
Abstract
Receptor tyrosine kinases (RTKs) are important players in various cellular processes, including proliferation, migration, metabolism, and neuronal development. EphB4 RTK is essential for the development of a functional arterial–venous network in embryonic and adult neoangiogenesis. To develop novel inhibitors of EphB4 that might have applications in severe diseases like cancer and retinopathies, assays need to be in place that resemble, in a most physiological fashion, the activation and downstream function of the kinase. In addition, such assays need to be amenable to high-throughput screening to serve efficiently the modern drug discovery processes in the pharmaceutical industry. The authors have developed an enzyme fragment complementation assay that measures the interaction of a downstream docking protein to the activated and phosphorylated full-length EphB4 kinase in cells. The assay is specific, robust, and amenable to miniaturization and high-throughput screening. It covers most steps in the activation process of EphB4, including ligand binding, autophosphorylation, and docking of a downstream interactor. This assay format can be transferred to other RTKs and adds an important cell-based kinase assay option to researchers in the field.
Introduction
EphB4 and its ligand, ephrinB2, are complementarily expressed in newly forming venous and arterial vessels, respectively. 5 Both are essential for vessel formation and vascular patterning during embryonal development. Mice defective in either EphB4 or ephrinB2 have a very similar phenotype, failing to develop a hierarchical blood vessel system resulting in embryonic lethality. 5 –7 EphB4 has also been implicated in hepatic cell function, 8 bone mineral metabolism, 9 and cell migration, and proliferation. 10,11 In addition, EphB4 has been shown to be overexpressed in a wide range of tumor cells and tissues, including cancers found in the ovary, 12 bladder, 13 prostate, 14,15 breast, 16 and colon. 17,18 These findings have qualified EphB4 as an interesting angiogenic target for antitumor and antiretinopathic therapies, and different approaches have been taken to target EphB4, including small molecules, 19 –25 peptides, 26 soluble extracellular domains, 11,27 –31 or functional blocking antibodies. 32 –34
On the molecular level, ligand–receptor interactions in the Eph family are characterized by clustering of the proteins at the membrane followed by bidirectional intracellular signaling events into both the Eph receptor expressing cell and the ephrin expressing cell. Binding of Eph receptors to ephrin ligands result in clustering of the Eph receptors and subsequent transphosphorylation at tyrosine residues of the juxtramembrane region and the kinase domain. This, in turn, generates docking sites for additional Src-homology (SH2) containing signaling proteins, for example, PTPN6, phospholipase Cγ, phosphatidylinositol-3-kinase, and other adapter proteins. 3,35,36 Classically, kinases have been screened using kinase domains as purified proteins accompanied with a biochemical detection of activity. Although these technologies enable sufficiently fast and cost-efficient testing of large compound libraries, these enzymatic kinase assays only represent one aspect of RTK signaling, omitting the extracellular interaction as well as the intracellular signal transduction processes. Utilizing whole-cell assays can add more physiologically relevant read-outs, but traditional technologies, such as Western blot or ELISA detection of phosphoproteins in cellular lysates have been hampered by their limited throughput in the past. 37 Much progress has been made over the last years with phospho-specific antibody-based detection systems like ALPHAscreen™, HTRF™, and Lanthascreen™. 38 In addition, label-free and subcellular imaging technologies can enable assays that monitor special intracellular events following kinase activation, for example, translocation or cellular morphology. 39,40
In the approach described here, we utilized the PathHunter enzyme fragment complementation (EFC) system 41,42 to develop a cell-based assay to monitor the activation and downstream signaling of EphB4 (Fig. 1). The PathHunter system is comprised of two weakly complementing fragments of β-galactosidase, a small 42aa peptide termed ProLink 43 and the complementing fragment termed enzyme acceptor (EA). When these fragments are coexpressed in cells, little or no enzyme activity is generated due to their reduced affinity. However, when the proteins are fused to two proteins that interact, the interaction of the two proteins forces the complementation of the enzyme effectively restoring the enzyme activity. 44 The PathHunter EphB4 assay detected small molecule kinase inhibitors and antibodies reflecting the expected pharmacology. The platform was further optimized for high-throughput screening through measures of assay reproducibility and solvent tolerance. These studies establish a novel cell-based assay for EphB4 and its suitability for the screening and characterization of inhibitors.
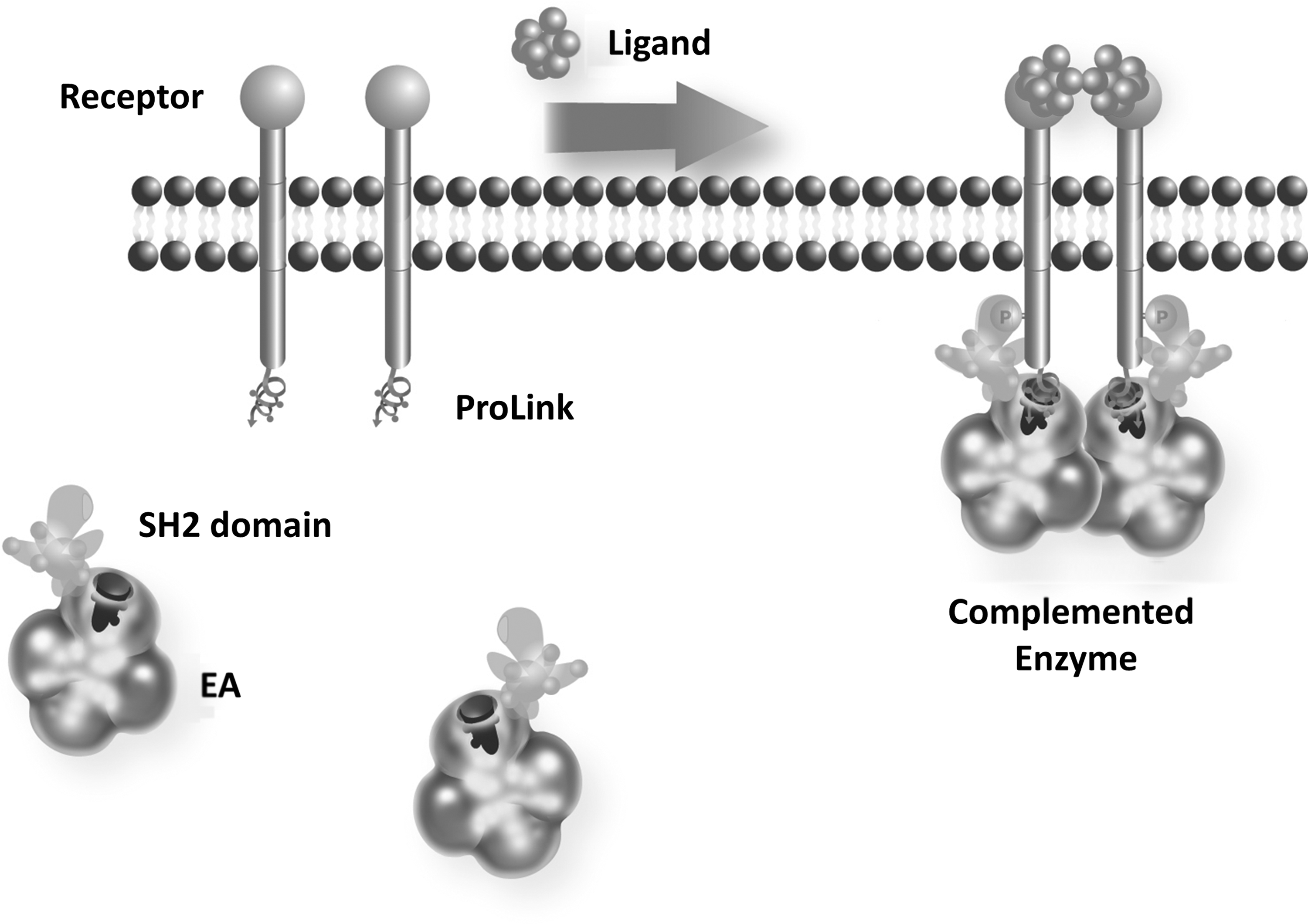
The enzyme fragment complementation (EFC) protein interaction detection system is comprised of two low-affinity enzyme fragments, called ProLink and Enzyme Acceptor (EA), that can be forced to complement when fused to two interacting proteins. To monitor receptor tyrosine kinase activation, the receptor is fused at its C-terminus to the ProLink peptide and the EA component fused to a phosphotyrosine binding domain (SH2). Ligand-mediated activation of the receptor initiates autophosphorylation providing a binding site for the SH2-EA fusion, forcing the complementation of the two enzyme fragments. The increase in enzyme activity is measured using a chemiluminescent substrate as a measure of receptor phosphorylation.
Materials and Methods
Cell Lines
U2OS cells (obtained from ATCC) were cultured in a maintenance medium, which was Eagle's minimum essential medium (MEME) supplemented with 10% fetal bovine serum, 100 μM L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin. The U2OS-EphB4-PTPN6 cell line was cultured in the above maintenance medium supplemented with 250 μg/mL hygromycin (Invitrogen) and 500 μg/mL G418 (GibcoBRL). Cells were routinely grown at 37°C in a humidified incubator in the presence of 5% CO2.
Vectors
EphB4ProLink fusion proteins were created by amplifying the respective cDNA, Accession Number MN_004444 via PCR. The forward primer consisted of a NheI site and the sequence ATG ACC ATG GTT GAC ACA GAG ATG, and the reverse primer consisted of an XbaI site and the sequence GAG TAC AAG TCC TTG TAG ATC TCC TG. The products were ligated into the N-terminus of the ProLinkvector (DiscoveRx). A similar strategy was also used to generate plasmids with EA expressing PTPN6 and the EphA5, EphA7, and EphB2.
Compounds
mEphB2-Fc chimera was purchased from R&D Systems. PP1 and PP2 were purchased from BioMol. Dasatinib was synthesized at Bayer Pharma AG. The antiEphB4 antibody was generated by MorphoSys AG using standard techniques. 45,46
Generation of Double Stable Cell Lines
Double stable cell lines were generated by viral transduction. Briefly, EphB4 and PTPN6 constructs were transfected into the packaging cell lines using the Fugene HD transfection reagent (Roche). After 48 h, the supernatant was collected, filter sterilized with a 0.45-μM filter, and stored at −80°C. Initially, the PTPN6 supernatant was used to transduce U2OS cells, which were then put into selection using 250 μg/mL hygromycin. After 1 week of selection, the PTPN6 pool was infected with the EphB4 viral supernatants and the pools were put into double selection using 250 μg/mL hygromycin and 500 μg/mL G418. After 1 week of double selection, the pools were tested for functional response to agonist, and then single-cell clones were made. A single clone was then selected and further characterized.
EphB4 Assay
See figure legends for detailed assay conditions and Supplementary Table S1 for the assay protocol table (available online at
To evaluate the effect of antagonists, the assay plate was set up as described above. After overnight incubation, the cells were initially treated for 1 h with the antagonist at 37°C. The cells were then induced with agonist and treated as described above.
Co-Immunoprecipitation
To further confirm the functionality of the EphB4-PK construct, we tested the cell line by co-immunoprecipitation and Western blotting. Initially, cells were plated at 1×105 cells/well in a six-well plate in the assay buffer (MEME with 0.1% BSA faction IV). The cells were incubated overnight at 37°C in a humidified incubator with 5% CO2. The cells were then treated with increasing amounts of mEphB2-Fc chimera for 1 h at 37°C, and then lysed by the addition of 100 μL of ice cold RIPA buffer. The lysate was then collected and treated with an anti-myc antibody to immunopreciptate EphB4-PK. The collected fraction was then fractionated by polyacrylamide gel electrophoresis using a 4%–20% gel at 150 V for 1 h. The gel was transferred by Western blotting and probed with an anti-phosphotyrosine antibody (Cell Signaling Technologies).
Kinase Assay with Purified EphB4 Kinase Domain
Human recombinant GST-tagged EphB4 intracellular kinase domain (Proqinase) was preincubated for 10 min at 20°C with a test compound (2 μL enzyme plus 50 nL compound in 100% dimethyl sulfoxide [DMSO]) in a buffer containing 25 mM HEPES (pH 7.3), 5 mM MnCl2, 2 mM DTT, 0.1 mM NaVO4, 1% (v/v) glycerin, 0.02% NP40, and ethylene diamine tetraacetic acid (EDTA)–free complete protease inhibitors (Roche). The kinase reaction was started by adding 3 μL of substrate solution resulting in final concentrations of 2.7 μg/mL biotinylated polyGAT (CisBio) and 10 μM ATP (Amersham) in 5 μL volume. The reaction (5 μL) was terminated after 10 min with 10 μL of a solution containing 50 mM HEPES, pH 7.0, 0.125% (w/v) BSA, 0.14 μg/mL PT66-Eu (PerkinElmer or Cisbio), 3.25 μg/mL streptavidin-XLent (CisBio), 75 mM EDTA, and incubated for 60 min. Homogeneous time-resolved fluorescence (HTRF) was measured in a Ruby star instrument (BMG) by excitation at 337 nm and emission at 615 nm and 665 nm. The ratio of the signal at 665 nm (specific FRET signal) divided by the signal at 615 nm (internal control) was calculated. These values were normalized using a positive control (with enzyme) and negative control (without enzyme). PP2 inhibition plots were evaluated using the 4-parameter nonlinear regression algorithm of Grafit 4 Software (Erithacus).
Results
Generation of the EphB4 Cellular Assay
To generate the EFC assay for EphB4 activation, fusion constructs were generated to express the full-length EphB4 fused to the 42aa ProLink peptide (Fig. 1). Since the ProLink tag is only 42aa, it is expected that this fusion will retain agonist-induced kinase activity. However, to directly confirm that the EphB4 fusion is phosphorylated upon activation with a ligand, we performed a co-immunoprecipitation experiment to detect the phosphorylated receptor (Fig. 2A). A dose-dependent increase in phospho-EphB4 with increasing amounts of mEphB2-Fc chimera was detected indicating that the receptor fusion catalytically active and capable of autophosphorylation in response to agonist.
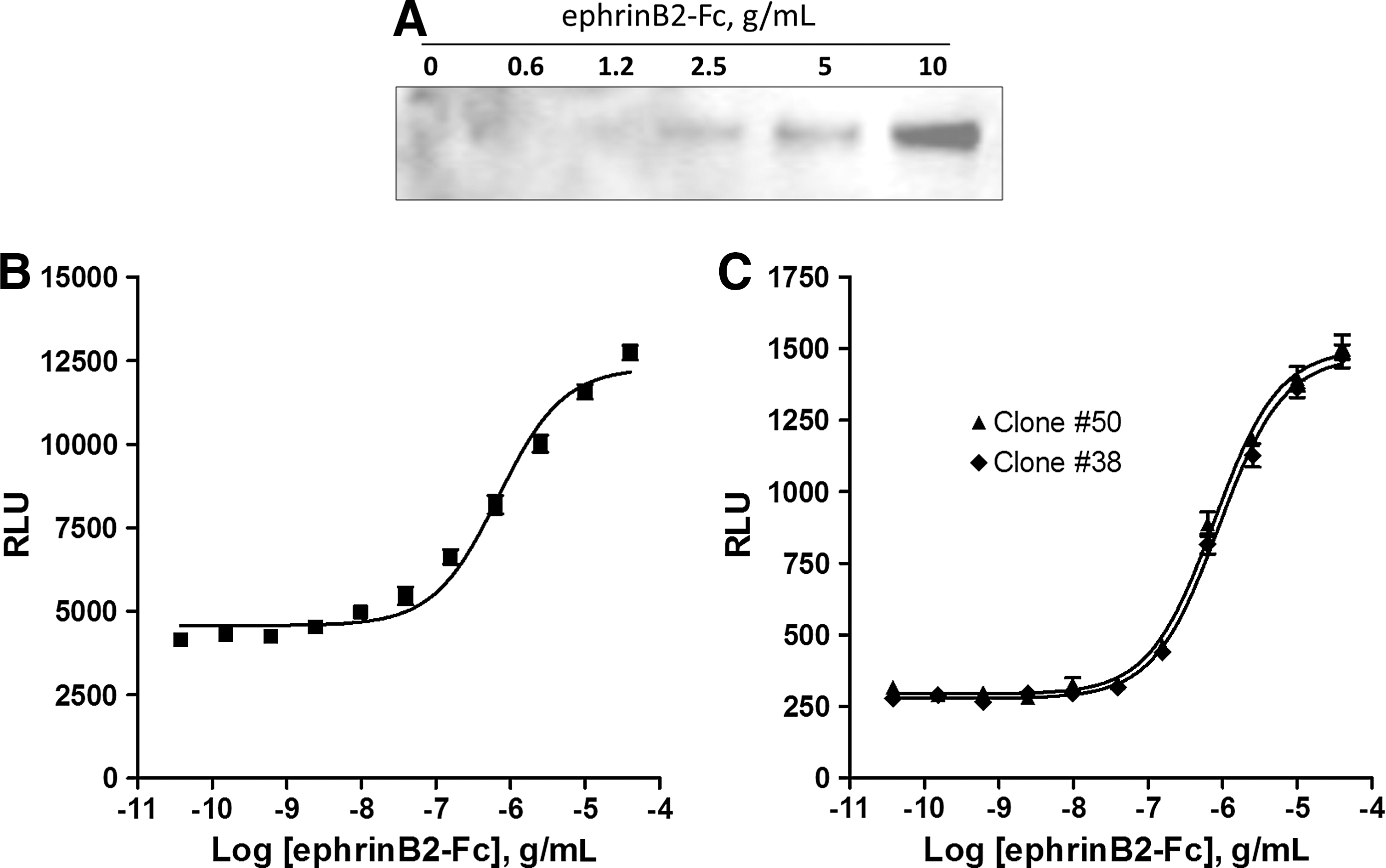
Agonist induced phosphorylation of the EphB4-ProLink fusion detected by Western blot and Enzyme fragment complementation. Cells expressing the EphB4-ProLink Chimera were stimulated with increasing concentrations of the EphrinB2-Fc ligand or left untreated. The EphB4 was immunoprecipitated and analyzed for tyrosine phosphorylation by Western BLOT
To complete the PathHunter system, the tandem SH2 domain from PTPN6, which is expected to specifically bind the phosphorylated EphB4 was cloned as an N-terminal fusion to EA. This construct was then introduced into the EphB4-ProLink stable pool. After selection in hygromycin for the EA fusion, the PathHunter response was assessed using the agonist, mEphB2-Fc chimera. After plating in 384-well plates, the cells were stimulated with mEphB2-Fc. The activation and phosphorylation of the EphB4-ProLink receptor should result in binding of the PTPN6-EA fusion and a subsequent increase in reporter enzyme activity. The forced complementation of the enzyme was measured using Pathhunter detection reagents in a single addition mix and read format (Fig. 2B). We have previously noted that assay performance and stability is enhanced in stable, clonal cell lines. To this end, we placed the cell pool expressing the fusion proteins into clonal dilution. Twenty-four clones were screened and two clones were identified with a robust assay performance (Fig. 2C). The EC50 of mEphrinB2-Fc chimera was very similar for all clones tested and in good concordance with published data of 250 ng/mL. 10
Pharmacological Characterization
To determine if the EFC format would be appropriate to identify antagonists, the assay was first tested with the ATP-competitive kinase inhibitors PP2 and Dasatinib. As shown in Figure 3A, preincubation of the cells with the ATP-competitive kinase inhibitors prevents the agonist-induced complementation. These results indicate that the small molecule inhibitors are preventing receptor phosphorylation as expected, demonstrating the ability of the system to detect small molecule kinase inhibitors. To further validate the results, the same compounds were tested in a biochemical activity assay with the purified catalytic domain of EphB4. As shown in Figure 3B, Dasatinib and PP2 displayed similar IC50 values in the cell-free assay (4 and 300 nM) compared to our cell-based assay (7 and 430 nM, respectively).
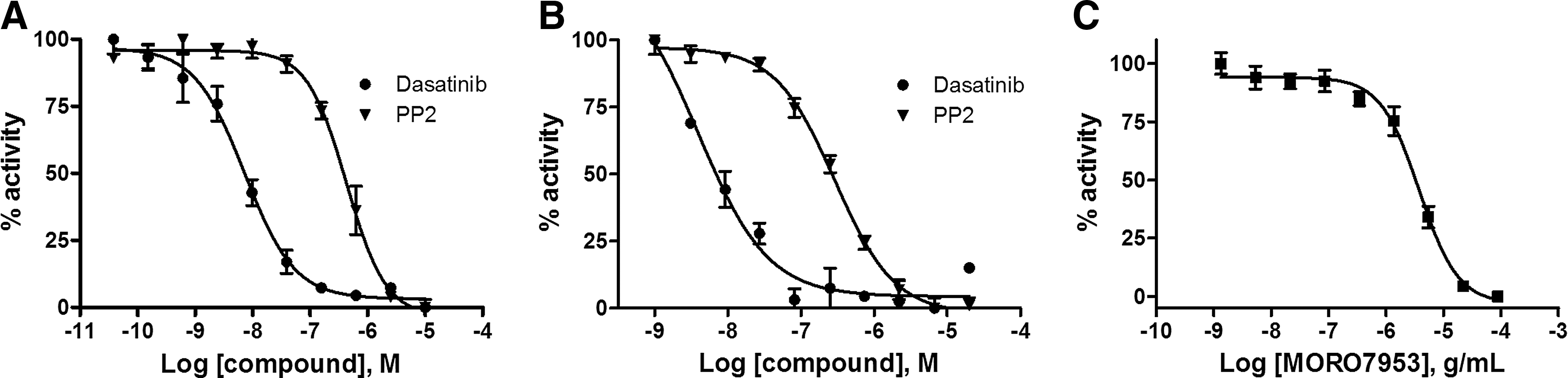
The EphB4 clones were tested for a dose-dependent inhibition using known small molecule antagonists and antibody-based ligand binding inhibitors. First, the EphB4 clone was tested using the known ATP-competitive inhibitors Dasatinib (●) and PP2 (▼) in the cell-based PathHunter EphB4 assay
A distinct advantage of using the full-length kinase and natural ligand-activated pathway is that non-ATP competitive inhibitors can be characterized, such as ligand binding or dimerization inhibitors. As a test, we preincubated the cells with a monoclonal antibody against the extracellular portion of EphB4 that is known to block ligand binding. We found a dose-dependent inhibition of the agonist induced receptor activation in our assay (Fig. 4C). The antibody was shown to reduce the activity down to the basal uninduced state, indicating that although the antibody is bivalent, it retains no agonist activity. These results highlight an important feature of the PathHunter assay that it is capable of detecting intracellular and extracellular modulators of kinase activity.
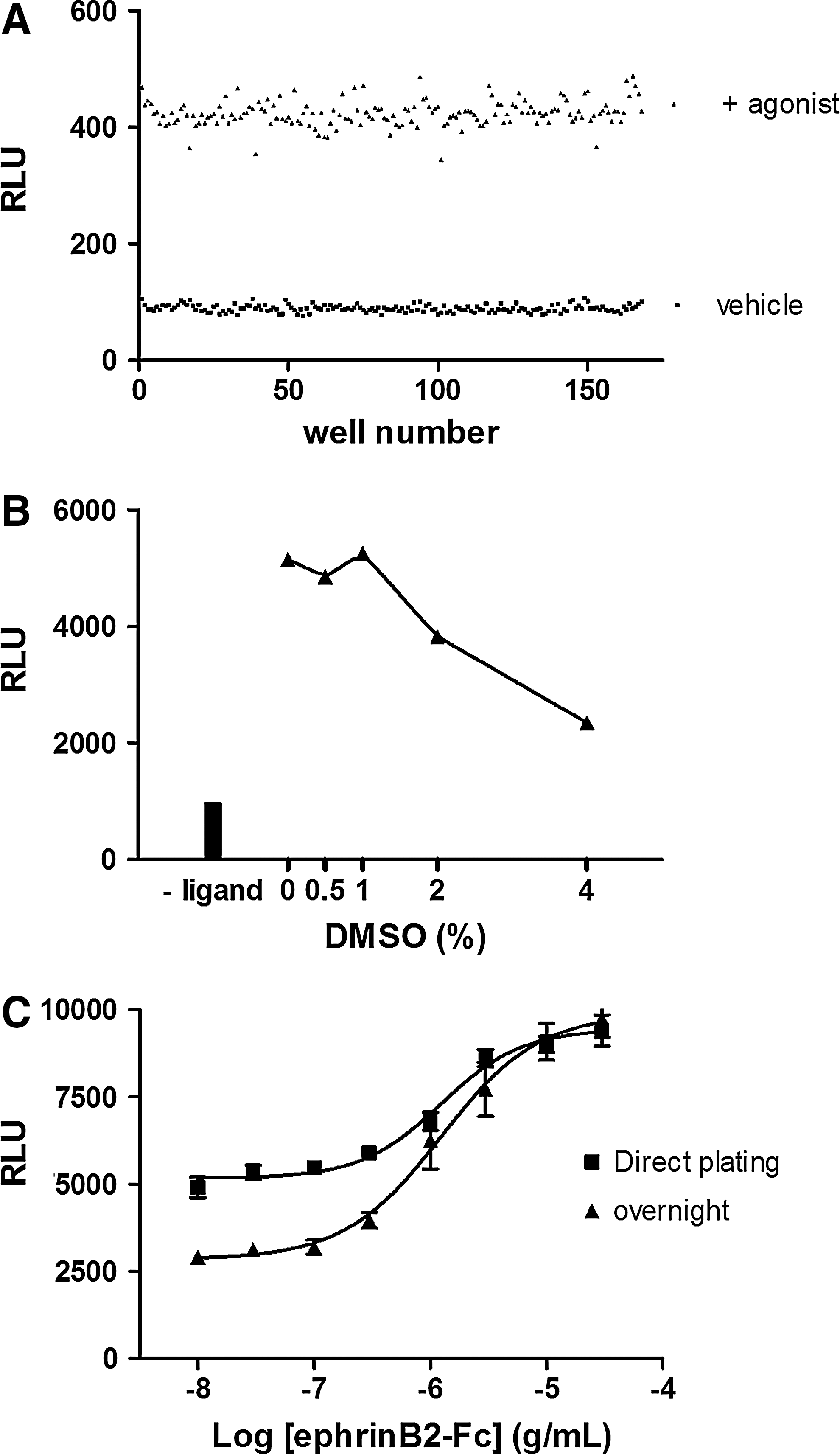
To determine the Z′ of the assay, the EphB4 clone was plated across a full 384-well plate. 168 wells were treated with vehicle (■) or with an EC80 of EphrinB2-Fc (▲). The Z′ calculation yielded a score of 0.74
EphB4 Assay Optimization for Library Screening
After initially characterizing the EphB4 assay, we proceeded to determine the compatibility of the assay with standard library screening protocols. Cell density optimization showed that there is very little difference in assay performance between 0.5, 1, and 2×104 cells per well in a 384-well plate with respect to signal to background ratio (data not shown). We selected 1×104 cells per well as our standard seeding density for the remaining experiments.
To assess the robustness of the assay, a full 384-well assay plate was tested by hand pipetting. Based on 192 wells stimulated with an EC80 of the agonist and 192 wells without stimulation, the Z′-factor was calculated to be 0.74, indicating that the EphB4 assay is a robust assay, which is capable of generating statistically significant data (Fig. 4A).
To further validate the suitability of the system for a variety of compound screening scenarios, DMSO tolerance was tested by exposing the clone to increasing amounts of DMSO with and without the ligand. DMSO concentrations up to 2% were well-tolerated with less than a 20% drop in signal (Fig. 4B), which is more than sufficient for routine screening applications. Finally, we tested the ability of the EphB4 assay to be performed directly from frozen cell stocks under two screening scenarios: direct plating and assay versus a 24-h recovery period before testing. Cryopreserved cells were thawed and either plated into a tissue culture grade assay plate for overnight incubation or directly into a nontissue culture grade plate for immediate processing. The cells were stimulated with agonist and luminescence was measured. As shown in Figure 4C, the agonist EC50 values in both approaches were comparable to the standard assay with cells from routine culture. The signal to background ratio of the frozen cell format was of high quality when cells were plated into tissue culture plates for 24 h. We observed a 50% reduction in the signal to background ratio with the direct format. These results indicate that the PathHunter assay can be used in a variety of frozen cell formats, but the optimal assay window requires overnight plating.
The sum of these experiments indicates that the PathHunter cellular kinase assay meets the performance metrics necessary for high-throughput screening. Since the biology behind the assay is the generic detection of tyrosine phosphorylation and is not specific for EphB4, we reasoned that the technology could be extended to other members of the Eph receptor family; EphB2, EphA7, and EphA5 were cloned as fusions to ProLink and coexpressed in U2OS cells containing an SH2-EA fusion (as in Fig. 1 for EphB4). The cells were tested for a response to agonist using the protocol developed for EphB4. Figure 5 shows that for each of these targets, the ligand-mediated activation of the kinase can be detected using the PathHunter technology. These results indicate that the PathHunter platform can be applied across a range of targets and since the assay conditions are similar to the EphB4 assay, we expect the assay properties that were outlined in this work to extend across the range of Eph receptors.
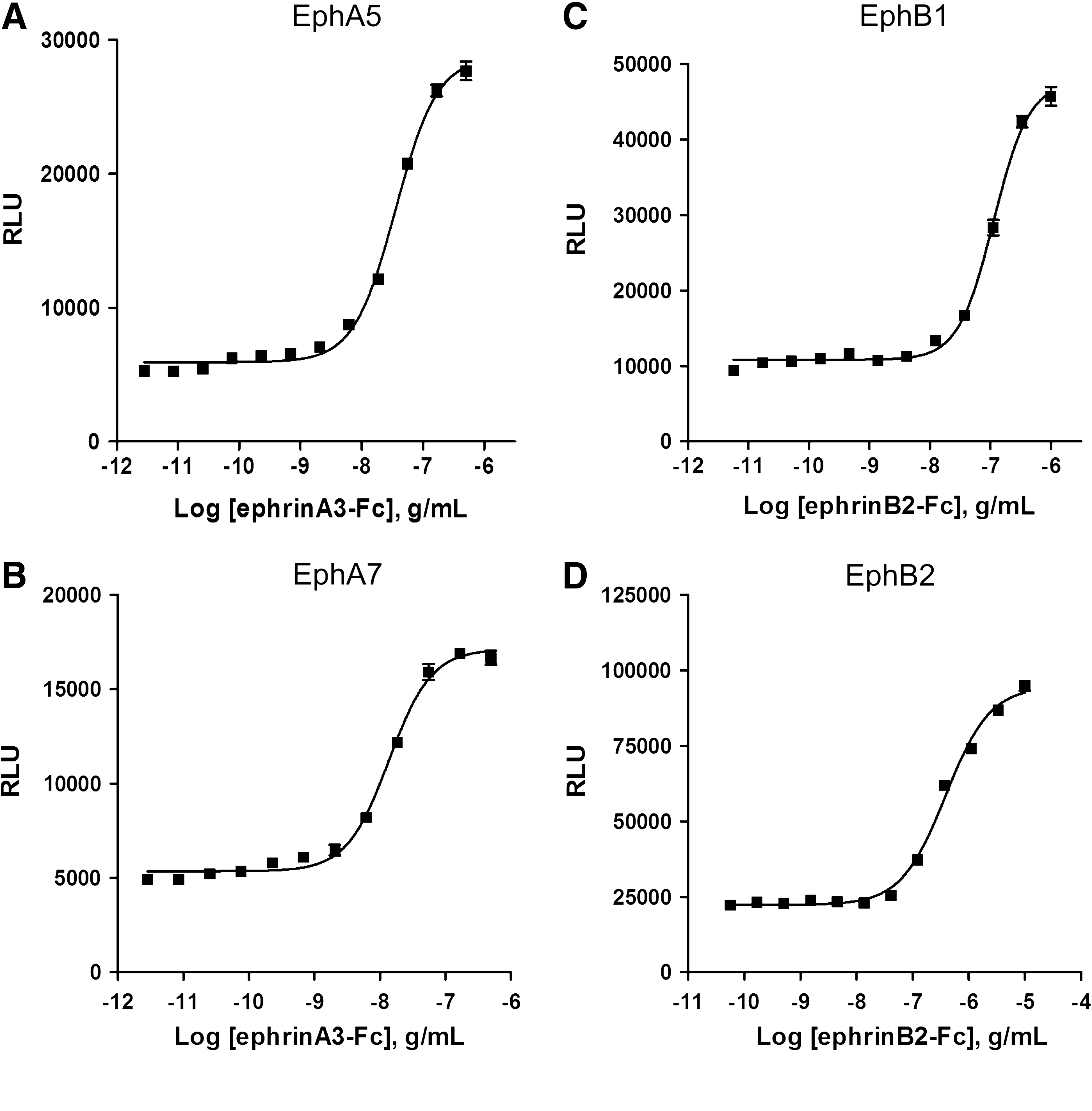
EphA5
Discussion
Cellular assays for kinase activity continue to play a major role in kinase drug discovery. As an adjunct or downstream biological activity tool, the addition of cellular hosts describes many important factors affecting compound function that cannot be obtained utilizing purified kinase reactions. These factors include membrane permeability, modulation of kinase structure due to binding of host-cell factors, and dynamic equilibrium between activated and nonactivated states. In addition, cytosolic protein, ATP, and salt concentrations, which can affect inhibitor function are difficult to recapitulate in vitro; thus, cell-based assays provide an important link between in vitro kinase inhibitor assays and functional activity in vivo.
The system described here was designed to provide a physiologically and pharmacologically relevant cell-based assay suitable for compound screening. We selected full-length kinases to maintain the protein in their natural cellular context and enable the discovery of novel inhibitors. Using the full-length EphB4, we show that inhibitory monoclonal antibodies as well as small molecule kinase inhibitors are readily detected with the expected pharmacology. The assay platform described here maintains the precision and tolerance necessary for the demands of small molecule library screening. The observed Z′ values, solvent tolerance, and ability to be run from cryopreserved stocks make this assay ideally suited for high-throughput screening applications. In addition, we show that the assay principle can be extended to a number of other Eph receptors indicating that this may be a useful platform for specificity and liability screening.
The assays described here preserve the natural, ligand-induced activation pathway, thus inhibitors that bind to the unactivated or activated form of the receptor can be detected as well as agonist or antagonist functions of therapeutic antibody candidates. Moreover, implementing cellular kinase assays as compound discovery tools provides the opportunity to find novel chemical matter in the form of ligand binding, dimerization inhibitors, and a wider range of potential allosteric modulators.
Measuring the interaction of EphB4 with downstream signaling partners provides a direct measure of kinase activity. This eliminates any signaling amplification steps that may obscure inhibitor pharmacology or lead to false positives from inhibition of downstream kinases. Direct target detection also provides a high level of assay specificity, since the two enzyme fragments must physically contact each other to generate the enzyme activity making the system ideally suited for library screening.
The advent of the PathHunter system enables cell-based, antibody-free screening of large numbers of compounds in a high-throughput screening–friendly assay. The system utilizes a stable chemiluminescent signal allowing batch processing and reducing the liabilities associated with fluorescent read-outs. The complementation reaction is wholly contained within the cells, making the detection straightforward using a single addition, mix-and-read protocol that is compatible with virtually all luminescent detection instruments. The EFC system is well established and highly scalable from 96- to 1,536-well formats providing a platform for ultra-high-throughput analysis of kinase and receptor biology.
Footnotes
Acknowledgment
The authors would like to thank Dagmar Zeggert-Springer for excellent technical assistance.
Disclosure Statement
T.W., W.F., and M.N. are employees of DiscoveRx.