
Abstract
We describe a cellular assay for detection of phosphorylation of endogenous proteins, whereby cells are seeded, treated, and assayed for modulation of phosphorylation in a single microplate well. The procedure is coupled to a rapid, one-wash sandwich enzyme-linked immuno-sorbent assay, enabling results to be obtained within 3–4 h from cell seeding. The assay was tested in two separate cellular systems, namely, HeLa and MCF-7 cells. When using the one-well protocol with Akt phosphorylation as a model, the response to a number of agonists was the same as the response obtained using cells treated in a separate microplate, using a conventional lysate transfer approach. The assay procedure was automated, and quantitative pharmacological data on three known inhibitors of the PI3-kinase signaling pathway was obtained within 4 h from seeding cells, with six dispense steps, and a single wash cycle. Thus, the protocol affords a reliable means of assaying for cellular signaling events in different cell types, and is amenable to automation.
Introduction
The conventional sandwich ELISA in particular has been a standard technique for detecting analytes, including phosphorylated analytes in complex cellular lysates, for several decades, and remains a mainstay among general bioanalytical techniques. 8 The ELISA's enduring popularity is based on characteristics such as sensitivity, reproducibility, and robustness in various biological matrices, which have allowed its application in many areas of analytical biochemistry, including both research and diagnostic applications. However, similar to Western blots and other common techniques used for cell signaling in research applications, ELISAs are time-consuming, require a high degree of handling throughout the procedure, and often take up to a day to complete a standard protocol.
More efficient means of probing protein phosphorylation have been developed, and techniques such as phospho-proteome analysis 9 –11 can be used to detect and quantify cellular phosphorylation on a global scale. However, more established techniques such as Western blot or ELISA remain important for the subsequent target-specific validation of findings derived from global analysis techniques. While there are a multitude of high-throughput techniques available for gaining information on protein phosphorylation, relatively few of these are amenable for the measurement of endogenous phosphorylation events in non-transfected cells. 12 Technologies for detecting endogenous phosphorylation events include high-content screening, 13,14 AlphaScreen®-based immunoassays 12,15 (PerkinElmer), time-resolved fluorescence resonance energy transfer-based immunoassays (CisBio), and electrochemiluminescence-based immunoassays (Mesoscale discovery). These methods typically require specialized reading equipment, and their use is therefore limited to laboratories with access to such equipment.
Here we describe a simple procedure for performing cell-based assays for detection of endogenous phosphoproteins, whereby cells are seeded and assayed in the same microplate. The procedure is coupled to a one-wash ELISA procedure, enabling a minimal degree of handling from cell seeding through to assay, and horse radish peroxidase (HRP)-based detection compatible with standard plate reading equipment accessible to most laboratories.
Using detection of Akt phosphorylation as a model system, we first tested the performance of the one-wash ELISA in assaying both recombinant protein and whole cell lysates, using a conventional approach; requiring the generation of cellular material in a separate well to the assay plate, and transfer of the lysate to another plate for assay. Next, we developed the single-well assay protocol, whereby cells were seeded, treated, and subsequently assayed in the same well, generating data comparable to that obtained when cells were cultured and treated in a conventional manner. Finally, the single-well assay was automated, and quantitative pharmacological data on PI3 kinase-signaling pathway inhibition was obtained.
Materials and Methods
Materials
All cell lines were from ATCC. All cell culture media and supplements, Hank's balanced salt solution (HBSS) and NuPage sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and apparatus, and conventional p-AKT Ser473 ELISA were from Life Technologies. AlphaScreen SureFire® phospho-AKT1/2/3 Ser473 assay kits and Proxiplate™-384 Plus microplates were from PerkinElmer. Anisomycin and insulin were from Sigma. Epidermal growth factor (EGF) and tumor necrosis factor-alpha (TNF-α) were from R&D Systems. Rapamycin, wortmannin, PI-103 hydrochloride, and LY294002 hydrochloride were from Tocris Bioscience. All ELISAONE ® assay kits were from TGR BioSciences. Tissue culture-treated 96-well plates and growth flasks were from NUNC. Recombinant active AKT1 was from Biaffin. Immunoglobulin G (IgG)-free bovine serum albumin (BSA) was from Jackson Immunoresearch.
Comparison of One-Wash ELISA, Conventional ELISA, and Homogeneous Assay Formats
Conventional ELISA
A recombinant human active AKT1 (rhAKT1) dilution series was prepared by diluting rhAKT1 to various concentrations in phosphate-buffered saline (PBS) containing 0.1% IgG-free BSA, and analyzed by conventional ELISA (Invitrogen) according to the kit instructions. Briefly, 100 μL/well of diluted analyte was added to the wells, sealed, and incubated at room temperature for 2 h, and then wells were washed four times in 300 μL with PBS/0.05% Tween20. After the wash cycle, 100 μL/well of AKT S473 detection antibody was added to the wells, and the plates were re-sealed and incubated at room temperature for 1 h. The wells were washed (4×300 μL), and 100 μL/well of anti-rabbit IgG-HRP conjugate was added to the wells, and the plates were re-sealed and incubated at room temperature for 30 min. The wells were washed (4×300 μL), and 100 μL 3,3,5,5-tetramethylbenzidine (TMB) was added to the wells and incubated for 30 min at room temperature, in the dark. The reaction was stopped by the addition of 100 μL/well of stop solution, and absorbance at 450 nm in the wells was measured using an Envision multilabel plate reader (PerkinElmer).
One-wash ELISA
An rhAKT1 dilution series was prepared as described above, and analyzed using a one-wash ELISA, according to the kit instructions. Briefly, 50 μL/well of diluted analyte was added to the wells, followed immediately by 50 μL/well of antibody mix. The plates were sealed, and incubated at room temperature for 1 h on a Ratek MPS1 plate shaker set to ∼350 rpm. The wells were washed (3×300 μL) and 100 μL of either fluorogenic HRP substrate 10-acetyl-3,7-dihydroxyphenoxazine (ADHP), or TMB, was added to the wells and incubated for 10 min at room temperature, in the dark. For TMB experiments, reaction was stopped by the addition of 100 μL/well of stop solution, and absorbance at 450 nm in the wells was measured. For ADHP experiments, fluorescence in the wells was determined using an Envision multilabel plate reader, fitted with a 535ex/590em filter set.
AlphaScreen SureFire homogeneous assay
rhAKT1 was diluted to various concentrations in 1× AlphaScreen SureFire lysis buffer, containing 0.1% IgG-free BSA. The diluted samples were transferred to a Proxiplate™-384 Plus assay plate (4 μL/well) and assayed using the AlphaScreen SureFire kit's instructions. Briefly, 5 μL/well of a mixture containing Reaction buffer, Activation buffer, and AlphaScreen Acceptor beads was prepared at a ratio of 40:10:1, and added to the analyte in the wells. The plates were sealed and covered from light, and incubated for 2 h at room temperature. Subsequently, 2 μL/well of a mixture of Dilution buffer and AlphaScreen donor beads, prepared at a ratio of 20:1, was added to the wells. The plates were re-sealed, covered, and incubated for a further 2 h. At the completion of the assay, signal in the wells was measured using an Envision multilabel plate reader.
One-Wash Assay Performance and Specificity in Whole Cell Lysates
Detection of AKT 1/2/3 in various cell lines
Various human (A431, A549, HEK293, HeLa, MCF7, PC3, U2OS) or mouse (C2C12, NIH3T3, RAW264.7) cell lines were grown in flasks to 70%–90% confluency, then lysed (Lysis mix supplied with the ELISAONE assay kits and prepared according to the manufacturer's instructions) with shaking for 10 min at room temperature. The lysates were adjusted to 200 μg protein/mL, and portions of lysate (50 μL/well) were transferred in duplicate to ELISAONE assay plates and analyzed for AKT 1/2/3 using the standard one-wash ELISA protocol described above.
AKT phosphorylation response in HeLa cells
HeLa cells were seeded into 96-well tissue culture microplates at a density of 30,000 cells/well in medium containing 10% fetal bovine serum (FBS), penicillin/streptomycin, non-essential amino acids, and sodium pyruvate, and cultured overnight at 37°C/5% CO2. The following day, the cells were serum-starved for 3 h by replacing the culture medium with 60 μL/well of the same medium without 10% FBS, and incubating at 37°C/5% CO2. The cells were subsequently treated for various amounts of time with 60 μL/well of 100 μg/mL insulin, 100 ng/mL EGF, 2 μg/mL anisomycin, or 100 ng/mL TNF-α, all diluted in serum-free medium containing 0.1% IgG-free BSA. Cell lysates were prepared by adding 30 μL/well of 5× Lysis Mix Concentrate (supplied with the ELISAONE assay kits and prepared according to the manufacturer's instructions), and shaking for 10 min at room temperature on a plate shaker. Lysates (50 μL) were transferred to the assay plate and analyzed for p-AKT 1/2/3, using the standard one-wash ELISA protocol described above.
Development of One-Well Assay Procedure
Cell density requirement
HeLa cells were grown in flasks to 70%–90% confluency, and harvested in trypsin-free cell dissociation solution, and resuspended at various concentrations in culture medium containing 1% FBS. These suspensions (30 μL) were added to the wells of an ELISAONE assay plate that had been rinsed with water to remove preservatives, to give final number of 20,000, 10,000, 5,000, or 2,500 cells per well. The plates were covered and incubated at 37°C/5% CO2 for 2 h, and then treated for 15 min with either 10 μL serum-free medium containing 0.1% IgG-free BSA, or 10 μL of the same medium containing 40 μg/mL insulin. The cells were lysed with 10 μL 5× Lysis Mix Concentrate, with shaking for 10 min at room temperature. Subsequently, 50 μL/well p-Akt1/2/3 Antibody Mix was added to the wells, the plates were sealed, and incubated at room temperature for 1 h on a Ratek MPS1 plate shaker set to ∼350 rpm. The wells were washed (3×300 μL) and 100 μL substrate was added to the wells and incubated for 10 min at room temperature, in the dark. Fluorescence in the wells was determined using an Envision multilabel plate reader, fitted with a 535ex/590em filter set.
AKT phosphorylation response in HeLa cells
HeLa cells were handled as described above, after seeding into rinsed ELISA assay wells at a density of 5,000 cells/well. The cells were treated for various amounts of time with 10 μL/well of 100 μg/mL insulin, 100 ng/mL EGF, 2 μg/mL anisomycin, or 100 ng/mL TNF-α, all in serum-free medium containing 0.1% IgG-free BSA. Cell lysates were prepared by adding 10 μL/well of lysis mix concentrate, and shaking for 10 min at room temperature on a plate shaker. Lysates were analyzed for p-AKT 1/2/3, as described above.
Automation and Validation of the One-Well Cell-Based Assay Protocol
The assays were automated with BioTek liquid-handling equipment. Various concentrations of cell lysates were prepared from insulin-treated MCF7 cells, ranging from 1 to 250 μg/mL. These lysates were analyzed using a p-p70S6K-specific one-wash ELISA, over a 4-day period in replicates of six. Analyte serial dilution and transfer to the assay plate were performed with a Precision™ XS Microplate Sample Processor. Antibody mix was subsequently added to the plates with a MultiFlo™ Microplate Dispenser, fitted with 5 and 10 μL cassettes, and dispensed using the low flow rate setting. The plates were covered and incubated for 60 min at room temperature with shaking. The plates were washed with an ELx50™ Microplate Strip Washer, using the pre-programmed NUNC Flat protocol provided by the manufacturer that includes an integrated 30-s soak. A Synergy™ HT Multi-Mode Microplate Reader was used to detect Raw Fluorescence Units, using a filter set of 540/25 nm for excitation and 590/20 nm for emission, and a constant sensitivity setting of 46, predetermined based on a high well test for the assay. A lysis buffer-only control was used for the zero wells.
Insulin-mediated AKT phosphorylation in MCF7 cells
Cells were harvested from 70%–85% confluent flasks of cells, and resuspended in HBSS containing 5% FBS. The cells (20 μL) were seeded directly into an ELISAONE microplate that had been pre-rinsed to remove preservatives, at a density of 10,000 cells/well. The cells were allowed to adhere for 2 h at 37°C. Following incubation, the cells were stimulated with 20 μL of HBSS/5% FBS, containing various concentrations of insulin for 15 min at 37°C. To stop the reaction, 10 μL of 5×lysis mix was added to the wells. The plate was subsequently incubated with shaking for 10 min to lyse the cells. Antibody Mix was added to the wells, and AKT phosphorylation in the wells was subsequently determined as described above.
Antagonist dose inhibition of insulin-mediated AKT phosphorylation in MCF7 cells
Various concentrations of wortmannin, PI-103, or LY294002 were prepared in HBSS containing 5% FBS and 4% dimethyl sulfoxide. Aliquots (20 μL) of these preparations were dispensed into an ELISAONE microplate that had been pre-rinsed to remove preservatives, followed by 10 μL of MCF-7 cells resuspended in HBSS, containing 5% FBS, to yield 7,700 cells/well in a final volume of 30 μL. The cells were incubated for 2 h at 37°C, then treated with 10 μL/well of 0.72 μM insulin, prepared in HBSS, containing 5% FBS, to yield a final concentration of 0.18 μM insulin in the well. The cells were incubated for 15 min at 37°C. To stop the reaction, 10 μL of 5× lysis mix concentrate was added to the wells. The plate was subsequently incubated with shaking for 10 min to lyse the cells. Antibody Mix was added to the wells, and AKT phosphorylation in the wells was subsequently determined as described above.
Results and Discussion
Conventional sandwich ELISA protocols can limit the throughput of samples that can be addressed during an experimental run, and its ability to be automated. To address some of these handling-associated limitations of conventional ELISA, we developed a protocol whereby all assay steps, including cell treatment steps, occur in a single well, thereby eliminating transfer steps. We coupled this methodology to a one-wash ELISA with a simple workflow, making the protocol amenable to automation and high-throughput applications.
The one-wash ELISAs we used here use an analyte-specific antibody pair, and HRP reporting, similar to conventional sandwich ELISA, but are distinguished by the solution-phase delivery of both analyte-specific antibodies. During the incubation, the immuno-complexes formed between the capture antibody, the antigen, and the detection antibody are bound to the plate surface via a specific ligand conjugated to the capture antibody. This is in contrast to conventional sandwich ELISAs, where an analyte-specific capture antibody is passively or covalently pre-immobilized to the solid phase, and the analyte and any subsequent detection antibodies are added in sequential incubations. ELISA kits in this format for detection of endogenous proteins are commercially available from several vendors (InstantOne ELISA; eBioscience, PhosphoTracer ELISA; Abcam, and ELISAONE; TGR BioSciences), and the validation of solution-phase ELISA for other target classes has been described elsewhere. 16
The key practical benefit of a solution-phase ELISA is the single assay incubation of 1 h, enabled by simultaneous addition and binding of all assay reactants, which can therefore reach equilibrium concurrently. This feature subsequently affords less aspiration and dispensing steps, less wash steps, and an overall shortening of assay time. Thus, the protocol is more suitable to higher experimental throughput and automation.
Validation of the One-Wash ELISA
Sensitivity relative to standard sandwich immunoassay techniques
We first examined sensitivity in comparison to both conventional ELISA, and the well-established AlphaScreen SureFire homogeneous assay format, to ensure that the one-wash ELISA format did not compromise sensitivity requirements needed to detect endogenous protein levels in cell-based assays. A dilution series of rhAKT1 was analyzed by the one-wash p-AKT ELISA, conventional p-AKT ELISA (both of which use HRP-based substrate readouts), or a homogeneous p-AKT sandwich immunoassay format (AlphaScreen SureFire; PerkinElmer), which uses bead-based detection. 12,15
The one-wash ELISA protocol required addition of 50 μL of sample and 50 μL of assay reagents in the assay plate, a single incubation for 1 h with shaking (∼350 rpm, Ratek MPS1), prior to plate washing and HRP substrate addition to the plate for detection, yielding a total assay time of 1:15 h. The conventional p-AKT ELISA required 100 μL of sample, three sequential assay incubations of 2 h, 1 h, and 30 min, each intervened by wash cycles, before the TMB addition, yielding a total assay time of around 4 h. The AlphaScreen SureFire p-AKT assay, which employs solution-phase antibody delivery similar to the one-wash ELISA, required a sample volume of 4 μL, and two sequential assay reagent additions/incubations of 2 h each, yielding a total assay time of 4 h. Despite the differences in assay length and protocol (summarized in Table 1), all assays afforded similar sensitivity, each in the low pg/mL range (Fig. 1), consistent with previous findings when comparing solution-phase and conventional ELISA. 16 The TMB-based ELISAs had a lesser dynamic range than either fluorescent or Alpha-based assays, and were capable of distinguishing around 3-logs analyte concentration, limited by the dynamic range afforded by absorbance measurements on most plate readers. The one-wash ELISA paired with the fluorogenic HRP substrate ADHP afforded an assay range capable of distinguishing around 4-logs analyte concentration, similar to that of AlphaScreen, and did not require the addition of a stop solution. Taken together, these data indicate that the solution-phase ELISA afforded equivalent sensitivity to standard assay techniques for the same analyte.
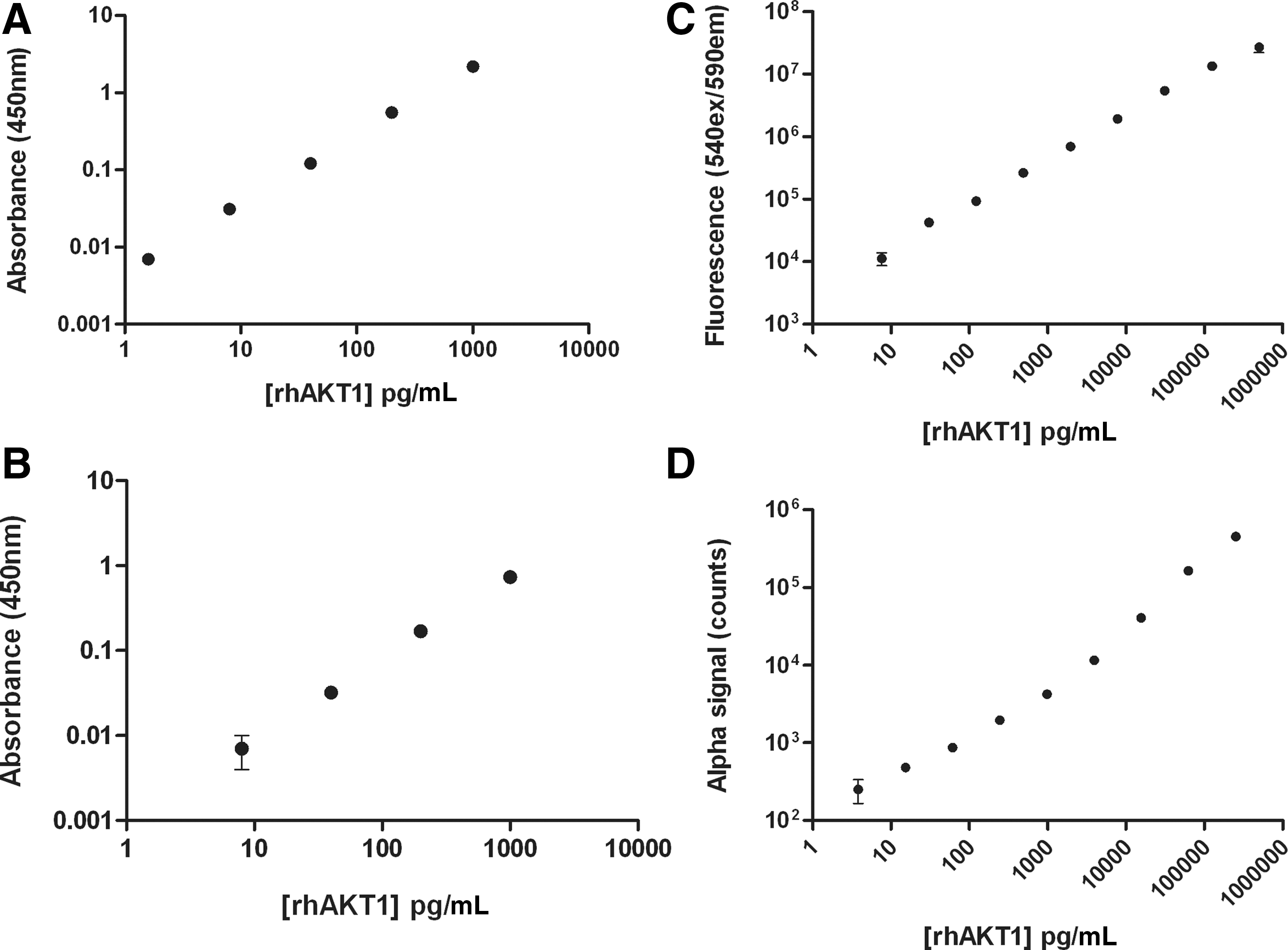
Comparison of one-wash ELISA sensitivity with traditional ELISA and homogeneous assay formats. The one-wash ELISA format
ELISA, enzyme-linked immuno-sorbent assay.
Performance and specificity on whole cell lysates
For use on whole cell lysates, we first examined the ability of the one-wash ELISA to detect total AKT 1/2/3 levels in a range of mouse and human cell lines. For this, cellular lysates at a concentration of approximately 200 μg protein/mL from several human cell lines (HeLa, HEK, MCF7, PC3, A431, A549, and U2OS) or mouse cell lines (NIH3T3, C2C12, and RAW 264.7) were prepared. Using the standard one-wash ELISA protocol (10 μg protein/well in a volume of 50 μL), AKT was detectable over buffer-only controls in all cell lines examined (Fig. 2A), although some differences in expression levels were observed between the different cell lines.

Performance of one-wash ELISA on cellular lysates.
Next we examined AKT phosphorylation in HeLa cells, which we found to express readily detectable levels of total AKT (Fig. 2A), to determine the specificity of the assay in response to a range of treatments. The cells were cultured in 96-well microplates, and stimulated for various lengths of time with four agonists that were expected to differentially stimulate AKT phosphorylation, namely EGF, insulin, anisomycin and TNFα. The cells were lysed, and a portion of lysate was transferred to a one-wash ELISA assay plate and analyzed for p-AKT using the standard protocol.
For each of the treatments, the AKT phosphorylation response that was observed was within expectations. 17 –20 In particular, AKT phosphorylation was induced by both insulin and EGF, with the other treatments unable to elicit a response (Fig. 2B). EGF induced a rapid and transient peak of AKT phosphorylation, while insulin induced a rapid and more sustained response. These data indicated the specificity of the one-wash assay for AKT phosphorylation. To ensure the cells were responding to the treatments as expected, a portion of the same lysate preparation was also analyzed for phosphorylation of ERK, NF-κB p65 and p38 MAPKα (summarized in Table 2). As the time course for each agonist was run in parallel on the same plate of cells, the relative strength of induction for each agonist was readily apparent. GAPDH levels were also monitored for all treatments, and did not significantly change over the course of the experiment (data not shown).
The peak response (S:B) was calculated by dividing the maximum raw signal by the raw signal observed in untreated cells. All data were averaged from three separate wells.
EGF, epidermal growth factor; n/r, no response; TNF, tumor necrosis factor.
Development of a One-Well, Cell-Based Assay
A procedure was developed to replace a conventional assay for cell-based protein phosphorylation, requiring overnight culturing of cells in a tissue-culture plate, and separate treatment and lysis prior to analysis of lysates on a separate assay plate. Based on our results on relative expression levels (Fig. 2A), for these experiments we focused on cell lines that expressed both relatively low (HeLa) and relatively high (MCF7) amounts of AKT to test utility of the assay across a range of cellular expression levels. For the procedure described here, all assay steps after cell harvesting were performed in the same microplate well (summarized in Table 3). An important pre-requisite for this protocol was rinsing assay plates prior to introduction of the cells, thereby removing preservatives and/or stabilizers in the wells. Without this step, cell viability was affected during the assay (data not shown). However, as the protocol does not require long-term cell culture or sterile conditions, washing was achieved quickly and effectively with a standard plate washer.
1. BioTek® MultiFlo™ automated dispenser with peri-pump cassettes.
2–4,6. MultiFlo automated dispenser with using low flow rate and the Liquid Handling Control software.
5. BioTek® ELx50™ microplate strip washer via onboard NUNC flat protocol integrated with a 30 s soak.
7. BioTek® Synergy™ HT Multi Mode microplate reader was used to detect Raw Fluorescence Units using a filter set of 540/25 for excitation and 590/20 for emission. Following an Auto Sensitivity test on a high well for the assay, a setting of 46 was used for all experiments.
n/a, not applicable.
The amount of HeLa cells required for an adequate assay window was first examined, using cell seeding densities ranging from 2,500 to 20,000 cells per well. Over this range of cells, a clear insulin-mediated AKT phosphorylation response was observed for all cell densities, with the amplitude of the signal increasing linearly with the number of cells/well (Fig. 3A). We also tested various concentrations of FBS in the seeding medium (data not shown). Under these conditions, levels of AKT phosphorylation in untreated cells increased with FBS concentrations, likely due to bovine insulin or IGF-1 present in the FBS activating the PI3-kinase pathway. The absence of any FBS in the seeding medium also stimulated AKT phosphorylation, and an FBS concentration of 1% in the seeding medium was found to be optimal, and was used for subsequent experiments.
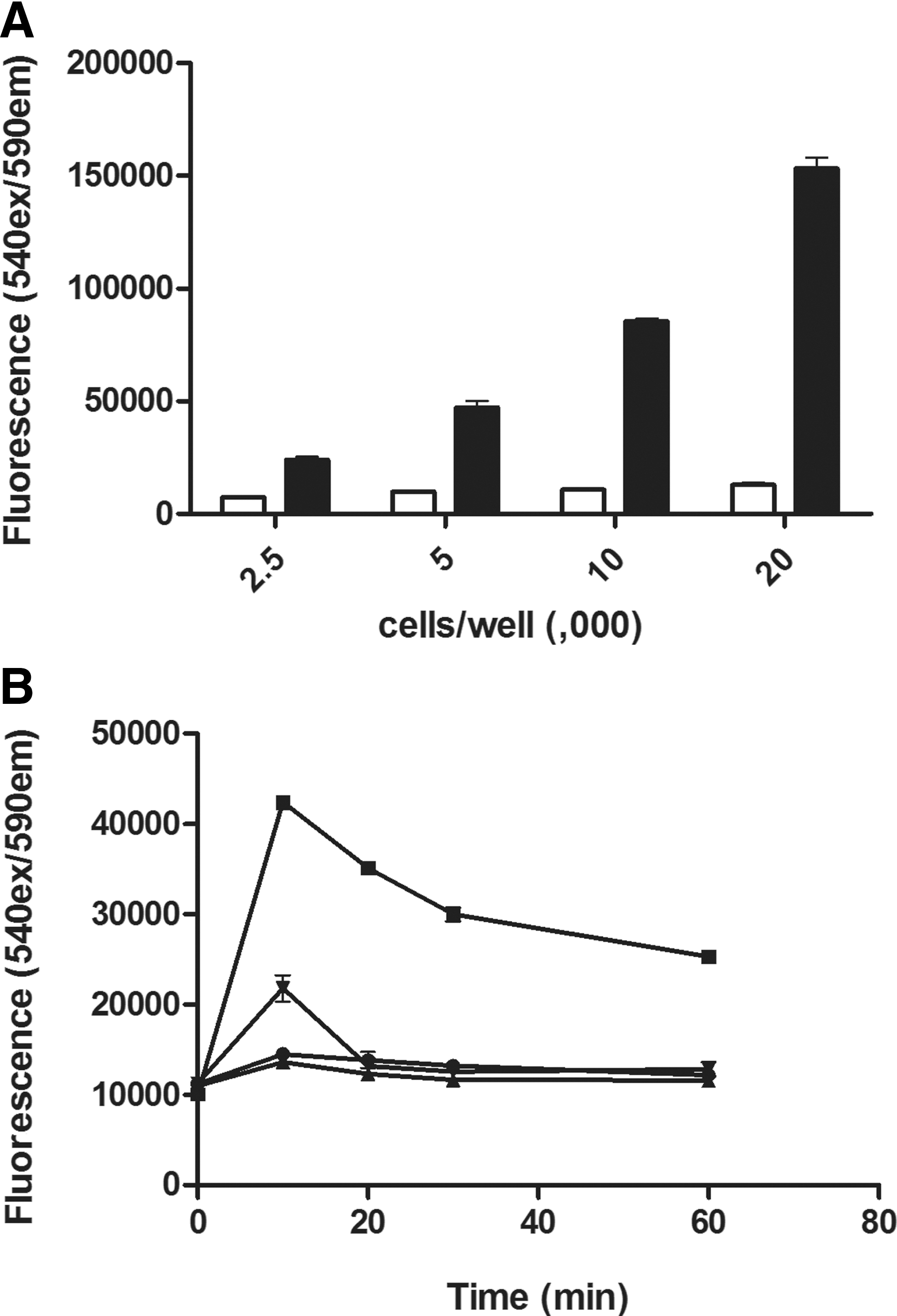
Determination of AKT phosphorylation using a one-well ELISA procedure: manual handling.
Next we examined the specificity of the AKT phosphorylation response to a range of treatments. The cells were stimulated for various lengths of time with the same four agonists used in the conventional lysate transfer experiments described above, and that were found to differentially stimulate AKT phosphorylation (EGF, insulin, anisomycin, and TNFα). Under these conditions, the response pattern observed was identical to the conventional transfer assay format derived from cells cultured overnight in tissue-culture treated microplates, indicating that for this pathway, the one-well assay protocol was not noticeably affecting cell response patterns (Fig. 3B).
Full Automation of the One-Well, Cell-Based Assay
Finally, we tested the one-well assay protocol under automation, to examine if the protocol could be successfully adapted for higher throughput requirements. All dispensing, incubation and washing steps of the standard protocol were automated with liquid handling and plate washing equipment using three side-by-side pieces of equipment, one of which was dedicated to sample preparation, one for dispensing assay reagents, and the other for washing the assay plate. The assay performance under automation was initially assessed using conventional lysate transfer using MCF7 cell lysates. Using cell lysates diluted to a concentration range over 2.5-logs, and run on four separate occasions, intra and inter-assay precision was calculated. Under these conditions, both inter- and intraplate variations of less than 6% for all analyte concentrations were observed.
Antagonist dose–response and IC50 pharmacology
For single-well experiments, MCF-7 cells were seeded directly into the one-wash ELISA microplate. Firstly, an insulin-mediated AKT phosphorylation dose–response curve was generated using the standard protocol described in Table 3, with an EC50 in the mid-nanomolar range, consistent with reported results. 21 The resulting insulin EC80 calculated for p-AKT (180 nM) was used in subsequent antagonist dose–response experiments. Three inhibitors known to modulate PI3-kinase pathway signaling (wortmannin, PI-103, and LY294002) were tested in the one-well assay format. For these assays, a total of six dispense steps, and one wash step, were required post cell-harvest (Table 4). The three inhibitors act upstream of AKT to inhibit PI3-kinase activity and consequently reduce AKT phosphorylation, and were used to assess assay specificity and performance. All three upstream inhibitors tested here reduced AKT phosphorylation to baseline levels, with fully inhibited giving signal comparable to buffer-only controls (Fig. 4). The rank order of inhibitor potency was wortmannin and PI-103 approximately equivalent (IC50 2.6 and 8.1 nM, respectively), with LY294002 approximately 100-fold less potent (IC50 495 nM). These responses are consistent with previous findings for these PI3-kinase inhibitors. 22 –24
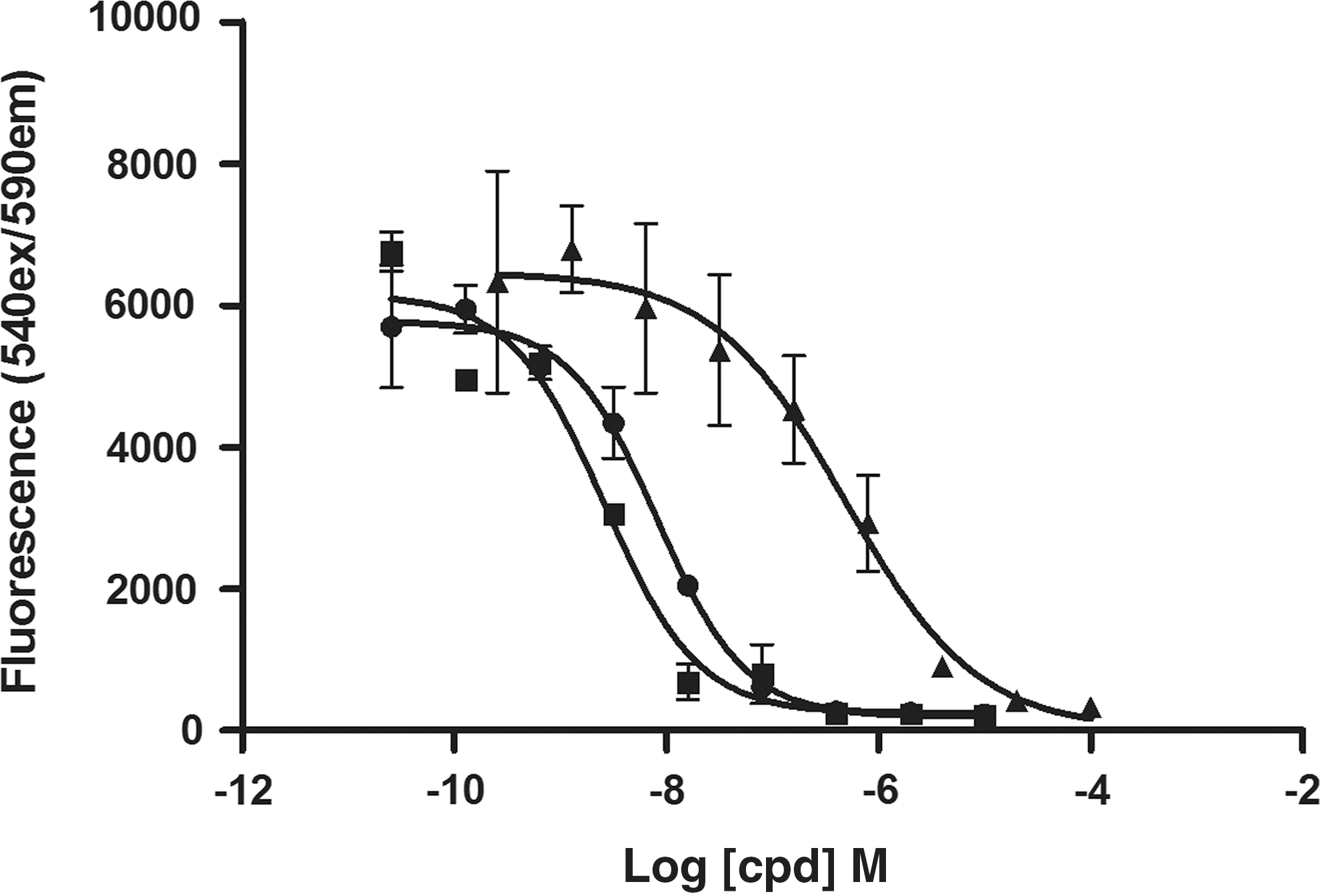
Determination of AKT phosphorylation using a one-well ELISA procedure: automated handling. MCF7 cells were harvested and seeded directly into one-wash ELISA microplates containing various concentrations of either wortmannin (■), PI-103 (•), or LY294002 (▲), and incubated for 2 h. The cells were treated with 180 μM insulin, and subsequently lysed and analyzed for AKT phosphorylation, using the one-wash ELISA protocol. The signal is representative of the mean±SD of three separate assay wells.
1. BioTek® Precision™ XS automated pipetting station was used for all compound serial dilutions and transfer to the assay plate.
2. BioTek® MultiFlo™ automated dispenser with peri-pump cassettes.
3–5,7. MultiFlo automated dispenser using low flow rate and the Liquid Handling Control software.
6. BioTek® ELx50™ microplate strip washer via onboard NUNC flat protocol integrated with a 30 s soak.
8. BioTek® Synergy™ HT Multi Mode microplate reader was used to detect Raw Fluorescence Units using a filter set of 540/25 for excitation and 590/20 for emission. Following an Auto Sensitivity test on a high well for the assay, a setting of 46 was used for all experiments.
Taken together, these results demonstrate practical application of using the one-well assay to generate pharmacologically relevant data, with minimal handling requirements and a total assay time of around 4 h after cell harvest. The cells used here, MCF7 and HeLa cells, are adherent cell lines; however, non-automated data generated in our laboratory using Jurkat cells (data not shown) indicates that the protocol works equally well with non-adherent cell lines, suggesting that the protocol should be widely applicable to cell signaling experiments that require high throughput capability.
Conclusions
Sandwich ELISAs are relatively cheap, and plate readers and washers required for performing these experiments are widespread. Here we have validated an immunoassay format, from which quantitative results can be obtained much more rapidly than conventional ELISA, and with less handling. The assay was shown to be highly amenable to automation, with good inter-assay reproducibility and performance. Based on this, a fully automated, one-well cell based assay procedure was developed and validated using insulin-mediated AKT phosphorylation in MCF7 cells. This assay format can facilitate greater experimental throughput than conventional ELISA, and offers an alternative in the toolbox for medium to high throughput applications.
Footnotes
Disclosure Statement
TGR BioSciences manufactures ELISAONE™ assay kits, used for the experiments described here. BioTek instrumentation was used for some of the experiments described here.