
Abstract
Gap junctions (GJs) are intercellular channels which are composed of the connexin family of proteins that allow electrical and chemical communications and synchronization in tissue ensembles. Evidence suggests that pharmaceutical modulators of these channels may have therapeutic potential or carry undesired liability. In this report, we exogenously expressed human connexin 43 (Cx43, GJA1) and demonstrated functionality in a 96-well flow cytometry assay detecting intercellular transfer of the calcein dye. We have designed a 384-well high-throughput method for detecting the transfer of calcium between HeLa cells expressing Cx43. In this assay, donor cells coexpress Cx43 and the α1A adrenergic Gα-coupled receptor, while recipient cells coexpress Cx43 and the cytoplasmic version of the calcium-sensitive luminescent protein aequorin enhanced by codon optimization (cytoAeq). The two cell populations were mixed, dispensed to 384-well plates, and incubated for 3 h to allow the formation of GJs. Activation of α1A by epinephrine in donor cells led to dose-dependent calcium increases in recipient cells, which were detected by measuring the intensity of aequorin luminescence. The response was dependent on the expression of Cx43 and inhibited by the GJ blocker 18α-glycyrrhetinic acid, suggesting Cx43 GJ-mediated activity. In a parallel experiment with capsaicin and the TrpV1 ion channel in place of phenylephrine and α1A, a similar magnitude of difference in the maximal calcium response was detected in both donor and recipient cells, suggesting that calcium is likely the permeant ion through the GJ. This assay may pave the way for high-throughput screening of GJ modulators for drug discovery.
Introduction
Of the 21 connexins found in humans, connexin 43 (Cx43, GJA1) is widely expressed in many cell types, and is the primary GJ-forming protein in the ventricular myocardium. Autosomal dominant mutations in Cx43 exhibit developmental disorders, resulting in a disease termed oculodentodigital dysplasia, while disruption of the gene causes lethal cardiac arrhythmias. 3 –5 Heterogeneities in Cx43 expression across the ventricular wall contribute to the development of re-entrant arrhythmias in failing hearts. 6,7 Other connexins are also implicated in disease. For example, mutations in Cx26 and Cx30 result in Keratitis–ichthyosis–deafness, a disease that is characterized by progressive corneal opacification, either mild generalized hyperkeratosis or discrete erythematous plaques, and neurosensory deafness. 8 Mutations in Cx32 cause X-linked Charcot-Marie-Tooth disease, which is characterized as motor and sensory neuropathy. 9
Multiple lines of evidence suggest that connexins could be targeted pharmacologically for treating many diseases. 10,11 For example, antiarrhythmic peptides, such as AAP10, have been shown to increase GJ conductance in cardiomyocytes and heterologously expressed Cx43 and Cx45 but not Cx40. 12 An orally available nonpeptide analogue of AAP10, gap-134, improves conduction and reduces atrial fibrillation/flutter in the canine sterile pericarditis model, 13 and is used in clinical trials for atrial fibrillation. 14 A synthetic peptide, Gap26, which mimics the first intracellular loop of Cx43, specifically blocks the Cx43 hemichannel activity, and protects the intact heart against ischemic injury. 15 The GJ inhibitor tonabersat is efficacious in cortical spreading depression, and may have potential value in migraine prophylaxis. 16 The arousal inducing effects of the drug modafinil were blocked by GJ antagonists in the rat, suggesting the drug's action may be by increasing electrical coupling in neurones. 17 More recently, mice deficient in Cx32 were protected against liver damage, acute inflammation, and death caused by liver-toxic drugs. The small-molecule inhibitor of Cx32, 2-aminoethoxydipenyl-borate, protected against liver failure and death in wild-type mice when coadministered with known hepatotoxic drugs. 18
Despite their well-recognized therapeutic potential and cardiac/neuronal safety liabilities, large-scale screening for GJ modulators remains largely uncharted territory, primarily due to the lack of high-throughput assay technologies. Although it might be simpler to assay hemichannel activity, the result is likely not predictive of GJs. A number of techniques have been used to study GJs by measuring signals passing between two neighboring cells. Most experiments measure the transfer of dyes, and a few quantify the conductance of naturally occurring second messengers and ions. For example, donor cells can be injected with a dye or caged cAMP, and the transfer to the recipient cells can be detected under the microscope. 19,20 A similar experiment can be performed to measure the flow of Ca2+ from donor to recipient cells that are injected with a Ca2+ sensing dye such as fura-2. 21 These assays are generally difficult to perform and of very low throughput. The only higher throughput assay reported to date was by Li et al., 22 which measured the flow of the negatively charged dye calcein from donor to recipient cells by high content imaging. More recently, Chen and Lee 23 and Ye et al. 24 described promising microfluidic chip technologies that are capable of selective focal loading of dyes and time-dependent quantification of GJ-dependant dye transfer. There has been no report of a high-throughput assay that measures the intercellular transfer of native components in the cell, such as Ca2+, inositol-1,4,5-triphosphate (IP3), or cyclic nucleotides.
Here, we report the development of an assay which employs codon-optimized apoaequorin expressed in recipient cells that detects the conductance of elevated intracellular Ca2+ from donor cells in a 384-well format. The assay could be potentially used to identify GJ modulators through high-throughput screening.
Materials and Methods
All reagents were from Invitrogen unless otherwise stated. Digitonin, uridine-5′-triphosphate (UTP), phenylephrine, capsaicin, 18-α-glycyrrhetinic acid, and Triton X-100 were from Sigma.
Recombinant Expression
The human Cx43 cDNA was amplified from a human brain marathon library, and the sequence is identical to NCBI reference NM_000165.3. The predicted peptide sequence of mitochondria-targeted apoaequrin was described by Stables et al., 25 with codon usage further optimized with GeneOptimizer and synthesized by GeneArt, achieving codon adaptation index of 0.99. Untagged apoaequorin coding sequence was amplified from this construct by a polymerase chain reaction. The predicted peptide sequences of human α1A adrenergic receptor and TrpV1 ion channel were identical to NCBI references NP_000671.2 and NP_542435.2, respectively. These cDNA fragments were placed under the control of the cytomegalovirus promoter in pFastBacMam-1 shuttle vector, transfected into SF9 insect cells, and BacMam viruses were generated according to Condreay et al. 26
Cell Maintenance
Cells were maintained in DMEM HAM's F12 medium supplemented with glutamax and 10% fetal bovine serum, at 37°C with 5% CO2, dissociated with TrypLE, and split 1/5 to 1/10 for propagation. BacMam viruses or control insect cell medium were added 24 h before the start of the experiment.
Immunocytochemistry
HeLa cells maintained in 96-well poly-D-lysine-treated microtiter plates (Becton Dickinson) were fixed with 300 μL of 4% paraformaldehyde made up in phosphate-buffered saline (PBS) for 15 min at room temperature. The fixative was removed, and the cells were made permeable by washing 3× with 200 μL of PBS/0.1% Triton X-100 for 10 min. The cells were blocked for 2 h at room temperature with 10% normal goat serum (Dako) in PBS containing 0.1% Triton X-100 and then incubated with 150 μL of a 1:400 dilution of anti-Cx43 antibody (Sigma) for 60 min at room temperature. The antibody solution was removed, and the cells were washed 3× over a 10 min period with 200 μL of PBS/0.1% Triton X-100. The cells were then incubated with a goat anti-rabbit AlexaFluor 488 (Invitrogen) antibody diluted 1:500 in 0.1% goat serum/PBS/0.1% Triton X-100 for 60 min at room temperature. The antibody solution was removed, and the cells were washed thrice with 200 μL of PBS/0.1% Triton X-100 over a 10 min period. 100 μL of PBS containing a 1:10,000 dilution of Hoechst (Invitrogen) solution was added and incubated for 10 min at room temperature. The staining solution was removed, a drop of Slowfade (Invitrogen) anti-quenching agent was dispensed into each well, and the fluorescence was examined on a microscope using a 40× objective.
Cx43 GJ Flow Cytometry Assay
U2OS osteosarcoma cells were pretransduced with the respective BamMam viruses for 24 h and harvested by TrypLE. Donor and recipient cells were diluted in PBS buffer and loaded with 1 μM calcein-AM or 1/200 dilution of CellVue (Sigma), respectively, for 20 min. Cells were then washed, resuspended in cell maintenance medium, mixed at a 1:1 ratio, and dispensed to 96-well flat bottom plates at 200,000 cells per well. After a 2 h incubation, cells were dissociated from the bottom of the plate by TrypLE, resuspended as single-cell populations in a cell maintenance medium, and profiled on an FC500 flow cytometer (Beckman Coulter) using laser excitations of 488 and 630 nm. The donor and recipient cells were identified by plotting the fluorescence emitted from two channels, 525 nm versus 675 nm (see Fig. 1C, D). The level of calcein present in the recipient cells was measured by the fluorescence at 510 nm.

Cx43 GJ Aequorin Assay
HeLa cells in culture were harvested by TrypLE and separated into donor and recipient cells. Respective BacMam viruses were added and left to incubate for 24 h, after which cells were harvested, resuspended in growth medium, and counted. The two cell populations were mixed at a 1:1 ratio and dispensed using a Multidrop Combi (Thermo) at 20,000 cell/well in black 384-well clear bottom plates (Greiner). Assay plates were incubated for 3 h under normal culture conditions to allow cells to adhere and form GJs. After incubation, the cells were washed and loaded for 3 h with 5 μM coelenterazine in HBSS (Sigma) with 0.1% Pluronic F68 and 1 mg/mL bovine serum albumin, pH 7.4, alongside any GJ blockers or dimethylsulfoxide control. The plates were subsequently transferred to a Lumilux platform (Perkin Elmer) for the online addition of receptor agonist and the detection of luminescent signal (Table 1). Data reported are integrated luminescence area under curve for a duration of 40 s after agonist addition. Dose-response curves were fitted using nonlinear regression with GraphPad Prism software.
Steps of the Connexin 43 Gap Junction Aequorin Assay
Step Notes
1. Donor: HeLa cells+Cx43 & α1A (or TrpV1); Recipient: HeLa+Cx43 & cytoAeq.
2. 20,000 cells in 50 μL per well in 384 well plate, in normal growth medium.
3. Aspirate off remaining medium/buffer.
4. HBSS+0.1% Pluronic F68, 1 mg/mL bovine serum albumin & 5 μM coelenterazine.
5. To 1 μL compound in dimethylsulfoxide, add 40 μL assay buffer, final dilution 1/120.
6. Coelenterazine loading+compound treatment, keep in the dark.
7. Agonists: phenylephrine for α1A; capsaicin for TrpV1.
8. Conducted by Lumilux.
cytoAeq, codon-optimized cytoplasm-targeted apoaequorin; PBS, phosphate-buffered saline.
Results
Exogenous Expression of Cx43 and Functional Validation by Flow Cytometry Assay
Cx43-expressing BacMam viruses were produced for this study. Cx43 protein expression was demonstrated by immunofluorescence using HeLa cells transduced with the virus. Highly fluorescent aggregates, similar to GJ plaques which are thought to be composed of approximately thousands of individual channels, 27 were revealed (Fig. 1A, B).
Functionality of Cx43 GJs was assessed by a 96-well flow cytometry assay. In this assay, both donor and recipient U2OS cells were transduced with Cx43 BacMam. Recipient cells were loaded with the red fluorescent dye CellVue, which binds lipid regions of the membrane and cannot be transferred through GJs. Donor cells were loaded with calcein-AM, which is not fluorescent but on entry to the cell, the acetomethoxy group is removed by esterases, leaving the highly fluorescent and hydrophilic free calcein molecules trapped in the cell. The dye can spread into neighboring cells through GJs 22 After coincubation of the two cell populations for 2 h in a 96-well plate, they were dissociated from the plate into individual cells, and analyzed by flow cytometry. The recipient cells were identified by the presence of CellVue red fluorescence emission at 675 nm (excited at 633 nm). The amount of a calcein green fluorescence emission at 525 nm (excited at 488 nm) in the recipient cells reflected GJ activity. Using cells transduced with Cx43 BacMam, a high amount of green fluorescence, with a median intensity of 33.5, was detected in CellVue-possitive recipient cells (Fig. 1C). This was significantly higher than the low basal median intensity of 0.78 from control experiments with un-transduced cells (Fig. 1D) or cells transduced with the same amount of BacMam expressing different targets (data not shown). Moreover, the transfer of calcein in Cx43-transduced cells was blocked by the GJ inhibitor 18-α-glycyrrhetinic acid (18αGA, data not shown). These results suggest that Cx43 forms active GJs in these cells.
Construction of Codon-Optimized Apoaequorin
We next looked at the possibility of configuring a high-throughput assay for screening Cx43 GJ modulators. A robust GJ assay requires the detection of minute changes in signals in the recipient cells, free from the influence of much larger signals of the donor cells. A good candidate is the apoaequorin protein from the jellyfish Aequoria victoria, which forms aequorin when bound to the cofactor coelenterazine. In response to Ca2+-binding, it catalyzes the oxidation of coelenterazine, emitting a luminescent light with a peak of 369 nm. Its ability to sense Ca2+ in a highly dynamic range and absolute dependence on exogenous expression of apoaequorin in mammalian cells for activity make it an ideal candidate for this purpose. 28 We envisaged that codon optimization may be required, 29 and cytoplasmic-targeting would be ideal to detect small Ca2+ movements in areas adjacent to the GJs.
A cDNA was, therefore, synthesized for wild-type apoaequorin, which is known to be expressed in the cytoplasm, 30 with optimized codon usage for mammalian expression (codon-optimized cytoplasm-targeted apoaequorin [cytoAeq]). Transduction with cytoAeq BacMam led to about 7, 12, 6, and 16 times higher luminescence than mitochondria-targeted aequorin (codon-optimized mitochondrium-targeted apoaequorin [mitoAeq]) in CHO, HEK293, U2OS, and HeLa cells, respectively (Fig. 2) after the addition of 100 μM digitonin, which causes Ca2+ entry into the cytoplasm by forming pores on the cytoplasmic membrane. The addition of the histone deacetylase inhibitor sodium butyrate significantly increased the response further (Fig. 2D and data not shown). The difference in luminescence was also highly significant when the cells were stimulated with G protein-coupled receptor agonists such as UTP and carbachol (Fig. 2A–C and data not shown). Expression of mitoAeq was previously shown to lead to significantly higher activities than the wild-type cytoplasmic aequorin without codon optimization. 25 Vernon and Printen 29 have reported that codon optimization increased aequorin activity by more than 20-fold. The level of response for cytoAeq over mitoAeq was, however, unlikely due to codon preference, as mitoAeq was also codon optimized and replacing with the sequence from cytoAeq led to little improvement (data not shown).

Comparison of the sensitivity of cytoAeq (cyto) and mitoAeq (mito) to cellular calcium on the Lumilux instrument. Cells were preincubated with cytoAeq or mitoAeq BacMam with or without sodium butyrate (NaB) for 24 h. Luminescence was measured after the addition of 100 μM digitonin, 10 μM UTP, or DMSO in : 10 μM UTP; ■: DMSO control. cytoAeq, codon-optimized cytoplasm-targeted apoaequorin; DMSO, dimethylsulfoxide; mitoAeq, codon-optimized mitochondrion-targeted apoaequorin; UTP, uridine-5′-triphosphate.
Development of a 384-Well Assay Measuring GJ-Mediated Transfer of α1A Receptor-Stimulated Ca2+ Increase by cytoAeq
We next assessed the feasibility of using cytoAeq to detect Cx43 GJ-mediated Ca2+ transfer between cells. The outline of the experimental design is illustrated in Figure 3. We selected the α1A adrenergic receptor due to the clean background in HeLa cells: cytoAeq Ca2+ response to phenylephrine was almost entirely dependent on receptor expression by BacMam (Fig. 4A). In this assay, both donor and recipient cells were transduced with Cx43 BacMam. Donor and recipient cells were also transduced with α1A or cytoAeq BacMam, respectively. The two cell types were mixed and dispensed into 384-well plates, along with coelenterazine, to allow GJ formation. On-line addition of phenylephrine elicited a Ca2+ response in the recipient cells detected by cytoAeq in a dose-dependent manner. The response was dependent on the exogenous expression of both Cx43 and α1A, and blocked by the GJ inhibitor 18αGA with IC50 of approximately 17 μM, while the response in the recipient cells elicited by activating the endogenous G-protein coupled receptor by UTP was insensitive to the inhibitor (Fig. 4B–D). Only a relatively small response to phenylephrine was observed with cells not transduced with Cx43, possibly due to low level endogenous GJ activities (Fig. 4B). These experiments suggest that the Ca2+ increase in the recipient cells was due to propagations through Cx43. GJ's signal:noise ratio was routinely 9–11 and Z′ was >0.4 for this assay.
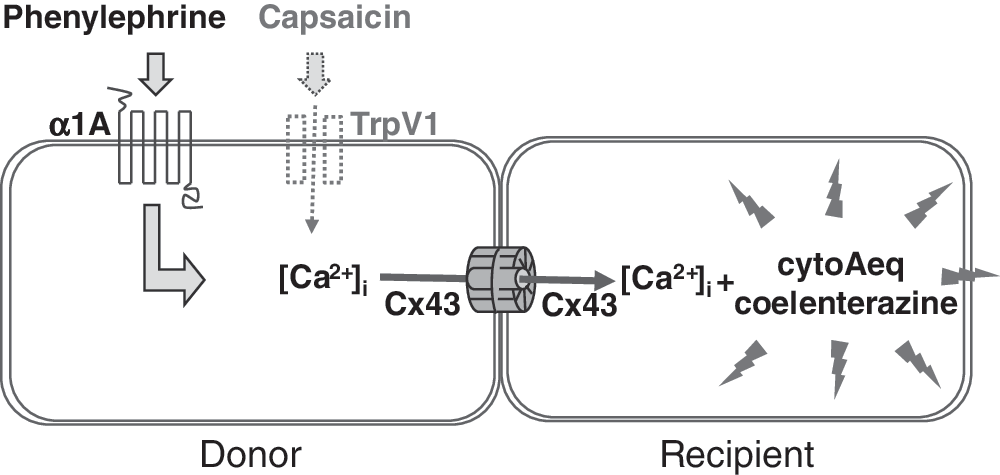
Diagram illustrating assay design. The donor cell coexpresses Cx43 and α1A or TrpV1, while the recipient cell coexpresses Cx43 and cytoAeq and was loaded with coelenterazine. Mixing the two populations allows GJs to form. The addition of epinephrine or capsaicin activates α1A or TrpV1 in the donor, leading to an increase in intracellular free calcium, which is spread into the recipient through the Cx43 GJs. Calcium in the recipient cell is detected by measuring the intensity of aequorin luminescence.

Detection of GJ-Mediated Transfer of TrpV1-Stimulated Calcium Response by cytoAeq
Activation of α1A adrenergic receptor causes elevation of the second messenger IP3 through stimulation of Gαq, which, in turn, elicits Ca2+ release from stores. 31 In order to assess whether Ca2+ or IP3 is passed through the Cx43 GJs, an alternative experiment was conducted using the TrpV1 ion channel, whose activation by capsaicin causes an influx of Ca2+ directly into the cytoplasm from the external medium (Fig. 3). The assay was conducted in an identical manner to that just described for α1A, except that donor cells were transduced with TrpV1 BacMam rather than α1A, and activated by capsaicin in place of phenylephrine. The capsaicin cytoAeq response was largely absent in un-transduced cells, but became pronounced in cells that express TrpV1, with a maximal response about three to four times higher than with phenylephrine for α1A (Fig. 5A). In the GJ transfer experiment, very high levels of 18αGA-sensitive cytoAeq luminescence were detected in the recipient cells after the addition of capsaicin (Fig. 5B). The maximal level of the response was also several times higher than with phenylephrine for α1A. Since the aequorin luminescence is reflective of the amount of intracellular free Ca2+, these results show that the amount of Ca2+ in the recipient cells is proportional to that in the donor cells, irrespective of the stimuli and the mechanism of Ca2+ entry. The most likely explanation for this observation is the direct transfer of Ca2+ ions rather than some intermediate factors such as IP3 into the neighboring cells through Cx43 GJs.

Discussion
Drugs affecting GJ activity are well recognized as having multiple therapeutic applications. Other compounds affecting a number of these targets may carry liability to drug development due to unwanted side-effects. To date, connexins as a target class are very much untapped in modern drug discovery largely due to the difficulties in designing robust assays for high-throughput screening of huge compound libraries. This type of assay requires highly specific reporters that have “zero” background in mammalian cells, as the chemical signals that get transferred to the recipient cells are likely much smaller than the donor, and are often overwhelmed by the background readouts in the donor cells. To our knowledge, before this article, high-throughput screening assays were not described for the connexin proteins that detect the intercellular transfer of naturally occurring cellular substances. Although it is possible to detect the transfer of dyes by alternative methods, by specifically identifying the recipient cells either by imaging 22 or by flow cytometry described in this article, they do not detect the transfer of native biological signals and require more complex equipment and analysis with compromised throughput.
Aequorin is perhaps ideal for this purpose. Compared with many of the traditional assays, it is not only highly specific for Ca2+ and noncytotoxic, but also easy to operate, associated with a very low cost, and often adaptable to a 1,536-well format. 28 While searching for a suitable reporter for a GJ assay, we have explored a number of versions of apoaequorin, the most suitable being the codon-optimized and gene-synthesized apoaequorin that is known to be targeted to the cytoplasm 30 The magnitude of increase in aequorin activity on codon optimization was in agreement with Vernon and Printen. 29 The difference between cytoAeq and mitoAeq was, nevertheless, surprising, suggesting that subcellular localization is also an important factor. Further experiments should be carried out to understand these differences and to assess their utilities beyond this article, for example, G protein-coupled receptors and ion channels. An alternative strategy has been explored by George et al., 32 who tagged apoaequorin to the connexin protein itself. In theory, it could be much more sensitive in picking up the Ca2+ at the site of action. However, these constructs may alter the maturation, trafficking, and assembly of the connexin protein. We also envisage that the aequorin response would be very small, as shown in their report. First, it is limited by the number of GJs, as the bulk of the unsuccessfully assembled connexin proteins are degraded after synthesis. 27 Second, the light-emitting reaction is irreversible, leaving the majority of the available enzymes fused to the connexins in the ground state due to cytosolic Ca2+ sparks during loading of the cofactor before the addition of agonists.
The assay described here can clearly detect an increase in cytoplasmic Ca2+ concentration in recipient cells immediately after activation of the Gαq-mediated α1A 7-transmembrane receptor and the TrpV1 ion channel in the donor cells. An interesting question is whether IP3 or Ca2+ or both are passed through the GJs. The magnitude of Ca2+ signal detected in the recipient cells was similar to that in the donor cells, irrespective of the mechanism of Ca2+ entry, suggesting that Ca2+ ions directly cross Cx43 GJs. It is interesting to note that the fold differences of maximal Ca2+ signal between capsaicin-TrpV1 and phenylephrine-α1A were broadly similar but not directly proportional—∼4–5-fold in recipient cells and ∼3–4-fold in donor cells (Figs. 4 and 5). This may be the result of differential Ca2+ uptake into intracellular stores at high and low concentrations, influenced by the slight time delay of Ca2+ traveling through the GJs. The slightly different luminescence reading time resultant from the time delay may also contribute to this difference.
Our data suggest that TrpV1 may be preferable to α1A as the Ca2+ stimulant, providing higher signal amplitude and signal-to-noise ratios and channeling Ca2+ directly into the cell. Further experiments should be conducted to ensure compound potency is not compromised and to compare the performance of the two assays in a screening environment.
Another interesting observation is the approximately 100-fold lower potency of both phenylephrine and capsaicin in the GJ assay than the receptor assay alone (Figs. 4 and 5). The reason for this is not clear. It is conceivable that within the very short duration between agonist addition and the luminescent detection reading, the proportion of calcium able to cross over to the recipient cells is limited.
As with most high-throughput methods in the screening environment, additional assays are required to filter out false-positive hits, and may be particularly necessary for this assay, owing to the involvement of multiple components. Off-target compound activities can be identified with receptor assays using cytoAeq, which should eliminate compounds that either affect any of these components or are nonspecific. Specificity filtering can be achieved with an alternative GJ channel in the same assay configuration. The remaining “specific” hits may affect the activity of GJs by a number of mechanisms, such as expression, channel assembly, and pore blockade. Further experiments should be designed to identify these mechanisms, such as high content imaging and dual-electrode recording. Our results suggest that the rate of Ca2+ transfer is an excellent indicator of GJ activity. Intracellular concentrations of Ca2+ are well within the dynamic range of aequorin sensitivity. Combined with the ability of cytoAeq in detecting cytoplasmic changes in free Ca2+ and miniaturization, this technology is ideally placed to conduct high-throughput screening for the discovery of GJ modulating drugs in the modern era.
GJ pores are of different sizes, specifically interact with cytoplasmic molecules in some way that results in selective permeability, and are regulated by a number of cellular processes such as Ca2+ and protein phosphorylation. 33 It is conceivable that Ca2+ is inhibitory to some connexins and may not always be a suitable permeant. Measuring the transfer of other components, for example, cAMP, cGMP, or dyes, may be a better option in those cases. Thus, it would be useful not only to assess the feasibility of the technology described in this article for other connexins, but also to develop a panel of “tool box” assays that unleash the full exploration of this coming-of-age target class for drug discovery.
Footnotes
Acknowledgments
The authors would like to acknowledge the following scientists from GlaxoSmithKline: Dr. Gareth Jones, Dr. Ceri Davies, Dr. Colin Glover, and Dr. Helen Lyons for scientific input in GJs; Dr. Steve Ratcliff, Dr. Robert Jepras, and Dr. Laura Dash for helpful technical discussions on GJ assays; Sandra Arpino and Dr. Jeff Clare for inputs in aequorin; and Alpa Mulji and James Fornwald for BacMam generation.
Disclosure Statement
No competing financial interests exist.