REPURPOSING AN AURORA INHIBITOR FOR A NEGLECTED TROPICAL DISEASE
Ochiana SO, Pandarinath V, Wang Z, Kapoor R, Ondrechen MJ, Ruben L, Pollastri MP: The human Aurora kinase inhibitor danusertib is a lead compound for anti-trypanosomal drug discovery via target repurposing.Eur J Med Chem2012 Jul 31 [Epub ahead of print]; DOI: 10.1016/j.ejmech.2012.07.038.
Abstract: New drugs for neglected tropical diseases such as human African trypanosomiasis (HAT) are needed, yet drug discovery efforts are not often focused on this area due to cost. Target repurposing, achieved by the matching of essential parasite enzymes to those human enzymes that have been successfully inhibited by small molecule drugs, provides an attractive means by which new drug optimization programs can be pragmatically initiated. In this report we describe our results in repurposing an established class of human Aurora kinase inhibitors, typified by danusertib (compound 1), which we have observed to be an inhibitor of trypanosomal Aurora kinase 1 (TbAUK1) and effective in parasite killing in vitro. Informed by homology modeling and docking, a series of analogs of 1 were prepared that explored the scope of the chemotype and provided a nearly 25-fold improvement in cellular selectivity for parasite cells over human cells.
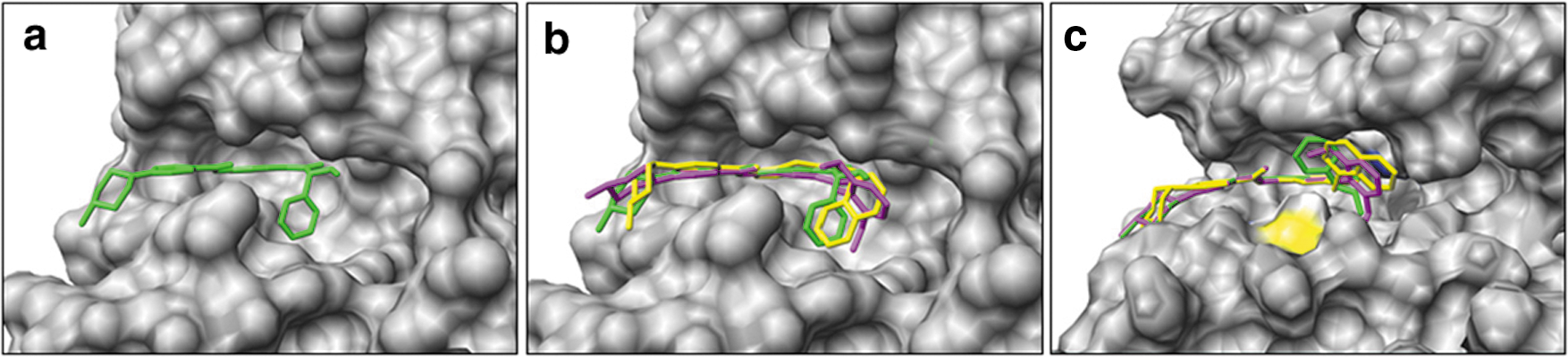
Commentary:The insect-borne Trypanasoma brucei parasite is responsible for human African trypanasomiasis (HAT). This disease is fatal if untreated and later stage infections have treatments in which the drug itself has a high mortality rate and toxicity. Patients are usually diagnosed in the later stages when the symptoms (lethargy, sleeping sickness, and/or coma) are more pronounced, and the parasite has moved from the bloodstream into the central nervous system. There are essential enzymes in the parasite that are homologous to human enzymes that have been successfully targeted by small molecule inhibitors for the treatment of other diseases, such as cancer. Trypanasomal Aurora kinase (TbAUK1) is an enzyme involved in the cell cycle that is essential to the parasite as determined by RNAi, and this enzyme is shared by human cells. Earlier work had shown that TbAUK1 was essential to infection in mice and that human Aurora kinase inhibitors, such as VX-680, could inhibit TbAUK1. Danusertib is a human Aurora kinase inhibitor that is in clinical trials as a treatment for a variety of cancers. Here danusertib was shown to inhibit both TbAUK1 and T. brucei growth. Analogs of danusertib were made in which the selectivity for inhibition of parasites versus human cells was improved 23-fold, although there was also a decrease in potency against trypanosomes. A homology model of TbAUK1 was created, and the pyrrolopyrazole danustertib and two analogs (compounds2and8) were docked into this model and compared with the crystal structure of human Aurora kinase with danusertib and three docked compounds (see
first figure
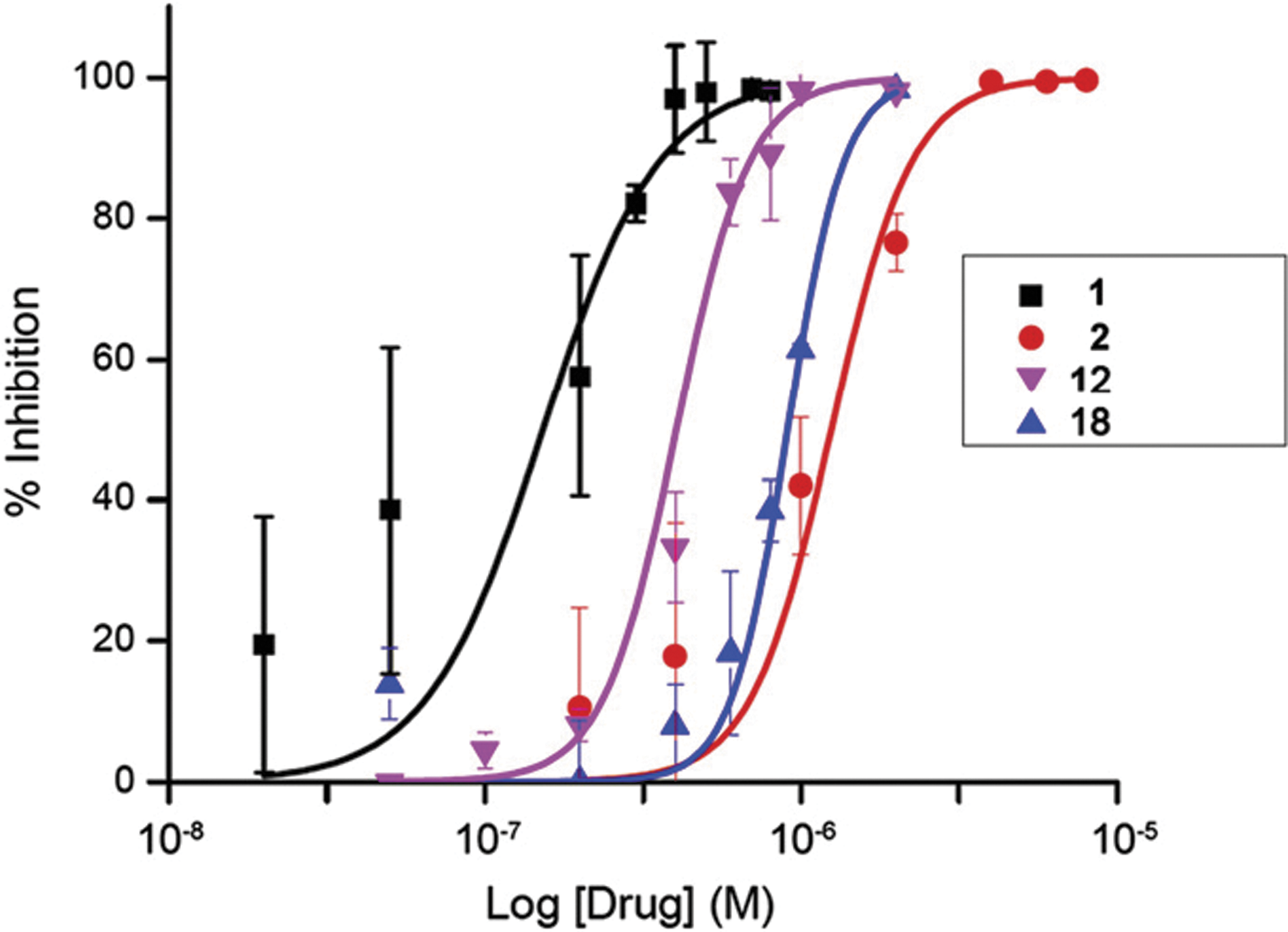
). The predicted conformation of the inhibitors differs significantly in the vicinity of Met113 (T. brucei)/Thr217 (human), which may be due to the bulkier Met sidechain in the parasite. The dose responses of danusertib (compound1), PHA-680632 (2), and compounds12and18were determined in a cell assay using T. brucei rhodesiense (see
second figure
). The best EC50 was 150 nM for danusertib. The rank order of the compounds from the docking and from the trypanosome cell assays correlated well. When considering targeting a parasite, one might consider focusing on enzymes that are essential and unique to the parasite. Targeting a protein with no known human homolog could potentially limit toxicity to human cells. Here, however, the opposite approach was used where an enzyme that is found in both parasites and humans was targeted. This strategy allowed the wealth of information available on the target and on compound inhibition of the target to be utilized. The authors were rapidly able to gain selectivity by exploiting residue differences in the TbAUK1 active site. The authors have shown that selectivity is possible, and it will be interesting to see whether compounds can indeed be found that have improved potency against the parasites, retain selectivity over human Aurora kinase, and have appropriate characteristics for in vivo applications. Contributed by Mindy I. Davis
Comparison of (a) the human Aurora A/danusertib complex (PDB ID: 2J50, danusertib; green) and the predicted conformation of compounds 2 (purple), 8 (yellow), and danusertib (green), (b) docked into human Aurora A and (c) in the TbAUK1 model. The colors in (c) are shown for side chain heteroatoms in Lys58 (blue) and Met113 (yellow). (For interpretation of the references to color in this figure legend, the reader is referred to the version of the original article available online at www.elsevier.com/locate/ejmech.)
Dose response of compounds 1, 2, 12, and 18 against T. brucei rhodesiense cultures. YTat1.1 BF cells were incubated for a 48-h period with varying drug concentrations. Cell viability was monitored with the Cell Titer Blue® assay. Values plotted are the average ± SD of two to three independent experiments, each done in triplicate.
TRIPPING THE LIGHT FANTASTIC: NEW REPORTER SYSTEM
Cheng KC, Inglese J: A coincidence reporter-gene system for high-throughput screening.Nat Methods2012;9:709–736.
Summary [no abstract]: Originally developed as sentinels of transcriptional activity to map the regulatory function of genetic elements, reporter gene assays have been extensively used in high-throughput screening (HTS) to identify chemical modulators of cellular pathways (Michelini E et al., Anal Bioanal Chem 2010;398:227–238). However, HTS chemical libraries consist of structurally diverse small molecules that frequently interact directly with the reporter, thus skewing data interpretation and complicating candidate selection. To distinguish compounds that target a biological pathway from those that interfere with a reporter, we designed a coincidence biocircuit based on the principle that it is easier to tell signal from noise when the signal is reported by two or more detectors. We conclude that coincidence reporter strategies rapidly discriminate compounds of relevant biological activity from those interfering with reporter function and stability, using a single assay platform. This study outlines an approach to improved use of reporter genes in HTS with numerous coincidence combination types and stoichiometries possible.
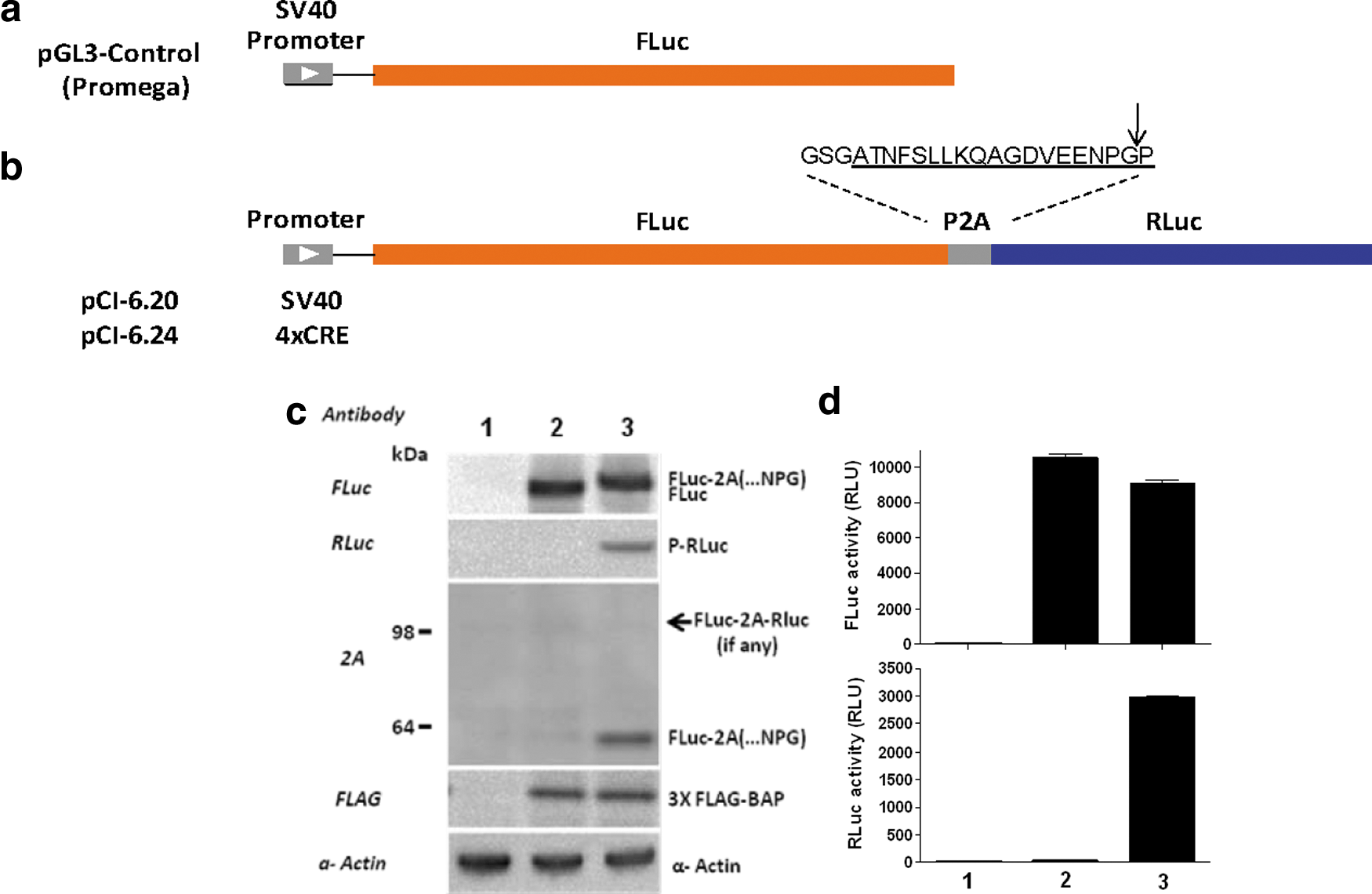
Commentary:Rapid assignment of compound activity to either wanted biological pathway/target modulation activity or unwanted interference with the assay system is critical to efficient progression of chemical matter arising from screens in chemical biology or drug discovery efforts. The most powerful assay for confirming that a compound's activity is due to relevant biological activity is an orthogonal assay. An orthogonal assay aims to replicate the exact same biology as the primary assay but differs in that a single component is switched, such as the method of detection, which shouldn't affect the targeted biology. Therefore, a positive result in an orthogonal assay provides evidence for target activity (e.g., the activity does not depend on the detection technology and is likely related to a mechanism relevant to the biological system under study). On the other hand, a negative result is a strong indication that the primary screen activity was due to the method of detection. An orthogonal assay is therefore a much more direct approach to addressing assay reporter-based artifacts then using counter-screens in which a negative result is the desired outcome because negative results can arise from a number of assay format differences (such as the level of the reporter expression, incubation times, and other modifications of assay components necessary to create the counter-screen). For reporter-gene assays (RGAs) that employ enzymes to generate the assay signal a large portion of compounds can interfere with the assay by acting as enzyme inhibitors, which can lead to many confounding effects that complicate the interpretation of the results. For orthogonal RGAs, the goal is to provide two reporters in which the enzymes differ in substrate requirements, enzymatic mechanisms, and inhibitory profiles. Ideally these reporters are produced in an isogenic background. The most optimal and efficient method to produce two orthogonal RGAs is to express the reporters in the same cell line. However, methods to express multiple reporters (such as the use of an internal ribosomal entry site [IRES] elements, bidirectional promoters, and coinfection with multiple vectors) are often plagued by inefficient and unpredictable expression of both reporters. For example, incorporation of an IRES sequence often results in more efficient expression of the upstream open reading (ORF) frame compared to the downstream ORF. This article provides a solution to efficient co-expression of multiple reporters (or any other set of proteins) by exploiting the mechanism by which polyproteins are cleaved to produce multiple proteins in Picornaviridae. The virus uses an 18-residue peptide sequence (so-called P2A peptide) that trips up the ribosome, causing translation to skip over this sequence and express the next ORF, thereby producing multiple separate proteins at near equimolar amounts. The authors validated this system using a construct wherein firefly luciferase (FLuc) and Renilla luciferase (RLuc) were separated by the P2A peptide sequence under control of a CRE response element (see
figure
). Coincidence expression of both reporters using this construct in HEK293 showed near equal expression of FLuc and RLuc as separate proteins with little detectable fusion protein (see
figure
). In the assay, activity from both reporters can be measured in the same assay well using standard detection reagents (e.g., DualGlo, Promega). A small library of known drugs was screened (the Library of Pharmacology Active Substance; LOPAC, Sigma) and it was demonstrated that compounds annotated as agonists of CRE-dependent signaling were identified as active with both FLuc and RLuc signals; however, compounds known to specifically inhibit either FLuc or RLuc were only found when measuring the activity of the reporter that was inhibited. Many reporter inhibitors actually lead to increase reporter signal in RGAs because reporter inhibitors can act in cells to stabilize the reporter enzyme, leading to increased enzyme levels that mimic gene activation. This counter-intuitive result can greatly complicate the interpretation of compound activity arising from RGAs, but the coincidence expression demonstrated here rapidly flagged these compounds, which were detected as active in only one of the reporter responses (see also www.reportergene.com). This system should greatly facilitate the production of orthogonal RGAs as well as other assay systems in which production of multiple proteins is required. Contributed by Doug Auld.
Design and characterization of FLuc-P2A-RLuc coincidence reporter biocircuit expression and function. Arrangement of elements in the (a) SV40-driven FLuc mono-reporter (pGL3-Control), and (b) the SV40-driven FLuc-P2A-RLuc dual reporter (pCI-6.20) and 4XCRE-driven FLuc-P2A-RLuc dual reporter (pCI-6.24). P2A amino acid sequence (underline) used in this construct is shown; arrow indicates ribosomal “skipping” site. (c) Western blot analysis showing the efficient expression of non-tethered reporters, where lane 1 is non-transfection control (transfection reagent only); lane 2, SV40-driven FLuc mono-reporter (pGL3-Control); and lane 3, FLuc-P2A-RLuc dual reporter (pCI-6.20). Note that co-transfection of 3XFLAG-BAP is to demonstrate the transfection efficiency was similar. (d) Bioluminescent output from mono FLuc reporter and co-expressed FLuc and RLuc using Dual-Glo reagent; lanes are the same as in (c). FLuc, firefly luciferase; RLuc, Renilla luciferase.
MEGA-SELECTION
Zhang H, Wilson IA, Lerner RA: Selection of antibodies that regulate phenotype from intracellular combinatorial antibody libraries.Proc Natl Acad Sci USA2012;109:15728–15733.
Abstract: A method is presented that uses combinatorial antibody libraries to endow cells with new binding energy landscapes for the purpose of regulating their phenotypes. Antibodies that are expressed in cells infected with a lentiviral combinatorial antibody library are selected directly for function rather than only for binding. The potential diversity space can be very large because more than one lentivirus can infect a single cell. Thus, the initial combinatorial diversity of ∼1.0 × 1011 members generated by the random association of antibody heavy and light chains is greatly increased by the reassortment of the antibody Fv domains themselves inside cells. The power of the system is illustrated by its ability to select unusual antibodies. Here, the selected antibodies are potent erythropoietin agonists whose ontogeny depends on recombination at the protein level of pairs of antibodies expressed in the same cell to generate heterodimeric bispecific antibodies. The obligate synergy between the different binding specificities of the antibody's monomeric subunits appears to replicate the asymmetric binding mechanism of authentic erythropoietin.
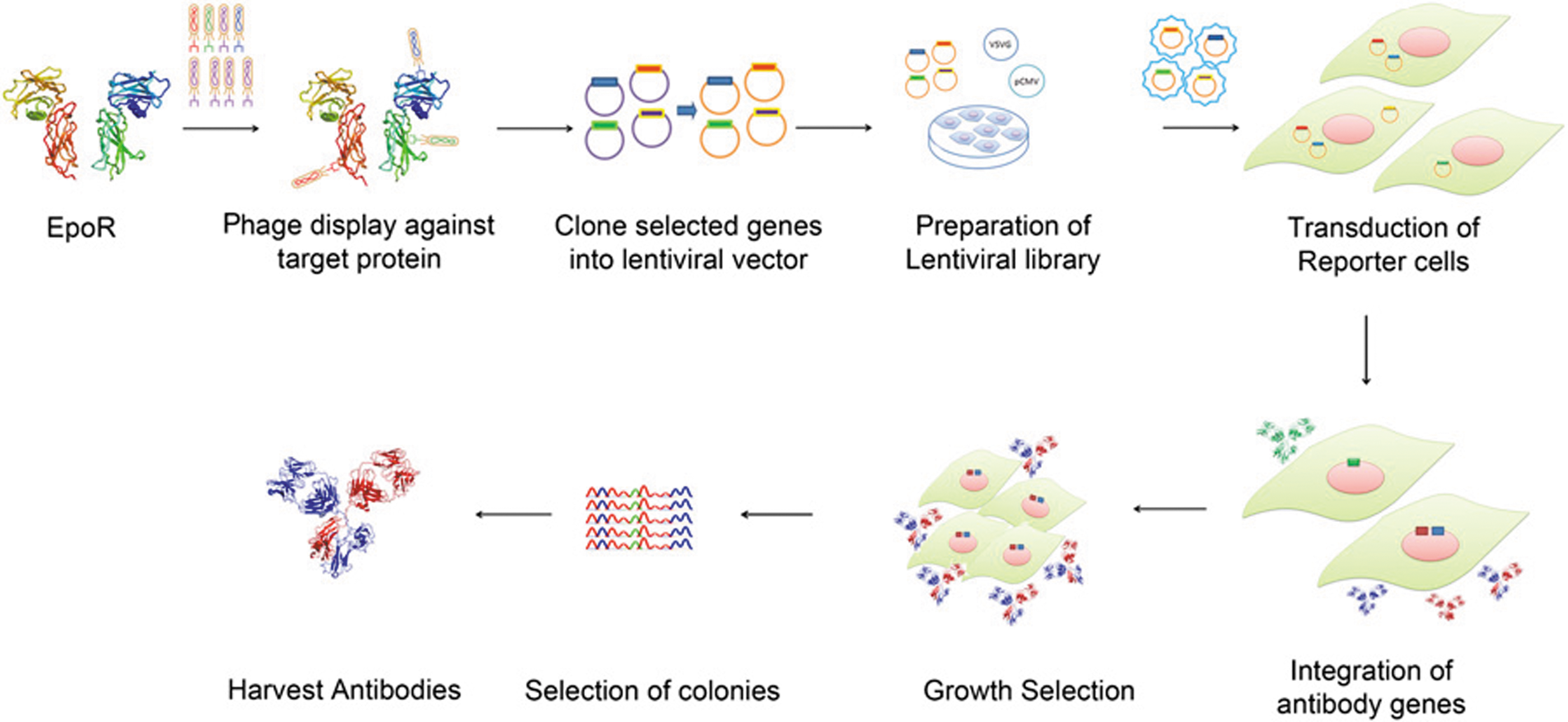
Commentary:The literature is replete with examples of highly potent and specific antibodies derived through the processes of phage display, in vitro maturation, and protein engineering. While development of high-affinity binders is becoming relatively routine, there are practically no methods to discover and improve antibodies that carry out relatively complex biological functions that extend beyond direct binding of an epitope: for example, catalytic antibodies exhibiting enzyme-like turnover and catalytic constants have been notoriously difficult to obtain; likewise, obtaining antibodies that act as receptor agonists, through complex set of interactions, has been very difficult. The primary reason for these difficulties has been that the steps of affinity selection and maturation have often been separated from the screening for biological function, the latter requiring the use of biology-specific mammalian model system. Zhang and colleagues present an elegant solution to the bottleneck by designing a combinatorial antibody library in lentivirus (see
figure
): after infection, multiple antibodies are expressed within each mammalian cell and are also secreted, allowing for multidimensional assessment of interactions and biological effects. Because more than one virus can infect a single cell, the initial diversity of the antibody population attained at the phage display level is retained and even enhanced upon the step of lenti infection and functional selection because of the possibility for further combinatorial associations of heavy and light chains derived from different antibody clones. In the example presented, the authors select directly for antibodies that operate as phenocopies of erythropoietin working through its receptor (EpoR). Phage displaying the initial combinatorial antibody library were enriched for EpoR binding through solution panning. The enriched pool was used to isolate the antibody genes and transfer them to lentivirus, which in turn were used to infect TF-1 cells overexpressing wild-type EpoR. The team was able to select directly high-quality agonist antibodies from cells expressing more than one antibody, with several distinct clones possessing high efficiency and acting through slightly different epitopes. The overall methodology presented in this work should be easy to adopt by others because it derives from readily available phage libraries and lentiviral vectors. Contributed by Anton Simeonov.
Scheme for selection of antibody agonists from combinatorial libraries. Antibodies that bound EpoR were selected from a combinatorial antibody library displayed in phage by affinity-based selection. The antibody genes from the selected phage were cloned into lentiviral vectors to allow phenotypic selections after infection of eukaryotic cells and integration of the antibody genes into the genome. The transduced cells were plated in methylcellulose agar such that the secreted antibodies were trapped around the cells producing them. The colonies that formed were harvested using a micromanipulator, and the antibody genes were recovered by PCR. The PCR products were cloned and sequenced, and the respective antibodies or antibody combinations were tested for their activity. The active antibodies were expressed in mammalian cells and purified for further characterization. PCR, polymerase chain reaction.
COLLOIDAL DRUGS
Owen SC, Doak AK, Wassam P, Shoichet MS, Shoichet BK: Colloidal aggregation affects the efficacy of anticancer drugs in cell culture.ACS Chem Biol2012;7:1429–1435.
Abstract: Many small molecules, including bioactive molecules and approved drugs, spontaneously form colloidal aggregates in aqueous solution at micromolar concentrations. Though it is widely accepted that aggregation leads to artifacts in screens for ligands of soluble proteins, the effects of colloid formation in cell-based assays have not been studied. Here, seven anticancer drugs and one diagnostic reagent were found to form colloids in both biochemical buffer and in cell culture media. In cell-based assays, the antiproliferative activities of three of the drugs were substantially reduced when in colloidal form as compared to monomeric form; a new formulation method ensured the presence of drug colloids versus drug monomers in solution. We also found that Evans Blue, a dye classically used to measure vascular permeability and to demonstrate the “enhanced permeability and retention (EPR) effect” in solid tumors, forms colloids that adsorb albumin, as opposed to older literature that suggested the reverse.
Commentary:Compound aggregation effects continue to lead to new surprises. Last year, compounds initially appearing as aggregators were found to form highly structured nanofibrils and lead to activation of procaspase 3 (see commentary “Aggregates get organized,” p. 498). Compound aggregation is now appreciated as a common artifact in biochemical assays. This article by Brian Shoichet's group examines the effect that aggregating compounds have in cell-based assays. The article examined seven anticancer drugs—bexarotene, crizotinib, fulvestrant, lapatinib, nilotinib, sorafenib, and vemurafenib—to determine if these formed colloidal aggregates. All these compounds were found to form large colloidal aggregates (>200 nm) in cell culture media at 37°C, even in the presence of serum albumin (see
figure
). In fact, these compounds were found to be highly disposed to aggregation, with three compounds (fulvestrant, nilotinib, and sorafenib) showing critical aggregation concentrations (CACs) <1 μM. Cell-based proliferation assays were developed in the presence and absence of 0.025% Tween-80 to measure the effect that either free compound or colloidal aggregates have on proliferation rate. Nonionic detergents such as Tween-80 are known to disrupt colloidal aggregates, and this study found that 0.025% Tween-80 did not inhibit the growth of the cells. Proliferation assays were performed for 72 hours in the presence and absence of detergent and although free drug (detergent present) inhibited cell growth by approximately 70%, no measureable effect of these compounds was observed in the absence of detergent. Therefore, contrary to compound aggregation effects in biochemical assay that leads to unwanted activity, compound aggregation in cell-based assays appears to lead to false negatives. Evans Blue is a dye used as a diagnostic reagent. Evans Blue was the first compound used to study the tendency of certain compounds to accumulate in tumor tissues more readily than in normal tissues (a phenomenon known as the “enhanced permeability and retention [EPR] effect”). This study showed that Evans Blue forms colloidal aggregates and sequesters serum albumin. Thermophoresis (a biophysical technique that provides a means to measure changes in hydrodynamic volume of proteins upon ligand binding, see Jerabek-Willemsen et al., Assay Drug Dev Technol 2011;9:342–353) was used to measure binding of Evans Blue to bovine serum albumin (BSA) labeled with Alexa Fluor 647. It was found that Evans Blue had no effect on the thermophoresis signal of the labeled BSA until the CAC was reached. Concentrations above the CAC of Evans Blue yielded large changes in the thermophoresis, which plateaued at a concentration corresponding to stoichiometric adsorption of BSA to the colloids. Addition of 0.01% Triton raised the Kd 50-fold. Therefore, although the EPR effect has been thought to be due to albumin binding the dye and leading to facilitated transport into tissues, this study suggests that albumin is sequestered by the colloidal particles of the dye, and it is this colloid–protein complex that yields the EPR effect. This article provides new insights into compound aggregator interferences and also describes experiments that specifically test for compound aggregation in cell-based assays. Contributed by Doug Auld.
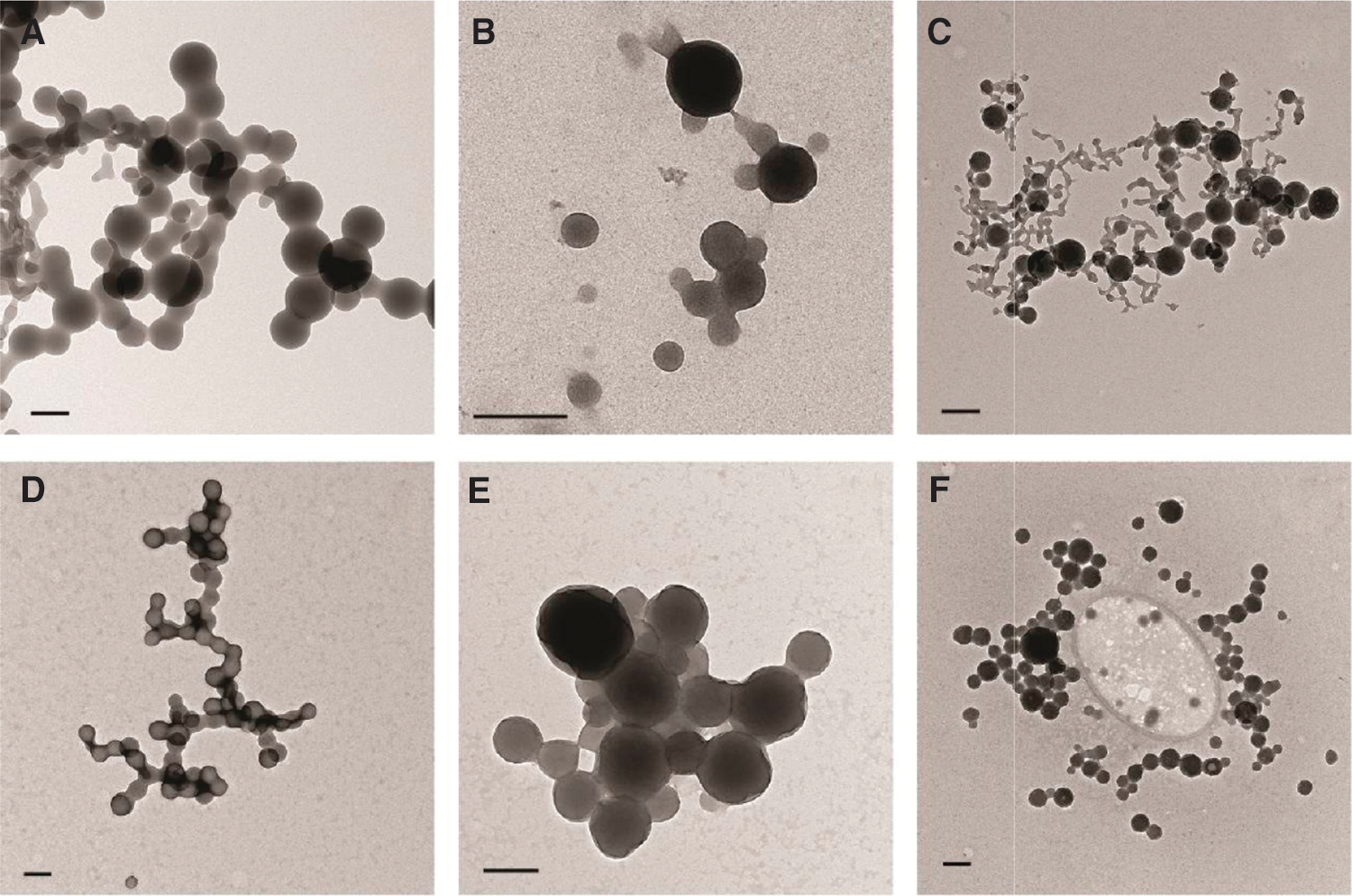
Transmission electron micrographs of aggregating drugs in phosphate buffer (top row) and 10% fetal bovine serum (bottom row): (A, D) fulvestrant, (B, E) lapatinib, (C, F) sorafenib. Bars represent 200 nm.
AGGREGATES GET ORGANIZED
Zorn JA, Wille H, Wolan DW, Wells JA: Self-assembling small molecules form nanofibrils that bind procaspase-3 to promote activation.J Am Chem Soc2011;133:19630–19633.
Abstract: Modulating enzyme function with small-molecule activators, as opposed to inhibitors, offers new opportunities for drug discovery and allosteric regulation. We previously identified a compound, called 1541, from a high throughput screen (HTS) that stimulates activation of a proenzyme, procaspase-3, to generate mature caspase-3. Here we further investigate the mechanism of activation and report the surprising finding that 1541 self-assembles into nanofibrils exceeding 1 μm in length. These particles are an unanticipated outcome from an HTS that have properties distinct from standard globular protein aggregators. Moreover, 1541 nanofibrils function as a unique biocatalytic material that activates procaspase-3 via induced proximity. These studies demonstrate a novel approach for proenzyme activation through binding to fibrils, which may mimic how procaspases are naturally processed on protein scaffolds.
Commentary:Inhibition by compounds that form aggregates has been characterized to be due to the formation of colloidal particles that sequester enzymes and prevent interaction with their substrates. Colloidal aggregates can be quite large and have irregular shapes. This article describes an unexpected phenomenon in which characterization of a compound leading to activation of procaspase-3 (1541, see
first figure
) revealed that the compound self-assembles to form large nanofirbrils. The possibility of1541forming a colloidal aggregate was investigated, but it was found that this compound behaved differently in standard tests for aggregates. Treatment of the zymogen procaspase-3 with1541results in activation, which involves a lag phase. Centrifugation and dialysis experiments demonstrated that1541formed particles ∼4 nm in size that interacted with procaspase-3. Further,1541was found to sediment with a solubility constant that was approximately the same as the AC50 for activation of procaspase-3. Investigation into the nature of the particles formed by1541included transmission electron microscopy (see
second figure
), which confirmed the formation of long thin nanofibrils by1541and coating of zymogen on the surface of the nanofibril. Caspase zymogens are activated by autoproteolysis and the nanofibrils effectively bind procaspase-3 protein on the surface, which increases the local concentration and leads to activation either by a cis or trans mechanism (see
first figure
); the specific mechanism is still not understood. Fibrous aggregates formed by amyloid-β proteins (Aβ) can lead to proenzyme activation such as the conversion of prekallikrein to kallikrein and the nanofibrils showed some resemblance to these fibrils. This prompted the investigators to generate fibrils from Aβ, and it was found that these Aβ fibrils also activated procaspase-3. Therefore, these observations may have relevance to Alzheimer's disease especially because neurotoxicity has been linked to caspase activity. The compound1541represents a unique class of compounds that mediate enzyme activity through self-assembling of nanostructures. Contributed by Doug Auld.
Model of procaspase-3 activation by 1541 nanofibrils. 1541 and analogues spontaneously self-assemble into nanofibrils. The fibrils bind directly to procaspase-3 to promote increased local concentration of the enzyme. Upon recruitment to the fibrils, procaspase-3 is activated to generate mature caspase-3.
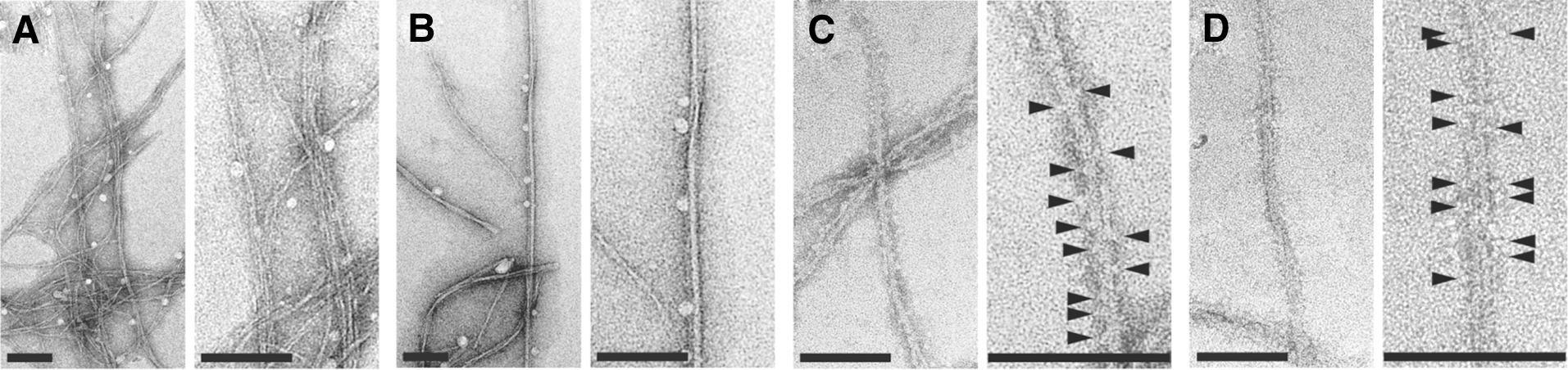
TEM images of 1541 nanofibrils with and without procaspase-3. (A) Negatively stained 1541 at 25°C shows bundles of very thin and flexible fibrils. (B) In contrast, 1541 at 37°C consists mainly of larger, less flexible fibrils. (C, D) 1541 fibrils decorated with procaspase-3 at (C) 25°C and (D) 37°C. The procaspase-3 molecules decorating the surface are indicated by arrowheads. Scale bars =100 nm.
HIJACKING A TRICK FROM CANCER CELLS TO CREATE A NOVEL ENGINEERED ENZYME WITH INDUSTRIAL UTILITY
Reitman ZJ, Choi BD, Spasojevic I, Bigner DD, Sampson JH, Yan H: Enzyme redesign guided by cancer-derived IDH1 mutations.Nat Chem Biol2012;8:887–889.
Abstract: Mutations in an enzyme can result in a neomorphic catalytic activity in cancers. We applied cancer-associated mutations from isocitrate dehydrogenases to homologous residues in the active sites of homoisocitrate dehydrogenases to derive enzymes that catalyze the conversion of 2-oxoadipate to (R)-2-hydroxyadipate, a critical step for adipic acid production. Thus, we provide a prototypic example of how insights from cancer genome sequencing and functional studies can aid in enzyme redesign.
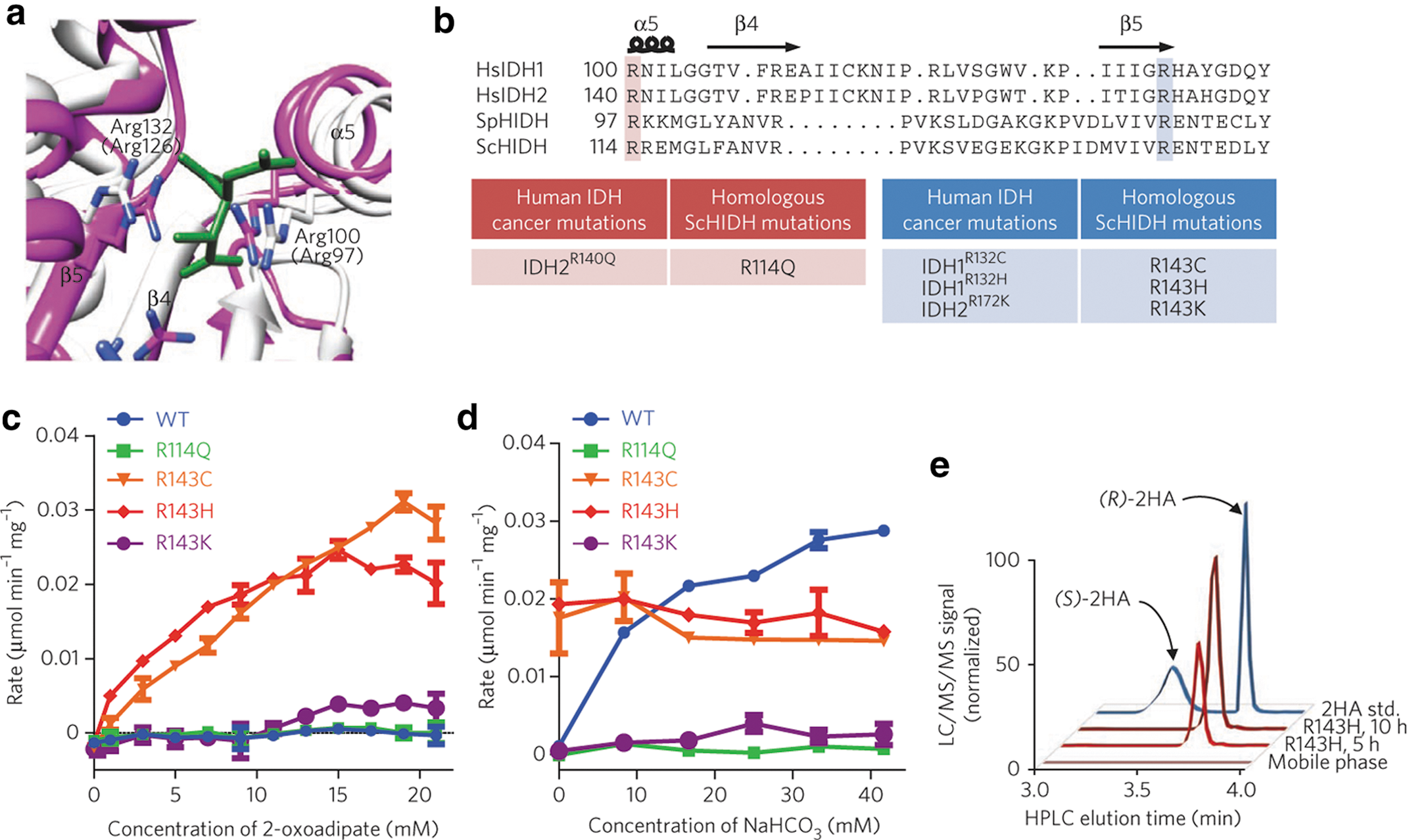
Commentary:Isocitrate dehydrogenases (IDHs) are important enzymes in metabolism that catalyze the NAD(P)-dependent oxidative decarboxylation of (2R,2S)-isocitrate to produce NADPH, α-ketoglutarate (α-KG, also called 2-oxoglutarate), and carbon dioxide. IDH1 and IDH2, which are NADP-dependent enzymes, were found previously to be mutated in acute myeloid leukemia, glioma, and glioblastoma. The most common site of mutation is at a crucial arginine (R132 in IDH1; HsIDH1R132) that is important for substrate binding and catalysis. When the arginine is mutated, there is a loss of function of the oxidative decarboxylation of isocitrate, but there is a novel gain of function for production of the oncometabolite (R)-2-hydroxyglutarate (2-HG) from α-KG. The authors took their cues from these cancers to mutate the homologous residues of Saccharomyces cerevisiae homoisocitrate dehydrogenase (ScHIDH) as determined by structure-based sequence alignment (R114Q, R143C, R143H, and R143K). They created mutant enzymes that convert 2-oxoadipate to (R)-2-hydroxyadipate rather than the wild-type activity of conversion of (2R,3S)-homoisocitrate to 2-oxoadipate (see
first figure
). The four mutant enzymes and the wild-type enzyme were compared in an enzyme assay in which the consumption of NADH was monitored in the presence of 2-oxoadipate (see
second figure
). The R143C and R143H ScHIDH enzymes had the highest activity, and the production of the (R)-2-hydroxyadipate product was verified by mass spectroscopy. Adipic acid is a widely used substrate for the synthesis of nylon, and there is an interest in finding cheaper and more environmentally friendly methods to produce it. 2-hydroxyadipate is a precursor to the production of adipic acid, and this neomorphic activity of mutant homoisocitrate dehydrogenase presents an enzymatic route to its production. It will be interesting to see whether these new mutant enzymes can be further improved by directed evolution and become useful in the production of nylon. Contributed by Mindy I. Davis
(a) Isocitrate dehydrogenases (IDHs) catalyze the NAD+- or NADP+-linked reversible oxidative decarboxylation of (2R,3S)-isocitrate to form 2-oxoglutarate and CO2. (b) HIDHs catalyze the NAD+-linked reversible oxidative decarboxylation of (2R,3S)-homoisocitrate to form 2-oxoadipate and CO2. In the reverse direction, HIDHs catalyze the reductive carboxylation of 2-oxoadipate with CO2 to form (2R,3S)-homoisocitrate. (c) In human cancer, IDH mutants such as HsIDH1R132H catalyze the noncarboxylating reduction of 2-oxoglutarate to (R)-2-hydroxyglutarate. (d) Analogous HIDH mutants could catalyze the noncarboxylating reduction of 2-oxoadipate to (R)-2-hydroxyadipate; that is, (R)-2-hydroxyadipate dehydrogenases.
(a) Superimposition of the active site for SpHIDH19 (white) onto HsIDH1 (Aktas & Cook, Biochemistry 2009;48:3565–3577) (pink; complex with isocitrate in green). Arg100 and Arg132 of HsIDH1 are shown, and corresponding residues for SpHIDH are shown in parentheses. (b) Alignment of HsIDH1, HsIDH2, SpHIDH, and ScHIDH. (c) Initial rate of NADH decrease catalyzed by the indicated ScHIDH mutant at varying concentrations of 2-oxoadipate and 300 μM NADH. WT, wild type. (d) Initial rate of NADH decrease catalyzed by the indicated ScHIDH mutant in the presence of varying concentrations of NaHCO3 as well as 15 mM 2-oxoadipate and 100 μM NADH. (e) LC/MS/MS chromatogram showing (R)-2-hydroxyadipate (2HA; Q1/Q3 m/z = 377/161) accumulation in a reaction containing ScHIDHR143H, 2 mM NADH, 2 mM 2-oxoadipate, and 500 mM HEPES at times indicated. A racemic (R/S)-2-hydroxyadipate standard (std.) and mobile phase are shown for reference. Unless otherwise specified, all reactions contained 20 mM MgCl2, 40 ng/μL of the indicated purified enzyme and 100 mM HEPES, pH 7.3. Data points are mean ± SD from n = 2 reactions. All plots are representative of three independent experiments.
DRUG DELIVERY THROUGH A “WET BALL OF SAND”
Korin N, Kanapathipillai M, Matthews BD, Crescente M, Brill A, Mammoto T, Ghosh K, Jurek S, Bencherif SA, Bhatta D, Coskun AU, Feldman CL, Wagner DD, Ingber DE: Shear-activated nanotherapeutics for drug targeting to obstructed blood vessels.Science2012;337:738–742.
Abstract: Obstruction of critical blood vessels due to thrombosis or embolism is a leading cause of death worldwide. Here, we describe a biomimetic strategy that uses high shear stress caused by vascular narrowing as a targeting mechanism—in the same way platelets do—to deliver drugs to obstructed blood vessels. Microscale aggregates of nanoparticles were fabricated to break up into nanoscale components when exposed to abnormally high fluid shear stress. When coated with tissue plasminogen activator and administered intravenously in mice, these shear-activated nanotherapeutics induce rapid clot dissolution in a mesenteric injury model, restore normal flow dynamics, and increase survival in an otherwise fatal mouse pulmonary embolism model. This biophysical strategy for drug targeting, which lowers required doses and minimizes side effects while maximizing drug efficacy, offers a potential new approach for treatment of life threatening diseases that result from acute vascular occlusion.
Commentary:Drug therapies being applied to very small target areas within the patient's body, such as obstructed blood vessels in stroke, inevitably suffer from toxic side effects and insufficient efficacy brought about by the active agent being distributed throughout the body while only being needed within a very confined area. Targeted drug delivery has been achieved through the relatively simple concepts of applying patches, pumps, or catheters that feed the drug to an area directly adjacent to the target, as well as through arguably more complex schemes, such as the design of “smart drugs” that carry an addressable or selectively unmasked chemical moiety that works through a specific molecular interaction. To address the particularly acute risk associated with the application of excessive doses of clot-busting drugs, Korin and colleagues designed a thrombolytic delivery system that makes use of the dramatic differences in physical characteristics between obstructed vessels and normal vasculature: fluid shear stress in obstructed areas was shown to increase locally by one to two orders of magnitude. The authors developed micro-sized particle aggregates that were close to the size of natural platelets; these were produced by spray-drying concentrated solutions of biocompatible and biodegradable poly(lactic-co-glycolic acid) to form micrometer-sized aggregates composed of small nanoparticles (
first figure
). The nanoparticles could be fabricated to incorporate fluorescent dye for tracking and an active drug ingredient. A key property of these nanoparticle aggregates was that they retained their integrity during administration and transport through regular blood vessels but burst into small nano-sized pieces upon experiencing the higher shear stress within the constrained vessels, thus releasing the drug through the sharp increase in the carriers' surface area (
second figure
): the authors described this process as “a wet ball of sand dispersing into individual grains when rubbed in one's hands.” The new delivery system was successfully demonstrated through the application of tissue plasminogen activator (attached to the nanoparticles through biotin-streptavidin chemistry) in several animal models of blood-clot resolution (injury-induced arterial embolism and pulmonary embolism). Contributed by Anton Simeonov.
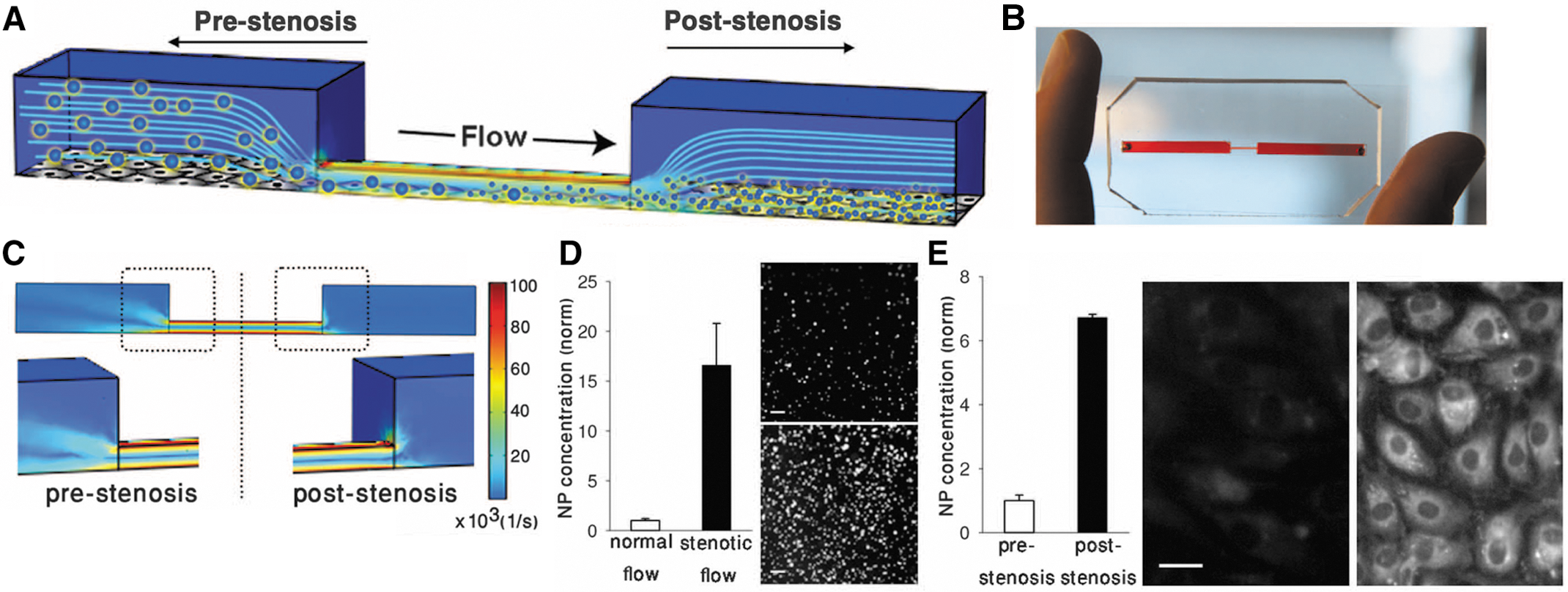
Microscale SA-NTs disperse into NPs only when exposed to pathological shear stresses. (A) Scanning electron micrographs of the microscale (∼2–5 μm) SA-NTs (left) and the PLGA NPs (∼180 nm) used to produce them (right). Scale bar, 2 μm. (B) Fluorescence micrographs demonstrating intact SA-NTs (top) and NPs dispersed after their exposure to 1000 dyne/cm2 for 10 min by using a rheometer (bottom). Scale bar, 10 μm. (C) Quantification of release of fluorescent NPs from the SA-NTs as a function of shear revealed that exposure to pathological levels of shear (≥100 dyne/cm2 for 1 min) caused large increase in the breakup of the microscale aggregates into NPs compared with physiological levels of shear (1 or 10 dyne/cm2; *p < 0.005). (D) CFD simulations comparing fluidic shear stress in a normal coronary artery (left) and a stenotic vessel with a 60% lumen obstruction (right). Left inset: The corresponding angiogram of the stenotic left coronary artery in a 63-year-old male patient. SA-NT, shear-activated nanotherapeutics; PLGA, poly(lactic-co-glycolic acid); NP, nanoparticles; CFD, computational fluid dynamics.
Shear-induced dissociation of SA-NTs and NP targeting under hemodynamic conditions in microfluidic devices. (A) A microfluidic vascular stenosis model showing how SA-NTs (large spheres) should remain intact in the prestenotic region but then break up into NPs (small spheres) when they flow through a constriction (90% lumen occlusion) and can accumulate in endothelial cells lining the bottom of the channel. (B) A photograph of the microdevice that mimics vascular stenosis fabricated in poly-dimethylsiloxane (PDMS). (C) CFD simulations of the microfluidic device shown in (B) demonstrating that a physiological inlet shear rate of 1000 s−1 (10 dyne/cm2) upstream from the constriction increases to a pathological level of ∼100,000 s−1 (1000 dyne/cm2) in the region displaying 90% lumen occlusion. (D) Graph shows a >10-fold increase in release of fluorescent NPs from SA-NTs when they are perfused through the channel shown in (B) compared with flow through an unconstricted channel (*p < 0.005). Fluorescent micrographs compare the NPs collected in the outflow from the control channel (top) versus the constricted channel (bottom). Scale bar, 2 μm. (E) Graph demonstrates that many more fluorescent NPs accumulate in endothelial cells lining the downstream area (poststenosis) of the constriction relative to an upstream area (p < 0.005). Fluorescence microscopic images show cells from regions before (left) and after (right) the constriction. Scale bar, 20 μm.
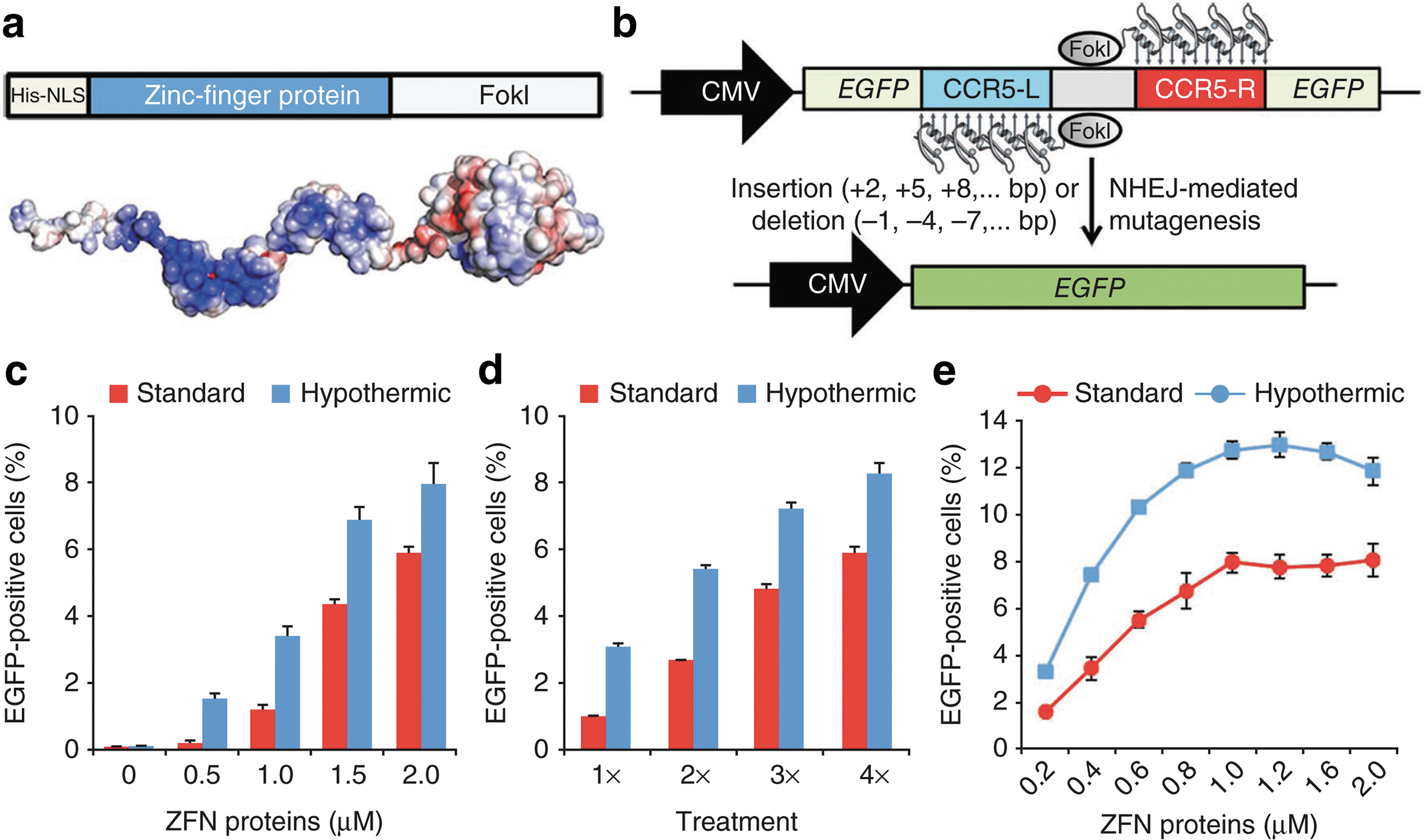
JUST ADD ZFN
Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF 3rd: Targeted gene knockout by direct delivery of zinc-finger nuclease proteins.Nat Methods2012;9:805–807.
Abstract: Zinc-finger nucleases (ZFNs) are versatile reagents that have redefined genome engineering. Realizing the full potential of this technology requires the development of safe and effective methods for delivering ZFNs into cells. We demonstrate the intrinsic cell-penetrating capabilities of the standard ZFN architecture and show that direct delivery of ZFNs as proteins leads to efficient endogenous gene disruption in various mammalian cell types with minimal off-target effects.
Commentary:The use of engineered zinc-finger nuclease (ZFN) to site-specifically modify an organism's genome has indeed taken off during the past few years; however, a major remaining limitation to the expansion of ZFNs into a clinical setting is that the viral vector delivery of these nucleases carries an associated danger of severe side effects due to the potential for insertional mutagenesis. While ongoing research has focused on making the ZFNs more precise and the delivery vectors safer, a better solution altogether may be to deliver the nuclease proteins directly into the cell, bypassing the vector. However, most proteins do not enter cells readily and are not easily amenable to standard transfection/transduction protocols typically applied to nucleic acid material. In the present work, the authors made a series of attempts to engineer ZFN fusions to known transductions domains, in order to facilitate the entry of ZFNs into cells, but repeatedly found that the engineered hybrids were extremely difficult to express. Then, surprisingly the authors discovered that unmodified ZFN proteins could enter cells completely on their own (see
figures
): the zinc-finger DNA-binding domain portion of these proteins carries a net positive charge, a factor that was proposed as the main driver for this newly discovered property of ZFNs. Importantly, during a detailed analysis of the efficiency and specificity of ZFN action, the team found that ZFNs delivered as proteins exhibited less off-target effects; this improved performance could be due to the shorter half-life of the protein delivered exogenously compared to proteins produced in-cell. The pilot study described here not only provides a proof-of-concept for the direct delivery of genome-editing agents into cells but also indicates that issues of off-target effects and toxicity could perhaps be better mitigated using this new delivery mode. Contributed by Anton Simeonov.
ZFN proteins are cell permeable and induce targeted mutagenesis in human cells. (a) Diagram of ZFN protein and electrostatic potential of the ZFN protein surface colored from dark red (–kT/e) to white (0 kT/e) to dark blue (+5 kT/e). (b) Schematic representation of the fluorescence reporter system used to evaluate cellular penetration of ZFN proteins. (c–e) Percentage of EGFP-positive cells as determined by flow cytometry in reporter cells treated with increasing amounts of ZFN proteins (c) or subjected to consecutive treatments with 0.2 μM ZFN proteins (d) or to three consecutive treatments with increasing amounts of ZFN proteins (e). Either standard or transient hypothermic conditions were used in all cases. Error bars, SD (n = 3). CMV, cytomegalovirus promoter; EGFP, enhanced green fluorescent protein; NHEJ, non-homologous end joining; NLS, nuclear localization signal; ZFN, zinc-finger nuclease.
Representative sequence analysis of the EGFP locus from isolated EGFP-positive cells. Multiple deletions (dashes) and insertions (lowercase) induced by NHEJ repair are aligned to the cleavage site (wild type [WT]).
LOOK, NO LABEL! LABEL-FREE MST
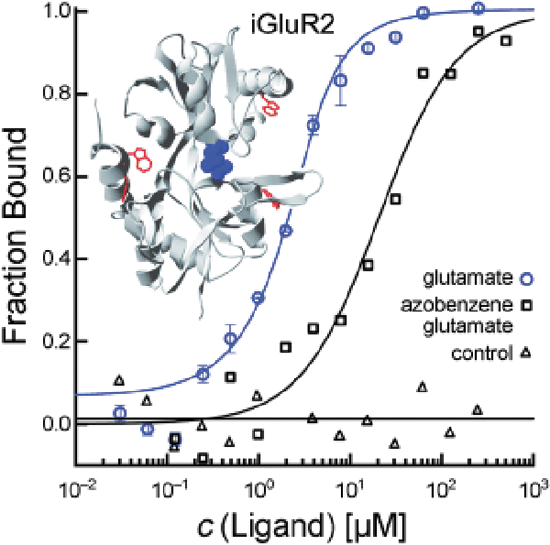
Seidel SA, Wienken CJ, Geissler S, Jerabek-Willemsen M, Duhr S, Reiter A, Trauner D, Braun D, Baaske P.: Label-free microscale thermophoresis discriminates sites and affinity of protein-ligand binding.Angew Chem Int Ed Engl2012;51:10656–10659.
Graphical Abstract: Look, no label! Microscale thermophoresis makes use of the intrinsic fluorescence of proteins to quantify the binding affinities of ligands and discriminate between binding sites. This method is suitable for studying binding interactions of very small amounts of protein in solution. The binding of ligands to iGluR membrane receptors, small-molecule inhibitorss to kinase p38, aptamers to thrombin, and Ca2+ ions to synaptotagmin was quantified.
Xu R, Ori A, Rudd TR, Uniewicz KA, Ahmed YA, Guimond SE, Skidmore MA, Siligardi G, Yates EA, Fernig DG: Diversification of the structural determinants of fibroblast growth factor-heparin interactions: implications for binding specificity.J Biol Chem2012 Sept 27 [Epub ahead of issue]; DOI: 10.1074/jbc.M112.398826.
Abstract:
Background. Heparan sulfate (HS) regulates the transport and signaling activities of fibroblast growth factors (FGF).
Results. The molecular determinants of the interactions of FGFs and heparin were identified.
Conclusion. There are clear molecular specificities determining the interactions of FGFs with the polysaccharide.
Significance. The expansion of the FGFs in metazoan evolution parallels the diversification of the specificity of their interactions with heparin.
Summary. The functions of a large number (>435) of extracellular regulatory proteins are controlled by their interactions with heparan sulfate (HS). In the case of fibroblast growth factors (FGFs), HS binding determines their transport between cells and is required for the assembly of high affinity signalling complexes with their cognate FGF receptor. However, the specificity of the interaction of FGFs with HS is still debated. Here, we use a panel of FGFs (FGF-1, FGF-2, FGF-7, FGF-9, FGF-18, and FGF-21) spanning five FGF subfamilies to probe their specificities for HS at different levels: binding parameters, identification of heparin binding sites (HBSs) in the FGFs, changes in their secondary structure caused by heparin binding, and structures in the sugar required for binding. For interaction with heparin, the FGFs exhibit KD values varying between 38 nM (FGF-18) and 620 nM (FGF-9) and association rate constants spanning over 20-fold (FGF-1, 2,900,000 M-1s-1, FGF-9, 130,000 M-1s-1). The canonical HBS in FGF-1, FGF-2, FGF-7, FGF-9, and FGF-18 differs in its size and these FGFs have a different complement of secondary HBS, ranging from none (FGF-9) to two (FGF-1). Differential scanning fluorimetry identified clear preferences in these FGFs for distinct structural features in the polysaccharide. These data suggest that the differences in heparin binding sites in both the protein and the sugar are greatest between sub-families and may be more restricted within a FGF sub-family in accord with the known conservation of function within FGF sub-families.
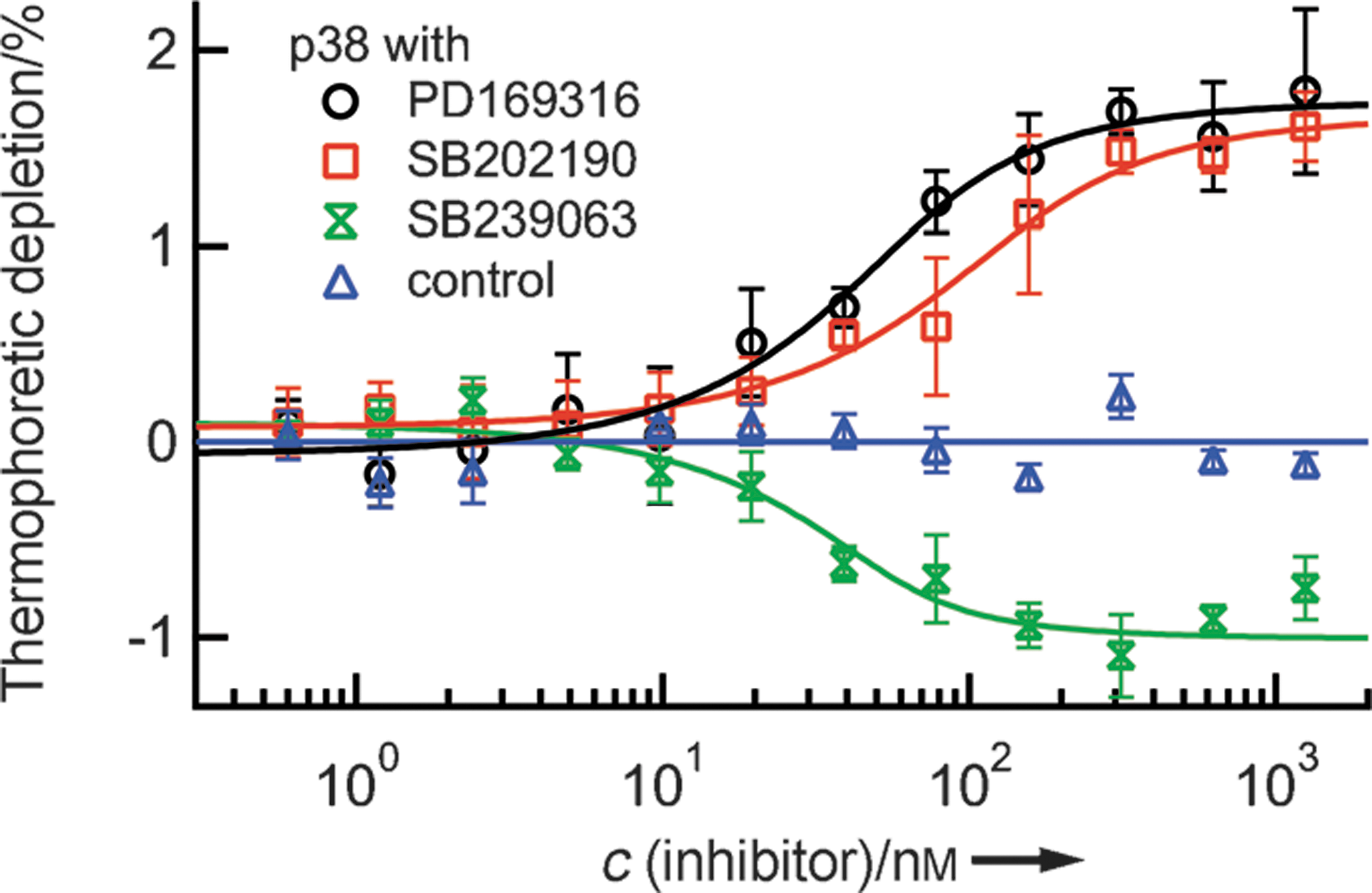
Commentary:Characterization of a biomolecular interaction usually ranges from obtaining basic binding parameters, such as binding affinities, to the understanding of more advanced binding signatures which can often provide additional insight on mode of action (MoA). Examples in the latter category include binding cooperatively and binding kinetics. The study of binding kinetics can quantify the concentrations of binding species as a function of time based on initial total concentration and rate constants. Association rates (“on rates,” Ka) and dissociation rates (“off rates,” Kd) are frequently measured by optical sensor-based techniques, such as surface plasmon resonance (SPR), and a recent article made a welcomed comparison on the performance on reporting Ka and Kd between SPR and a TR-FRET based assay (LanthaScreen Eu kinase binding assay; Mason et al., Assay Drug Dev Technol 2012;10:468–475). The concept of binding cooperativity has been featured in our August 2012 issue's “Literature Search and Review” (see “Evaluation of binding cooperativity by MST,” Assay Drug Dev Technol 2012;10:306–309), and it entails the interaction among different binding sites, provided that the binding sites for specifically studied ligands have already been known. Herein, two different articles are highlighted because they reported on two different methods that provided different levels of information on the differentiation of binding sites that were available on protein targets of interest. In the work done by Seidel et al., the authors successfully applied a true label-free version of microscale thermophoresis (MST) for the characterization of small molecule binding to a membrane receptor and a kinase. Compared to standard MST in which one binding partner has to be labeled with a fluorophore through covalent coupling, label-free MST takes advantage of protein intrinsic fluorescence, which is usually dominated by the presence of tryptophan (Trp). As a result, polarity of the local environment and proximity of other residues are main factors to influence fluorescence emission peak and strength. Using the nonactivated form of mitogen-activated protein kinase (MAP kinase) p38 that contains five Trp residues, two structurally similar p38 inhibitors were found to exhibit the same directions in their thermophoretic profiles while a structurally different p38 inhibitor was found to exhibit opposite thermophoretic profile with respect to the former two compounds. Specifically, relative to the unbound protein, the former two compounds led to less depletion for the protein complexes (positive MST) and the latter compound resulted in greater depletion for the protein–compound complex (see
first figure
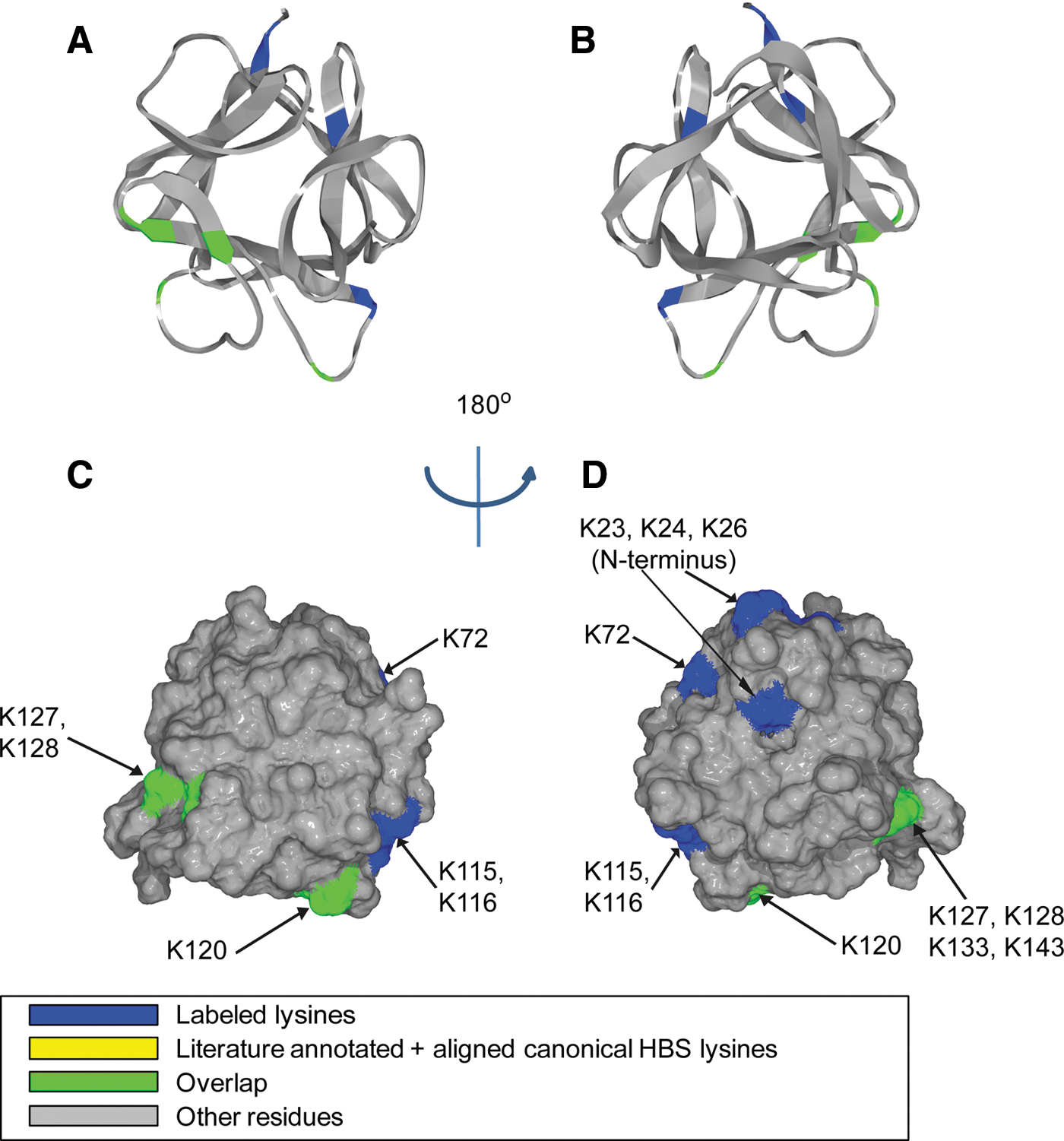
). In addition to the close agreement obtained between the affinities derived from label-free MST and previously published inhibitory activities, MST presented its potential for binding site discrimination through the direction of molecular movement under a controlled temperature gradient. In order to obtain a full picture of the inhibitor MoA, additional characterization, such as functional assays or competition assays, could be considered for further studies. Several criteria need to be considered when applying label-free MST: autofluorescent buffer components should be avoided because they could interfere with protein Trp emission measurement; protein concentration should be chosen at the lowest possible level at which its fluorescence still exceeds background fluorescence significantly. When the titrated binding partner also possesses Trp (such as another protein) or strong autofluorescence (such as certain small molecules), label-free MST can be difficult to perform or careful control experiments are required to remove the fluorescence contribution from the titrated partner. Depending on the number and local environment of Trp residues, protein consumption can vary from case to case, and this could limit the detection range by at least one order of magnitude compared to the standard MST approach. Nevertheless, the applicability potential of MST, be it standard or label free, has been on a steep ascension since its commercialization. Label-free MST complements standard MST because some proteins may suffer from labeling by losing activity. The ability of MST to answer advanced kinetics questions awaits further investigation and evaluation. In a separate work done by Xu et al., the authors applied standard MST to evaluate binding affinities for a panel of fibroblast growth factors (FGFs) with heparan sulfate (HS) and its derivatives. The Kds obtained from standard MST were derived at equilibrium, and they consistent with Kds derived from binding kinetics parameters, Kon and Koff, validating the set-up of both heterogeneous and solution-based methods. In order to gain a deeper understanding of the molecular determinants for the binding interactions between FGFs and heparin, Xu et al. also utilized a number of complementary techniques. In particular, they highlighted the usage of a validated “Protect and Label” approach for the identification and characterization of heparin binding sites (HBS) in a family of FGFs. Specifically, lysine side chains exposed to solvent when protein was bound to heparin was first protected by sulfo-N-hydroxysulfosuccinimide acetate,and was subsequently labeled with biotin after protein dissociated from heparin. Thus, biotinylated peptides could be further identified by tandem mass spectrometry (MS) after digestion, providing fingerprints for HBS. For example, in addition to a canonical HBS that had been recapitulated using this method, two other secondary sites were found for FGF1 (see
second figure
). Through this approach, additional HBS has been recognized for a number of FGFs that were studied. Heparin binding induced protein structural changes, structural elements required in heparin derivatives for binding, and binding specificities were further studied using circular dichroism spectroscopy and differential scanning fluorimetry. With due optimization of the reaction conditions, this Protect and Label approach presents a good level of sensitivity and selectivity towards lysine-containing binding sites. Followed by MS analysis, the method has the potential to be transformed into high-throughput formats. One pitfall of the method is its inability to recognize non–lysine-containing binding sites, and novel protein modification reagents that could specifically target different amino acids are welcomed. Contributed by Wendy Lea.
Screening of small-molecule kinase inhibitors. Three selective inhibitors were successfully tested for binding to the nonactivated form of MAP kinase p38α (c = 100 nM). Corresponding to structural differences, the binding of SB202190 and PD169316 has the opposite effect on the thermophoretic movement compared to SB239063. SB202190 binds with a Kd value of 48 ± 21 nM. The upper limits of affinity for PD169316 and SB239063 were determined as 33 nM and 20 nM, respectively. These results are in good agreement with previously reported values. Thermally denatured p38α did not show binding (control). MAP, mitogen-activated protein.
Position of biotinylated peptides in FGF-1 (residues 22–154) identified by structural proteomics mapped onto the predicted 3-dimensional structure (PDB 2ERM; Canales et al., FEBS J 2006;273:4716–4727). Labeled peptides are colored in blue, and peptides overlapping with literature-annotated and aligned canonical HBS lysines are colored in green (DiGabriele et al., Nature 1998;393:812–817; Pellegrini et al., Nature 2000;407:1029–1034). (A, B) Ribbon diagram. (C, D) Corresponding molecular surface. FGF-1 is shown using schematic representation. (B, D) 180° back view of (A) and (C). HBS, heparin binding site.