
Abstract
Several different segments of the gp41 N-heptad repeat coiled coil have been constructed using N-terminal bipyridyl modification of composite peptides and inducing trimerization by adding ferrous ions. These metallopeptides act as receptors in fluorescence-binding assays with corresponding fluorescently labeled C-peptide probes. The FeII coordination complex quenches C-peptide fluorescence upon binding, and reversal of quenching by a small molecule inhibitor can be used to obtain the inhibitor-binding constant. A total of 10 peptide pairs targeting 25–46 residue segments of the coiled coil were constructed, with C-peptide probes of different lengths and binding affinities. The result is a suite of assays for exploring binding in the mM to nM range to any desired region of the coiled coil, including the hydrophobic pocket (HP), extended regions on either side of the pocket, or a region associated with T20 resistance mutations. These assays are high-throughput ready, and could be used to discover novel compounds binding along various regions of the gp41 coiled coil groove. They were used to evaluate a sub-μM low molecular weight fusion inhibitor, resulting in the finding that the molecule bound specifically to the HP and attained its potency from a low off-rate.
Introduction
The gp41 coiled coil has been considered an important target for fusion inhibitors, based on peptides studies, which show that CHR peptides are highly effective fusion inhibitors. 6 –8 These include T20, currently the only FDA-approved fusion inhibitor. 9 Small molecule fusion inhibitors have been sought for many years, but with limited success, having only μM potency against fusion. 10,11 They have chiefly targeted a known hydrophobic pocket (HP) on the coiled coil. However, in many cases, there is little experimental evidence confirming the exact binding site and no way to test inhibitors against different sites along the coil. Finding two inhibitors that bind in adjacent sites could enable a fragment-based drug design approach, in which a linker connecting two adjacent-binding fragments can have a multiplicative effect on their binding affinity. 12
In previous work, we have demonstrated the use of metal–bipyridyl coordination to stabilize a segment of the gp41 coiled coil representing the HP, 13 as well as an extended receptor targeting adjacent sites and high-affinity interactions. 14,15 This study expands on the analysis of inhibitors detected with these assays, and introduces a new assay that focuses on the N-terminal end of the coiled coil, a site prone to development of resistance mutations in viruses exposed to T20. 16 C-peptide probes have been designed for fluorescence intensity assays that interrogate the NHR subsections. Adjusting C-peptide length tuned both assay selectivity and the range of detectable affinities. Small molecule inhibitors could easily be detected by competitive inhibition, as we have shown previously. Comparison of the results with the known biological activity of peptide segments of the CHR 17 and small molecules 18 confirmed the relevance of these biochemical screening assays for detection of fusion inhibitors. The ease and rapidity of the assays lends them to implementation in a high volume format. Utilization of the assays could result in identification of novel fragments or small molecules binding at specific subsites, with the possibility of linking them to generate high-affinity nonpeptide inhibitors. This concept has been applied for the derivation of C-peptide inhibitors of HIV-1 fusion containing multifunctional domains. 2,17
Materials and Methods
Materials
Peptides were prepared by solid-phase synthesis (Biosynthesis, Inc.). Small molecules were prepared according to previously published procedures. 18 Fluorescein (FL) and Lucifer yellow (LY) were purchased from Molecular Probes (Invitrogen) as iodoacetamides and covalently attached through standard cysteine disulfide chemistry according to the manufacturer's instructions. Labeled peptides were purified by high pressure liquid chromatography using acetonitrile: H2O with 0.05% trifluoracetic acid.
Circular Dichroism Experiments
Circular dichroism (CD) studies were performed on a DSM20 CD spectrophotometer from On-Line Instruments Systems, Inc., using 20 μM solutions of peptides in a 15 mM Tris-acetate buffer, pH 7.0, at 25°C.
Fluorescence Intensity Measurements
Fluorescence intensity was measured in 384-well low-volume plates (Greiner Bio-one) on a Spectramax M5 plate reader (Molecular Devices). Env# concentration was determined in 6 M Gdn.HCl from the absorbance at 291 nm, using the extinction coefficients of bipyridine (ɛ291=12,000) plus the calculated contribution of any Trp and Tyr residues in the peptide sequence (
Assay Protocol Table (C18-e2.0LY/FL, C21-e6.0LY)
1. Greiner low volume tapered black/clear 384-well plates; P=30 nM Cn-#FL or 400 nM Cn-#LY in 0.02% Tween-20, 25 mM Tris, 25 mM acetic acid, pH 7.0, B=buffer (no Tween).
2. R=7.2 μM Fe(env2.0)3 or 6 μM Fe(env5.0)3 or 6 μM Fe(env6.0)3 with C21-e6.0LY in a buffer (concentration of monomer peptide).
3. Determine assay bounds F max, F min and Z′.
4. To determine KI, use serial dilution.
5. Measure immediately and at 15 min intervals for up to 1 h, to allow for equilibration.
6. Fractional fluorescence=(Fs±σs)/<Fc>, where<Fc>is the average value of the control fluorescence in odd wells, and Fs is the fluorescence of the sample. Large increase in control values with compound concentration indicates compound interacts with probe, for example, by micelle formation.
DMSO, dimethyl sulfoxide; LY, lucifer yellow; FL, fluorescein.
Assay Protocol Table (C32-e5.0FL, C28-e5.1LY/FL, C21-e6.0FL)
1. Greiner low volume tapered black/clear 384-well plates; P=30 nM Cn-#FL or 400 nM Cn-#LY in 0.02% Tween-20 in buffer.
2. To determine KI, use serial dilution.
3. B=25 mM Tris, 25 mM acetic acid, pH 7.0.
4. R=150 nM Fe(env5.0)3 with C32-e5.0FL, 3 μM Fe(env5.0)3 with C28-e5.1FL/LY, 2 μM Fe(env6.0)3 with C28-e5.1FL/LY, 1.5 μM Fe(env6.0)3 with C21-e6.0FL in buffer (concentration of monomer peptide).
5. Measure immediately and at 15 min intervals for up to 1 h, to allow for equilibrium.
6. Fractional fluorescence=(Fs±σs)/<Fc>, where<Fc>is the average value of the control fluorescence in odd wells, and Fs is the fluorescence of the sample. Large increase in control values with compound concentration indicates compound interacts with probe, for example, by micelle formation.
Results and Discussion
Peptide Design
The array of overlapping segments used to scan the full-length coiled coil is shown in Figure 1. NHR peptide env2.0 contained the residues 565LLQLTVWGIKQLQARIL581 of the HP, as previously described, 13 and env3.0 14 and env4.0 were extended to include a region either C-terminal or N-terminal to the pocket, respectively. Env5.0 and env5.1 extended the N-terminal region even further, 15 and env6.0 contained just the N-terminal end of the coiled coil, without the HP. This region binds to the N-terminal two-thirds of T20. The env# peptides were modified with an N-terminal 5-carboxybipyridine (bpy) group and a linker sequence GQAV. Three bidentate bpy groups formed an octahedral complex with transition metal ions, stabilizing the trimeric form of the env# receptors.

Helical NHR and CHR domains of the gp41 extracellular region, showing the location of env# constructs and assay C-peptide probes listed in Table 3. For simplicity, only one NHR and CHR helix is shown from the trimer. The model shown is from protein data bank structures 2X7R
22
(light gray) and 1IF3
39
(gray). Residues at i, i+4, and i+7 positions of the CHR are indicated in yellow (hydrophobic) or orange (polar). Env# peptides are bipyridylated and studied complexed to 1/3 stoichiometry Fe(II) (shown in purple and light purple for env6.0). C-peptides are labeled with a fluorophore at a C-terminal cysteine (fluorescein shown for C21-e6.0 in red). Env peptides contain the wild-type sequence, while C-peptides are mutated to increase helicity and affinity (see text). A known hydrophobic pocket binding small molecule,
Figure 1 also depicts CHR peptides that were synthesized, aligned antiparallel to the NHR peptides, and labeled according to the number of residues and the cognate receptor. C18-e2.0 covered the HP and N-terminal adjacent region. C28-e5.1 and C21-e6.0 mimicked portions of T20 and did not contain HP-binding residues. C32-e5.0 and C39-e5.1 spanned close to the full length CHR. A fluorescent marker, LY or FL, was attached to each CHR peptide probe through a C-terminal cysteine, so that NHR–CHR binding could be followed by fluorescence quenching. The sensitivity of this experiment depended on the alignment of the fluorophore and metal complex, and C-peptides were designed with the fluorophore <18 Å from the metal. It was found that C-peptides designed for the receptors env5.1 and env6.0 were also quenched by env5.0, increasing the versatility of assay pairing.
Sequences of the peptides are indicated in Table 3. The wild-type (HXB2) sequence was used for all NHR peptides, to maintain receptor integrity for inhibitor detection, but C-peptides contained several helix-stabilizing modifications on the solvent-exposed face, based on studies demonstrating their effect on enhancing affinity. 8,19,20 Several C-peptide sequence variations were tested, with the sequences shown providing a good combination of high helicity and enhanced solubility.
NHR (env#) and CHR (Cn-e#) Assay Peptides
# is a model number that refers to a particular construct of the NHR coiled coil; C-peptides are labeled by the number of residues n and a matching env#. C-peptides are fluorophore labeled at a C-terminal cysteine. Amino acids forming key interactions at a and d positions of the heptad repeat are indicated in bold. B is α-aminoisobutyric acid. T20 is included for reference, but is not an assay peptide.
aDetermined by CD, using molar residue ellipticity of −33,500 degree·cm−2·dmol−1 for 100% helix.
bIncrease in helix content for the complex of C-peptide with cognate env# compared to the sum of component spectra.
NHR, N-heptad repeat; CHR C-heptad repeat; n.d., not determined.
NHR Trimer Formation
Metal binding to bipyridylated NHR peptides (env#) generally resulted in stabilization of the coiled coil, evidenced by an increase in helical content to 50% or higher in CD spectra and a 208 nm:222 nM peak intensity ratio of 1.0, which indicated α-helix bundle formation. 21 An exception was the peptide env4.0, which did not give a well-structured helical coiled coil upon metal binding and subsequently performed poorly in fluorescence experiments. An apparent helical content >100% was observed for the peptide env5.1 both in the absence and presence of Fe2+. This was an indication of aggregation that was not ameliorated by metal binding, demonstrating a size limit of the self-assembly process in generating discreet trimers above a certain concentration. The CD results were confirmed by fluorescence studies (see below).
The receptor Fe(env6.0)3 defined the region of gp41 at the base of the coiled coil, immediately adjacent to the fusion peptide proximal region that interacts with lipid membrane. A recent crystal structure of an extended segment of the gp41 ectodomain 22 revealed a loss of helical periodicity in the N-terminal region that is included in Fe(env6.0)3 (Fig. 1). This did not appear to affect the formation of a well-formed trimer, although it may explain the lower helical content observed compared to other well-formed receptors. Fe(env6.0)3 is the segment of the gp41 coiled coil to which T20 binds, including the residues 544LLSGIVQQQ552 that are known to make important energetic interactions with T20. 16 Mutations within these residues are strongly associated with resistance to T20, 23 –25 and a mutation of the T20 residue N656→L that binds to this segment abrogates fusion completely. 26,27 This receptor target could be used to interrogate small molecule replacements of T20. It is also straightforward to modify the receptor for small molecule discovery to include common T20 resistance mutations.
CD Studies of Six-Helix Bundle Formation
A change in the CD signature for a mixture of receptor and C-peptide compared to the sum of individual spectra reflected complex formation. Example CD spectra are shown in Figure 2 for the peptide pairs Fe(env5.1)3/C39-e5.1, Fe(env5.1)3/C28-e5.1, and Fe(env6.0)3/C21-e6.0. An increase in helicity accompanied formation of the complex between cognate peptides Fe(env5.1)3/C39-e5.1 and Fe(env6.0)3/C21-e6.0, with the large negative molar residue ellipticity at both 222 and 208 nm indicative of six-helix bundle (6HB) formation. 21,28,29 Thus aggregation of Fe(env5.1)3 did not preclude C-peptide binding. In Figure 2B, association of a truncated peptide C28-e5.1 with Fe(env5.1)3 resulted in a distorted CD curve relative to that of the typical helix bundle. C28-e5.1 is missing the residues that bind in the HP, which is likely to lead to a combination of aggregation and an unstable six-helix structure. This effect has been observed in previous studies using T20 as the C-peptide. 17,30

Circular dichroism spectra for assay pairs using the long receptor Fe(env5.1)3
Fluorescence Studies of 6HB Formation
Dissociation constants (KD) between peptide pairs were determined by direct titration of the Fe(env#)3 receptor into a constant concentration of fluorescently labeled probe peptide. 13,15,29 Peptides were labeled with two different fluorescent probes, LY and FL. Figure 3 demonstrates fluorescence quenching for several receptor/peptide pairs, and the fit to an equilibrium 1:1 binding model. The fluorescence quenching experiment with Fe(env5.1)3 confirmed that it aggregated above 0.1 μM, with the concomitant loss in binding sites reflected in an increase in fluorescence at receptor concentrations exceeding 0.1 μM. The KD's for all assay pairs are reported in Table 4. The assay pair Fe(env3.0)3/C27-e3.0 (KD=1.2±0.2 μM) has been discussed elsewhere 14 and is not evaluated further here.
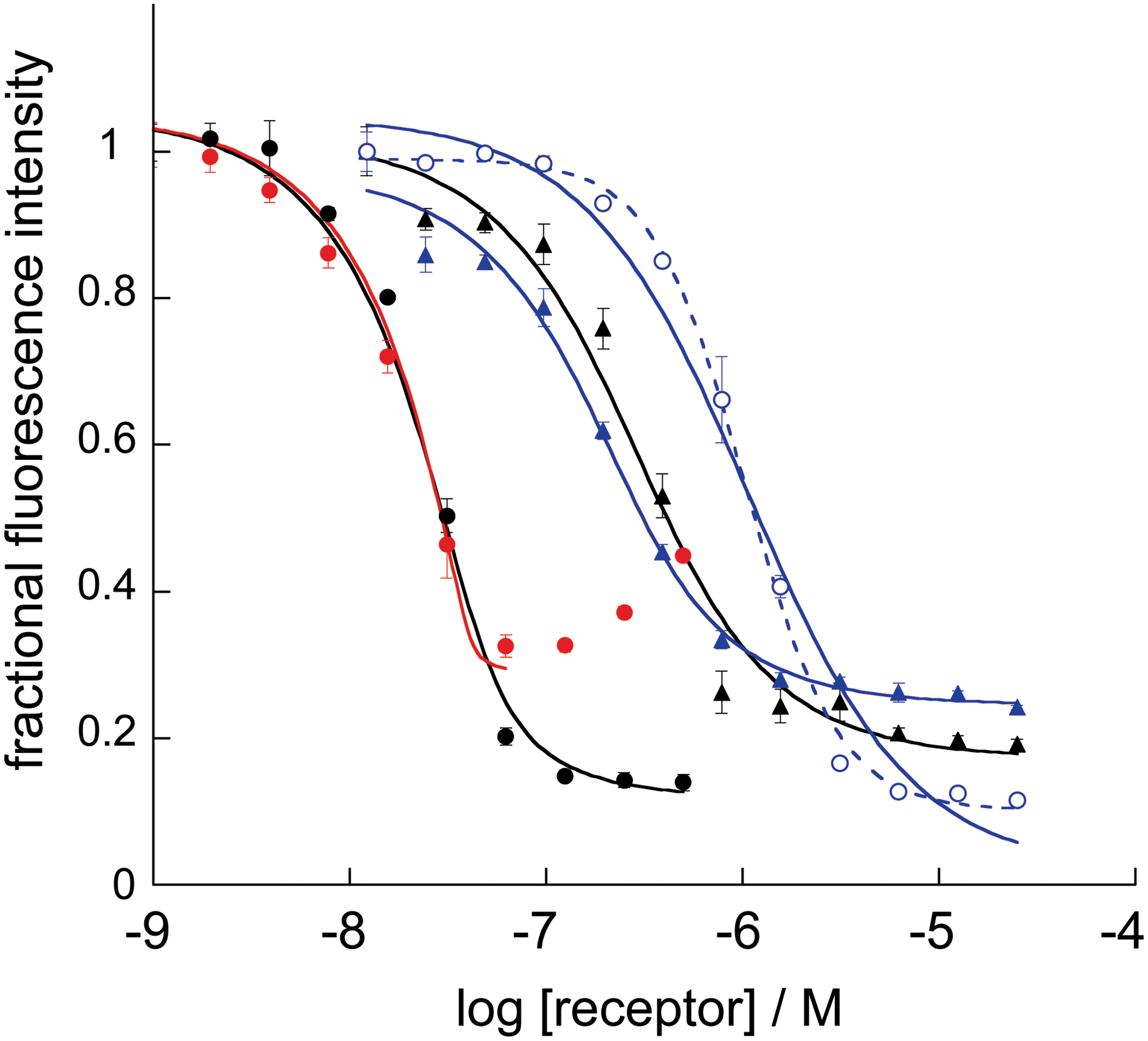
Fluorescence-detected binding curves for env#/Cn-e# pairs depicted in Figure 1. Receptors used were env5.0 (black), env5.1 (red, left), or env6.0 (blue, right) and C-peptides used were C32-e5.0FL or C39-e5.1FL (•), C28-e5.1LY (▴) or C21-e6.0LY (○). Solid lines indicate fitting to a 1:1 binding model; the dashed line is a fit to IC50. LY, lucifer yellow; FL, fluorescein. Color images available online at
Dissociation Constants (in μM) of Receptor–Probe Pairs Used in Fluorescence Assays
Receptor peptides are named env followed by a version number, probe peptides are labeled Cn followed by a version number. Experiments were repeated in quadruplicate and on at least two different days for each assay pair. The standard deviation is shown. Empty cells correspond to noncompatible pairs.
aEstimated from the IC50 (see text).
n, number of residues.
Inhibition constants were determined for unlabeled peptides, with the expectation that the observed KI for a peptide inhibitor should equal the KD of that peptide as a probe, barring any interference from the fluorescent group. This was found to be the case with one exception (see below). KI's are reported in Table 5. Fe(env5.1)3 was not generally used as a receptor in inhibition experiments due to aggregation at the required concentrations. In the typical experiment, a probe and an inhibitor were premixed before adding a receptor, although in moderate affinity assays (KD ∼ 1 μM), rapid equilibrium was also achieved by adding the inhibitor to the premixed receptor and probe. KD and KI data enabled us to determine the energetic contribution of various subsites to 6HB formation, stability, and inhibition. KD, or KI in self-inhibition, is expected to reflect both the enthalpic contribution to binding energy from the length and characteristics of the helix–helix interface and the entropic penalty associated with C-peptide helical content changes (Table 3). 14 KI in heterologous assays, on the other hand, could provide information about the contribution of a subsite in stabilizing the longer ectodomain.
Inhibition Constants (in μM) for C-Peptide and Small Molecule Inhibitors Using Several Assay Pairs
Inhibition constants obtained by competitive inhibition using the indicated metal-chelated NHR peptide Env# and fluorescently labeled CHR-peptide probe. In brackets is the standard deviation. Except where indicated, both probes gave the same KI. Empty cells indicate no or poor overlap of the peptide inhibitor with an assay probe.
aNonspecific binding, IC50 in italics, sharp rise (Hill coefficient >2.5).
The HP Plus About Five Residues Immediately Upstream was the Only Significant Hotspot Mediating the Helix–Helix Interaction
C32-e5.0 had 50-fold increased affinity over C28-e5.1 for Fe(env5.0)3, highlighting the importance of the HP-binding region, in agreement with previous binding and mutagenesis studies.
31
–33
C21-e6.0, directed to the region of the NHR that readily mutates upon exposure of the intact virus to T20, had 10-fold reduced affinity compared to C28-e5.1, indicating that important interactions with the NHR involved the segment 637N
Inhibition data in heterologous assays confirmed the expanded HP interaction with C18-e2.0 as the most significant hotspot mediating 6HB formation. Thus, C18-e2.0 was able to inhibit the much longer Fe(env5.0)3–C32-e5.0FL interface at about the same KI as for self-inhibition, while peptides corresponding to other segments (C28-e5.1, C21-e6.0) had significantly reduced ability to compete with C32-e5.0FL compared to inhibition of self.
Interactions Between NHR and CHR at the Base of the Coiled Coil were Energetically Significant, but Could not Inhibit 6HB Formation
The peptide pair Fe(env6.0)3 and C21-e6.0 representing the base of the 6HB formed a complex with a 1 μM KD/KI, a reasonably strong association, involving both hydrophobic and polar interactions (Fig. 1). The NHR segment 540QARQLL545 provided for a two-fold increase in binding potency with peptides C21-e6.0LY or C28-e5.1LY, as could be seen by comparing KD's obtained with Fe(env6.0)3 or Fe(env5.1)3 with those obtained with Fe(env5.0)3. The viral fusion-inactivating N656L mutation on the CHR 26 reduced affinity of C21-e6.0 by a factor of 7 for receptor Fe(env6.0)3, 7.6 for receptor Fe(env5.1)3 (data not shown), and 25 for receptor Fe(env5.0)3. Differences in the effect of the mutation appeared to be due to compensatory interactions of C21-e6.0 with 540QARQLL545.
The reduction of potency of C21-e6.0 in heterologous assays was particularly striking, since it was unable to displace either C32-e5.0FL or C28-e5.1FL/LY from Fe(env5.0)3 at concentrations up to 200 μM. It displaced C28-e5.1FL/LY from Fe(env6.0)3 with six-fold reduced KI compared to inhibition of self, and was unable to displace C21-e6.0FL (Fig. 4). There is an overwhelming evidence from structural and mutagenesis studies that the NHR–CHR interaction spanned by Fe(env6.0)3–C21-e6.0 forms part of the 6HB core, and plays a critical role in gp41 function. 22,27 Thus, while there is clear evidence for NHR–CHR stabilizing interactions in this region, our data and other studies 36 suggest they do not appear to play a central role in disrupting or preventing 6HB formation.
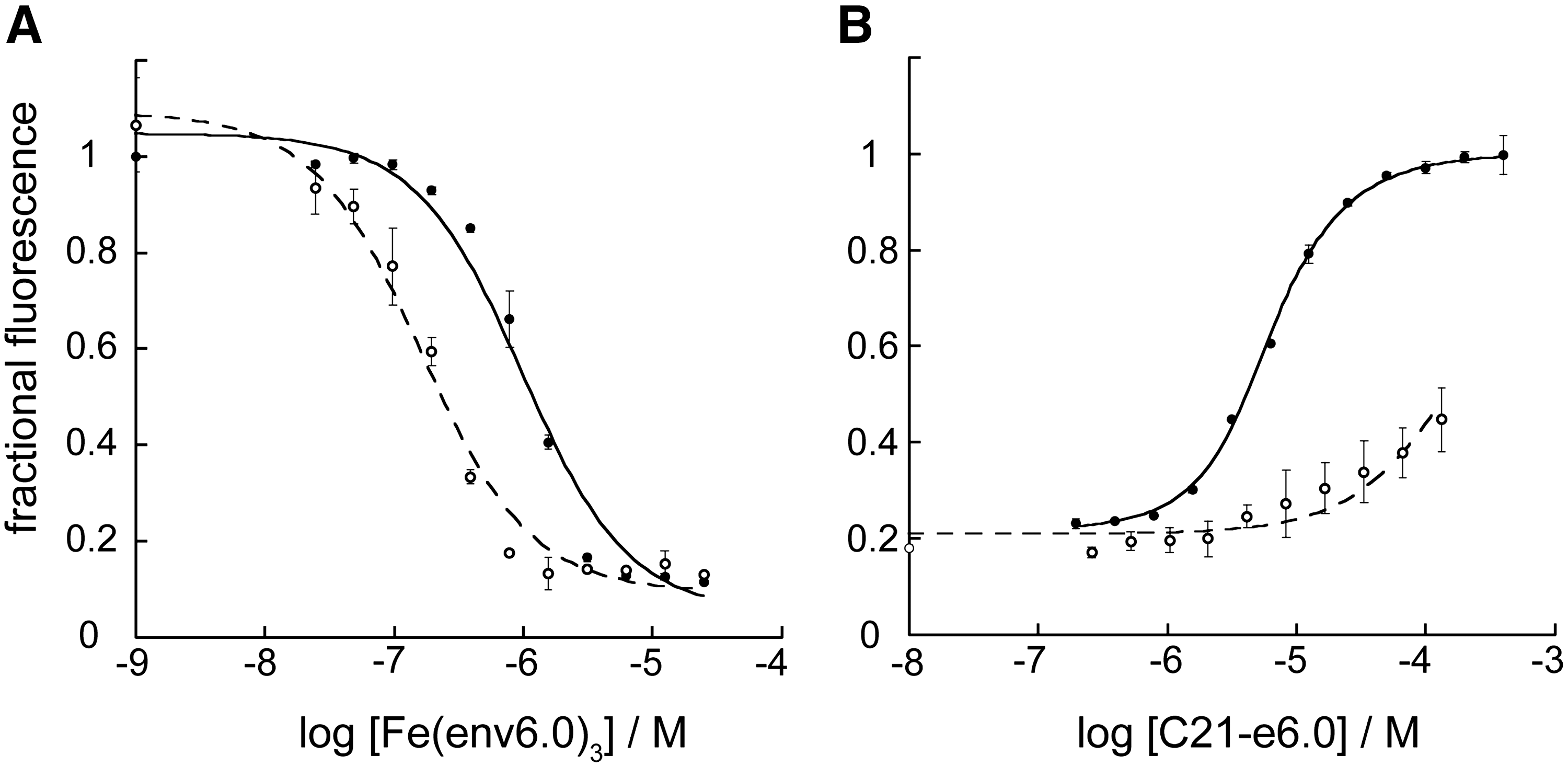
Dissociation and inhibition constant determination for the assay pair Fe(env6.0)3/C21-e6.0.
The Base of the Coiled Coil Showed Conformational Variability and Flexibility
Effects including nonspecific hydrophobic interactions and deviations from 1:1 binding in constructs containing residues at the base of the coiled coil were suggestive of conformational flexibility in this region. The segment 540QARQLL545 was implicated in nonspecific binding of the hydrophobic C-terminal FL group, which interfered in assays with Fe(env5.1)3 or Fe(env6.0)3, resulting in an observed KD of FL-labeled peptides three-fold lower than the KD for the corresponding LY-labeled peptides (Fig. 4A; Table 4) or the KI of unlabeled peptides (Table 5). No association with free FL was observed. It was further found that unlabeled C21-e6.0 could not displace C21-e6.0FL from Fe(env6.0)3, while readily displacing C21-e6.0LY (Fig. 4B), or either probe peptide from the receptor Fe(env5.0)3. Interestingly, FL-labeling resulted in a KD closer to the 100 nM KI observed for T20 binding to Fe(env6.0)3. T20 showed mid-nM inhibition in all assays except Fe(env2.0)3–C18FL, which was to be expected, since T20 does not contain HP-binding residues. The hydrophobic tail of T20 binds to the fusion-peptide proximal region, which is not included in env6.0. Therefore some nonspecific hydrophobic effect was occurring with T20 as well.
The nonspecific binding resulted in deviations from normal 1:1 binding with C21-e6.0FL and to a lesser extent C21-e6.0LY. Binding curves displayed sharp transitions with a Hill coefficient >1.5 (e.g., Fig. 4A). The fluorescent label appeared to be a principle factor in inducing the effect, since unambiguous 1:1 binding was observed in the inhibition data for C21-e6.0 (Fig. 4B). The effect was observed only for the 21-residue peptide, where nonspecific interactions could likely contribute a significant proportion of the affinity. Some type of distortion is necessary to explain the fluorophore interaction, since it was designed to point away from the NHR–CHR interface in a purported six-helix structure. The observed cooperative binding transition may be associated with conformational variation from the expected six-helix structure. A recent crystal structure of full length ectodomain, including the membrane proximal external region (MPER), indicated that splaying and loss of the NHR helical structure occurred in this region. 22 It is possible that FL could partially mimic the MPER in inducing a structural change.
High-Affinity Small Molecule Binding was Specific to the HP and Suggested a Dependence of Potency on Off-Rate
Assay performance was evaluated for potency and specificity with several confirmed low to sub-μM HP-binding inhibitors.
18
Three of these,
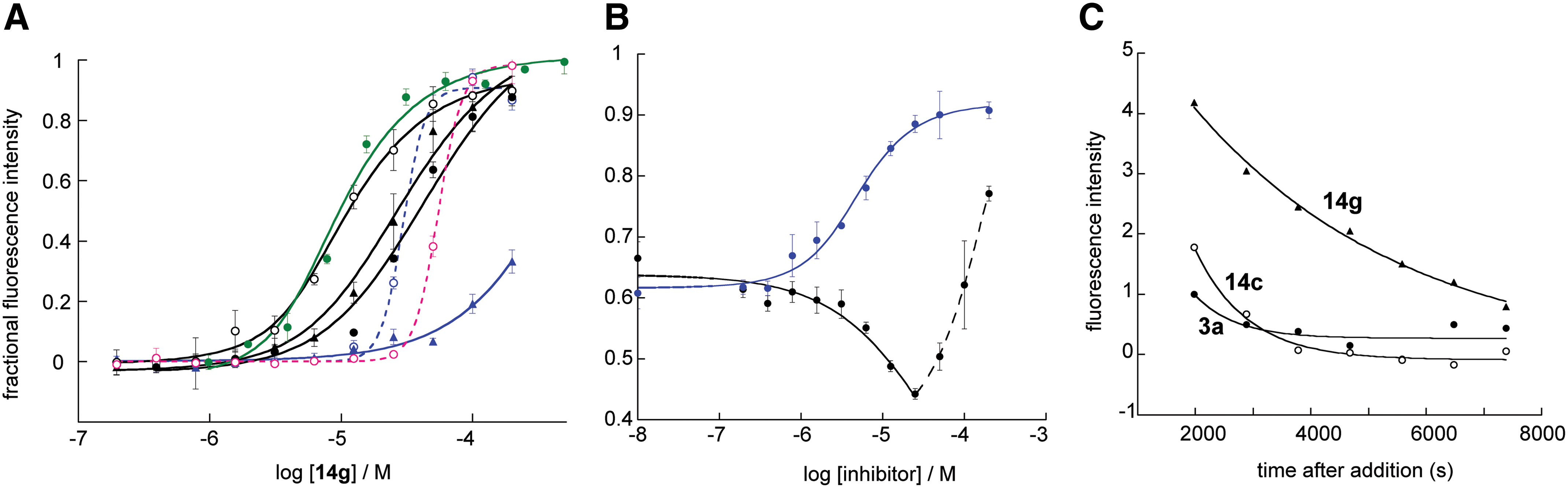
The apparent inhibition by
Having ruled out multiple-site binding for
Conclusions
This study demonstrated a suite of assays for probing binding and segmental activity of the coiled coil domain of HIV-1 gp41. The assays encompass HP or T20 subsite detection, as well as full-length 6HB coverage. Dissociation and inhibition constants for peptides and small molecules accurately reflected both affinity and specificity provided the CHR probe peptide had a complete cognate-binding site on the Fe(env#)3 receptor. The assay Fe(env6.0)3/C21-e6.0LY, simulating the T20-binding site where resistance mutations are common, is the newest addition to our repertoire for examining gp41. The 1 μM KD indicates that it can be used to detect inhibitors in the low μM to mM range that have a unique binding site apart from the HP. Experiments here demonstrate that use of the probe peptide in its LY-labeled form will be preferential over FL-labeling.
Importantly, many of the biophysical results that were obtained matched very well with observed biological data. The assays revealed that the HP was the sole hotspot mediating the NHR–CHR interaction, conferring significant stability to the 6HB. The first five residues of T20, which are critical for antiviral activity, were also necessary for high-affinity binding to the coiled coil. Peptides containing a known inactivating mutation N656L displayed a dramatic loss of potency. Evidence for a break in the rigid helical structure toward the base of the coiled coil was obtained, both from the inability of a C-peptide from this region to disrupt 6HB formation and from indication of conformational flexibility upon probe binding. The assay representing the 6HB segment at the base of the coiled coil showed unique features compared to the other assays, including interference of the FL group in binding, resulting in a cooperative binding transition, inability of the unlabeled peptide to compete with its FL-labeled counterpart, and nonspecific binding of hydrophobic compounds. The overall agreement between known biological properties and observed binding properties of the C-peptides confirmed the accuracy of the metallopeptide receptor models in representing the intact gp41 NHR trimer.
Evaluation of a sub-μM small molecule fusion inhibitor revealed that it was able to effectively inhibit the high-affinity interaction between a 40-residue NHR construct and a 32-residue CHR peptide. Small molecules that were not able to inhibit this interaction in the low to sub-μM range were found to have ≥10-fold weaker activity against cell–cell fusion, even if they had similar ability to disrupt an 18-residue CHR peptide binding in the HP. This property is demonstrated here with just three inhibitors, but recent results (unpublished) have indicated that it applies more broadly and may be a unique identifying property of effective low molecular weight fusion inhibitors. The mechanism of extended NHR–CHR inhibition appears to be associated with lower off-rate of the small molecule ligand, with binding specificity restricted to the HP. The assay suite could be used to aid in the discovery of alternative ligands, which bind adjacent to the pocket and which could be tethered to HP-binding inhibitors, leading to a significant increase in potency.
Footnotes
Acknowledgments
This work was supported by NIH grants NS066469 and GM087998. I thank Dr. Richard Shafer, UCSF, for use of the CD machine and Dr. G. Zhou for providing the low molecular weight compounds tested in the assays. Molecular graphics images were produced using Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the NIH (National Center for Research Resources grant 2P41RR001081, NIGMS 9P41GM103311).
Disclosure Statement
No competing financial interests exist.