag probes for studying endocytosis and recycling. ACS Chem Biol 2012;7:464–469.
Abstract: Site-specific labeling of cellular proteins with chemical probes is a powerful tool for live cell imaging of biological processes. One popular system, known as the SNAP-tag, is based on an engineered variant of the 20-kDa DNA repair protein O6-alkylguanine- DNA-alkyltransferase (AGT) that covalently reacts with O6-benzylguanine (BG) and can be derivatized with a number of reporter groups. For studying the endocytosis and recycling of cell surface proteins, the covalent nature of BG binding to the SNAP-tag is problematic, since removing excess noninternalized probe from the cell surface is not feasible. Here we describe a modification of the SNAP-tag technology that permits the rapid release of fluorescently labeled probes from the cell surface without affecting the population of labeled molecules sequestered within endosomes. This simple yet effective approach allows quantitative measurements of endocytosis and recycling in both imaging and biochemical assays and is especially useful when studying endosomal dynamics in live cells.
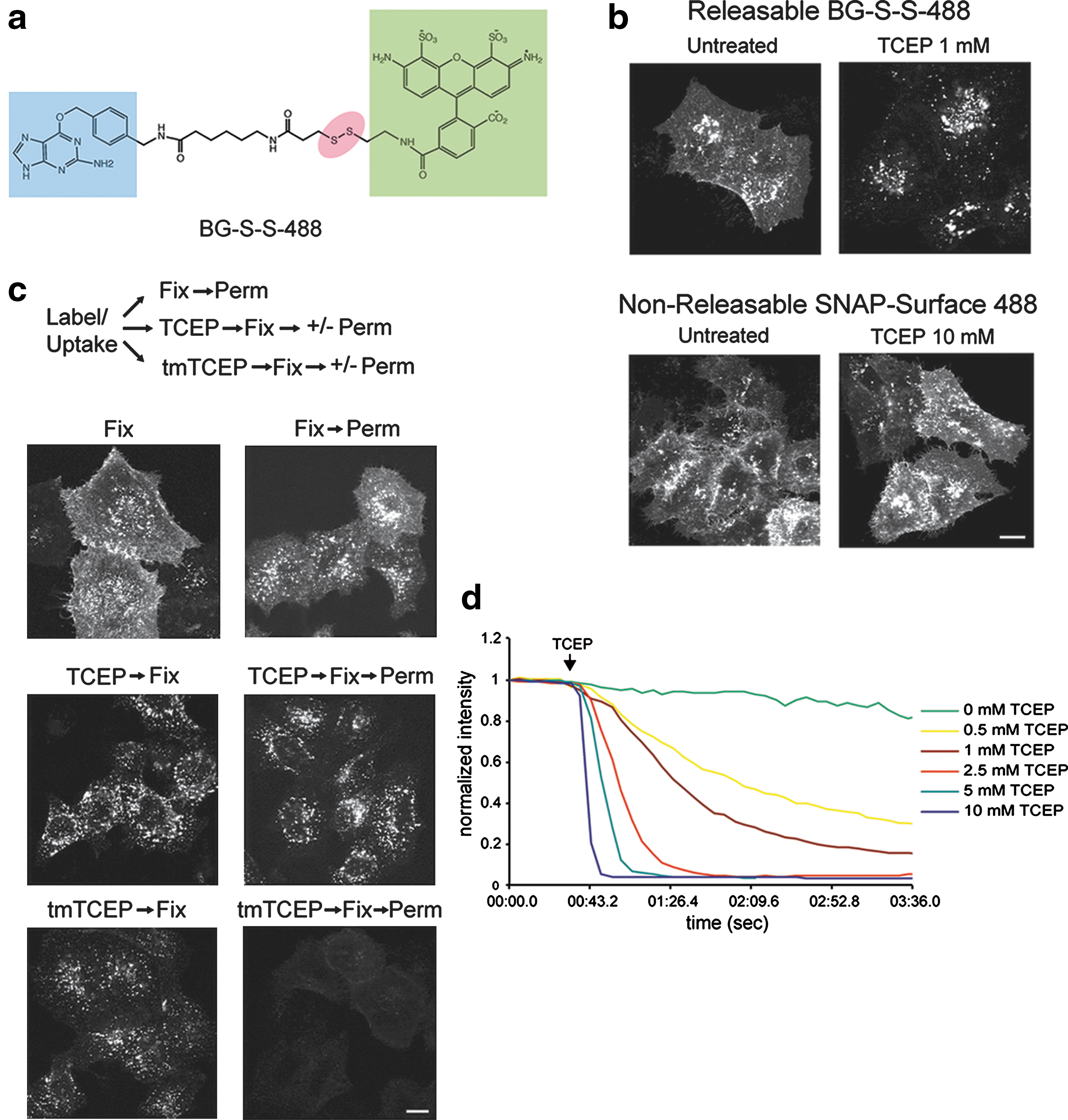
Commentary:Protein fusions to the DNA repair protein O6-alkylguanine-DNA-alkyltransferase (AGT), so-called SNAP tag, have been used to construct a variety of assays. Assays for endocytosis have typically involved high-content imaging approaches using green fluorescent protein (GFP)-tagged membrane receptors. The work described here uses an O6-benzylguanine (BG) linked to a fluorophore via a disulfide bond. Labeling of a cell surface receptor fused to a SNAP-tag allows monitoring endocytosis by removing the surface bound fluorophore with a cell-impermeable reducing agent such as tris(2-carboxyethyl)phosphine (TCEP).Following TCEP treatment of cells, only the internalized fluorophore is monitored (see figure). The endosomal environment is thought to be generally nonreducing, and so the internalized BG-S-S-488 probe is not cleaved in the endosome. To test this, the authors used a cell permeable form of TCEP (a trimethyl ester analog, tmTCEP). No free dye could be extracted from cells containing endosomes with SNAP-β2-adrenergic(ADR) which were labeled with BG-S-S-488 when TCEP was used; however, free dye was extracted from these cells when tmTCEP was used. Therefore, the normal environment of endosomes is apparently nonreducing. Endocytosis and recycling can be quantified with this method because the same cells can be used to measure the total fluorescence from labeled receptors on the surface (before treatment with TCEP) as well as the internalized fluorescence after treatment with TCEP. A two-color assay is also shown in this article in which an RFP-tagged β2-arrestin, and either SNAP-β2ADR or SNAP-NK1R are imaged. Agonist treatment shows translocation of the RFP-β2-arrestin2 to the cell membrane, and following TCEP treatment the co-localization of the receptors into endosomes could be measured. With this method different populations of receptor/β2-arrestin complexes can be studied. The assay can be applied to any cell surface protein and could be expanded to study other internalization processes such as phagocytosis as well. Contributed by Doug Auld.
Characterization of a releasable SNAP-tag probe. (a) Schematic of BG-S-S-488, with O6-benzylguanine (blue), Alexa Fluor 488 (green), and disulfide linkage (red) highlighted. (b) Comparison of sensitivity of BG-S-S-488 versus SNAP-Surface 488 (New England Biolabs) to TCEP in HeLa cells transfected with SNAP-β2ADR. Scale bar, 10 μm. (c) Comparison of the ability of unesterified TCEP versus tmTCEP to cleave intracellular BG-S-S-488. Free label is extracted by TX-100 from endosomes in cells incubated with tmTCEP. Scale bar, 10 μm. (d) Fluorescence time course showing sensitivity of BG-S-S-488 to different TCEP concentrations in live COS cells transfected with SNAP-β2ADR. Measurements were taken in the absence of endocytosis, so that only cell surface fluorescence was quantified.
Allosteric Inhibitors of HIV-1 Integrase to Delay Resistance
Demeulemeester J, Tintori C, Botta M, Debyser Z, Christ F. Development of an AlphaScreen-based HIV-1 integrase dimerization assay for discovery of novel allosteric inhibitors. J Biomol Screen 17:618–628.
Abstract: In recent years, HIV-1 integrase (IN) has become an established target in the field of antiretroviral drug discovery. However, its sole clinically approved inhibitor, the integrase strand transfer inhibitor (INSTI) raltegravir, has a surprisingly low genetic barrier for resistance. Furthermore, the only two other integrase inhibitors currently in advanced clinical trials, elvitegravir and dolutegravir, share its mechanism of action and certain resistance pathways. To maintain a range of treatment options, drug discovery efforts are now turning toward allosteric IN inhibitors, which should be devoid of cross-resistance with INSTIs. As IN requires a precise and dynamic equilibrium between several oligomeric species for its activities, the modulation of this equilibrium presents an interesting allosteric target. We report on the development, characterization, and validation of an AlphaScreen-based assay for high-throughput screening for modulators of HIV-1 IN dimerization. Compounds identified as hits in this assay proved to act as allosteric IN inhibitors. Additionally, the assay offers a flexible platform to study IN dimerization.
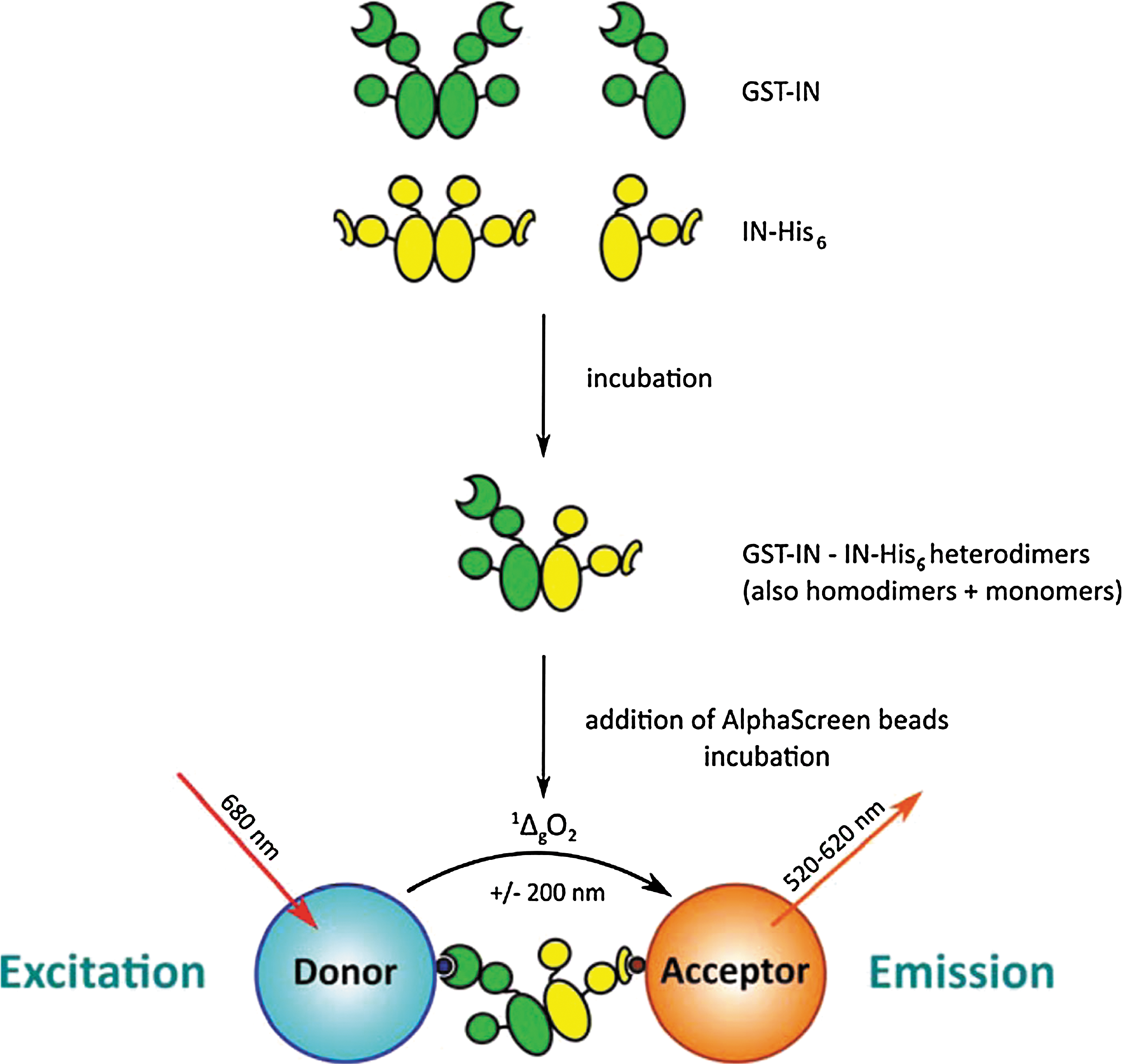
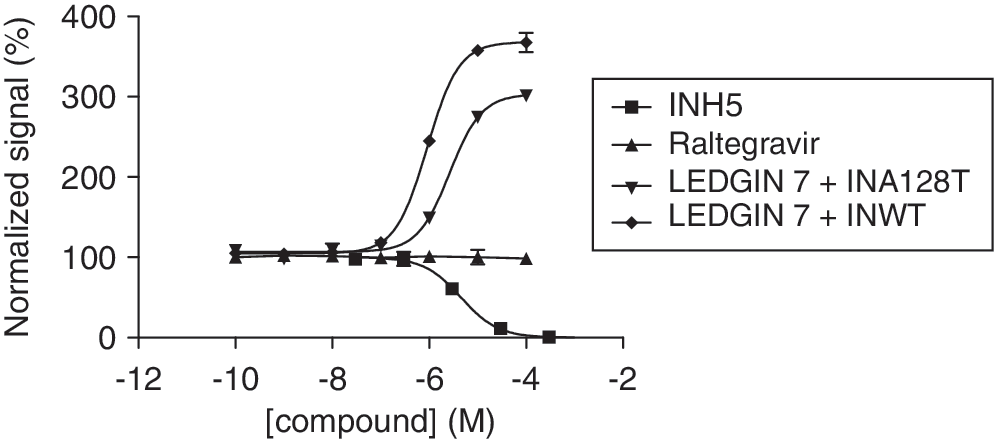
Commentary:HIV has a very high rate of mutation, which allows it to develop resistance to the drugs used to treat it. Drug cocktails can slow the rate of progression of the disease by attacking the virus from multiple angles but new types of drugs are still needed. Raltegravir (MK-0518) was approved in 2007 as an inhibitor of HIV-1 integrase (IN), which is an enzyme that enables the HIV DNA to integrate into the host DNA. This molecule and the two follow-on drugs currently in clinical trials for this target bind to the complex of viral DNA and IN and displace the catalytic adenosine. Due to the same mechanism of action, these three drugs have significant cross resistance. The authors describe their efforts to target IN via an allosteric mechanism, which would be expected to have different resistance mechanisms. IN has different functions, including 3′ processing and strand transfer, that occur during the different oligomeric states of IN (dimeric and tetrameric, respectively). IN is in equilibrium between monomeric, dimeric, tetrameric, and higher order oligomeric states. The authors sought to find modulators of IN dimerization by using the bead-based AlphaScreen technology (see the first figure) in a 384-well plate format. IN was labeled with GST or His6, and then GST-targeted donor beads and His6-targeted acceptor beads were used. The assay was allowed to equilibrate for 180 minutes to yield a stable population of His6- and GST-tagged heterodimers. Only in the presence of a protein–protein interaction between the two differentially tagged IN monomers would the distance-dependent energy transfer occur from the donor to the acceptor beads. Compounds that disrupt the dimerization would reduce the signal from the acceptor bead, while those that enhance the interaction would increase the signal from the acceptor beads. As expected, Raltegravir, which operates at the catalytic site, does not show an effect in the AlphaScreen assay, and INH5 and LEDGIN 7, which are known to function at the dimer interface, show the expected effects (see second figure) of inhibiting or enhancing the dimerization, respectively. The authors screened a very small library and identified compounds that were active in the AlphaScreen assay that also confirmed in an enzyme-linked immunosorbent assay that measures IN catalytic activity. Another group had previously reported the development of a homogeneous time-resolved fluorescence-based assay (HTRF) (Tsiang et al., Biochemistry 2011;50:1567–1581) and observed an effect of dithiothreitol on dimerization, which was recapitulated here. It will be interesting to see how druggable this dimer interface is when a larger library is screened. It would also be interesting to see whether the same true positives would be identified by the HTRF and AlphaScreen technologies from a larger library and whether there would be a difference in the number of false positives. Contributed by Mindy I. Davis.
Overview of the integrase (IN) dimerization AlphaScreen assay. Glutathione S-transferase (GST)-tagged IN (green) is mixed with His6-tagged IN (yellow) at the desired concentrations. Incubation at 4°C allows monomer exchange between dimers and the establishment of a steady-state population containing GST- and His6-homodimers as well as GST/His6-heterodimers. When Ni2+-chelate acceptor and glutathione donor AlphaScreen beads are added, the heterodimers will bring both beads into close proximity. Upon irradiation with 680 nm laser light, a photosensitizing phthalocyanine in the donor beads will produce large amounts of singlet oxygen (1ΔgO2). Due to the short half-life of this species (±4 μs), it can only diffuse about 200 nm before returning to the ground state. If, however, acceptor beads are nearby because of interactions of the biomolecules on their surface, the 1ΔgO2 will reach these beads and react with thioxene-based groups on the inside. Energy transfer to anthracene-based and then rubrene-based groups will ultimately result in emission of light between 520 and 620 nm: the AlphaScreen signal. Changes in the population of heterodimers (e.g., due to the presence of modulating compounds) will, upon addition and incubation with the beads, result in an altered output signal and can be picked up.
Titration of control compounds. The compounds INH5, LEDGIN 7, and raltegravir were titrated in the optimized IN dimerization assay against wild-type (WT) integrase (IN). Results were normalized by setting the no-compound and no-protein controls to 100% and 0%, respectively. Whereas INH5 acted as an antagonist with an EC50 of 4.5 μM, LEDGIN 7 behaved as an agonist in this assay with an EC50 of 0.67 μM. MK-0518 did not show any effect at all. When LEDGIN 7 was titrated against IN carrying the A128T resistance mutation, its EC50 was increased approximately fourfold to 2.6 μM, reflecting a reduced ability to stimulate IN dimerization.
Blood Flow as Diagnostic Marker
Wood DK, Soriano A, Mahadevan L, Higgins JM, Bhatia SN. A biophysical indicator of vaso-occlusive risk in sickle cell disease. Sci Transl Med 2012:4:23ra26.
Abstract: The search for predictive indicators of disease has largely focused on molecular markers. However, biophysical markers, which can integrate multiple pathways, may provide a more global picture of pathophysiology. Sickle cell disease affects millions of people worldwide and has been studied intensely at the molecular, cellular, tissue, and organismal level for a century, but there are still few, if any, markers quantifying the severity of this disease. Because the complications of sickle cell disease are largely due to vaso-occlusive events, we hypothesized that a physical metric characterizing the vaso-occlusive process could serve as an indicator of disease severity. Here, we use a microfluidic device to characterize the dynamics of “jamming,” or vaso-occlusion, in physiologically relevant conditions, by measuring a biophysical parameter that quantifies the rate of change of the resistance to flow after a sudden deoxygenation event. Our studies show that this single biophysical parameter could be used to distinguish patients with poor outcomes from those with good outcomes, unlike existing laboratory tests. This biophysical indicator could therefore be used to guide the timing of clinical interventions, to monitor the progression of the disease, and to measure the efficacy of drugs, transfusion, and novel small molecules in an ex vivo setting.
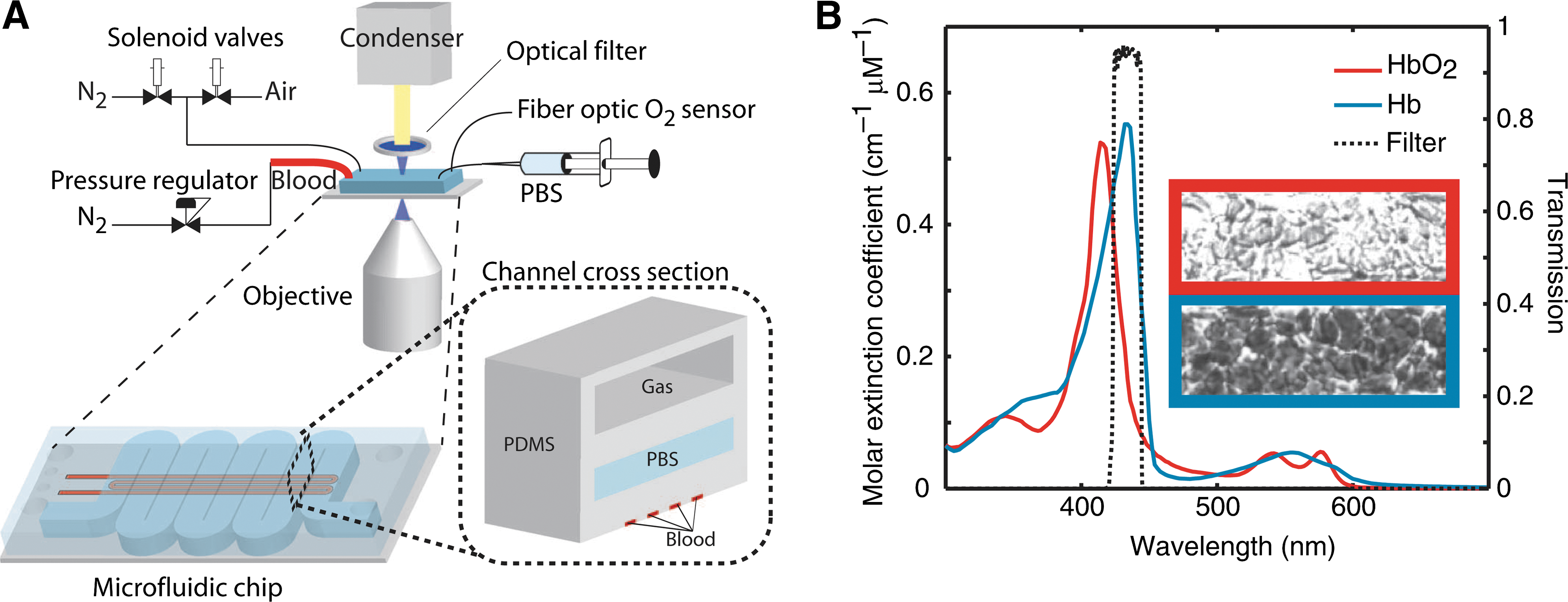
Commentary:The article by Wood and colleagues highlights the validation of a simple physical parameter associated with blood-flow properties as a diagnostic marker for increased risk in sickle cell disease. In an era when low-abundance protein analytes and combinations of single-nucleotide polymorphisms are increasingly being developed for diagnostic purposes, simpler (bio)physical parameters risk being overlooked. Examples of such parameters already driving clinical decisions include blood pressure, heart beat, and basic imaging. The present study of a sickle cell disease prognostic parameter marks a mini-revival in this area, with the authors presenting a microfluidic device that measures the change in blood flow characteristics upon deoxygenation. Typically, the status of sickle cell patients is evaluated through the measurement of the ratio of hemoglobin S to fetal hemoglobin, but this blood chemistry result does not provide for a strong enough prognostic marker to assess the risk of the patient developing vaso-occlusion crisis, a state associated with a dramatic change in blood flow characteristics due to the “jamming” of capillaries through the change in shape and rigidity of red blood cells. Instead of looking for molecular markers to assess the risk of vaso-occlusion, the authors opted to measure the blood flow properties directly. In the device, capillary channels carrying the blood sample were coupled with gas reservoirs (first figure). When whole blood was flowed through the device, the gas composition of the adjacent channel could be controlled to provide different levels of dissolved gasses in the bloodstream, imitating the transition of the blood cells from large blood vessels to the capillaries; in turn, the changes in hemoglobin polymerization state (driven by the changing level of dissolved oxygen) and the concomitant change in red blood cell rigidity could be measured through the changes in the resistance of the blood sample to pressure-driven flow. The device was initially validated by comparing blood flow resistance changes as a function of oxygen percentage for samples from healthy and sickle cell disease donors. The testing was then extended to compare samples from patients considered to have different levels of sickle cell disease severity: samples from patients who had been treated extensively (through transfusions and hydroxyurea administrations) during the past 12 months showed dramatically different flow resistance profiles when compared to those from patients considered to have a mild case of sickle cell disease (i.e., not having been treated intensively during the past 12 months). This patient stratification through flow characteristics measurement was then used in expanded survey of 23 severe-case and 6 mild-case patients which also showed that the new biophysical parameter correlated well with clinical outcome for the two patient strata. Finally, a new experimental treatment to manage sickle cell disease with the small molecule agent 5-hydroxymethyl furfural was evaluated through the use of the new microfluidic device (second figure). Patient samples treated with the agent showed a large and rapid improvement in their blood flow characteristics, indicating that the device is likely to be of utility in monitoring the treatment progress with new drugs. Contributed by Anton Simeonov.
Microfluidic device for studying sickle cell blood flow conductance. (A) The device comprised three layers (inset): artificial capillaries for blood flow, a hydration layer with PBS, and a gas reservoir. Blood was driven under constant pressure bias, controlled by a digital pressure regulator. Two solenoid valves controlled the gas (N2 and air) in the top chamber. A fiber optic probe was used to measure the oxygen concentration in the gas reservoir. The device was illuminated through an optical filter whose transmission band (434 ± 17 nm) was centered on an absorption peak for deoxyhemoglobin. (B) The absorption peak of hemoglobin shifts in deoxygenated conditions, making deoxygenated RBCs appear dark and oxygenated RBCs appear transparent. Qualitative measurements of hemoglobin oxygen saturation were made using the intensity of transmitted light. PBS, phosphate-buffered saline; RBC, red blood cell.
Rate of conductance decrease is modulated by a small molecule. (A, B) Oxygen data from gas reservoir (top) and conductance data (bottom) are shown for an untreated severe sample (A) and the same sample treated with 10 mM 5HMF (B). Oxygen data are shown as measured in the gas reservoir (dashed line) and in the blood channel (open circles), the latter measured by RBC intensity. (C) Rates of conductance decrease [open circles in (A, B)] are quantified. *P < 0.05, Mann–Whitney nonparametric analysis. Data are means ± SD of at least five independent deoxygenation cycles for both samples. All conductance values are scaled by the mean HbA blood conductance [∼1.7 μm s−1 mm Hg−1 (0.013 μm s−1 Pa−1)], and time is scaled by the time scale for hemoglobin deoxygenation (∼10 s). Rates of conductance decrease are scaled by mean HbA blood conductance divided by the time scale for hemoglobin deoxygenation (C*HbA/τdeox). 5HMF, 5-hydroxymethyl furfural; HbA, hemoglobin A.