
Abstract
Small ubiquitin-like modifier (SUMO) belongs to the family of ubiquitin-like proteins (Ubls) that can be reversibly conjugated to target-specific lysines on substrate proteins. Although covalently sumoylated products are readily detectible in gel-based assays, there has been little progress toward the development of robust quantitative sumoylation assay formats for the evaluation of large compound libraries. In an effort to identify inhibitors of ubiquitin carrier protein 9 (Ubc9)-dependent sumoylation, a high-throughput fluorescence polarization assay was developed, which allows detection of Lys-1201 sumoylation, corresponding to the major site of functional sumoylation within the transcriptional repressor trichorhino-phalangeal syndrome type I protein (TRPS1). A minimal hexapeptide substrate peptide, TMR-VVK1201TEK, was used in this assay format to afford high-throughput screening of the GlaxoSmithKline diversity compound collection. A total of 728 hits were confirmed but no specific noncovalent inhibitors of Ubc9 dependent trans-sumoylation were found. However, several diaminopyrimidine compounds were identified as inhibitors in the assay with IC50 values of 12.5 μM. These were further characterized to be competent substrates which were subject to sumoylation by SUMO-Ubc9 and which were competitive with the sumoylation of the TRPS1 peptide substrates.
Introduction
Ubc9 is the sole E2 carrier protein in the SUMO pathway and is distinct from ubiquitin E2 proteins in its ability to specifically recognize and promote trans-sumoylation of SUMO onto substrate lysines even in the absence of an E3 enzyme. 2 A substrate consensus sequence of Ubc9-dependent sumoylation has been identified as ΨKxE/D bearing the conserved target lysine, and where Ψ represents large hydrophobic amino acids and x is any amino acid. A surface patch on Ubc9 surrounding Cys-93 binds directly to this consensus sequence in RanGAP, p53, and c-Jun substrates. 3 –5 SUMO E3 ligases bind to both SUMO-Ubc9 and protein substrate and facilitate the transfer of SUMO from SUMO-Ubc9. 1 RanBP2, one of the few known SUMO E3 ligases, has been shown to form a stable complex with RANGAP1, SUMO-1, and Ubc9 in an optimal orientation to enhance SUMO conjugation. 6,7
The biological effects of sumoylation are diverse, impacting the functions of substrates involved in nuclear trafficking, genomic stability, chromosome integrity, and gene expression. 8 The nucleus is the main site of sumoylation where the primary targets are transcription factors and coregulators. 9 In contrast to ubiquitinylation, which regulates protein turnover, sumoylation appears to modulate protein functions directly by various mechanisms, including alteration of protein-protein interactions in transcriptional complexes, and by affecting the stability of transcription factors within these complexes. 9,10
There is emerging evidence that increases or decreases in protein sumoylation can contribute to multiple diseases, including cancer. 11 Sumoylation is known to modulate the function of certain target proteins which have previously defined roles in disease. For example, sumoylation of transcription factor 4 (TCF4) has been shown to affect β-catenin-dependent gene expression in the Wnt signaling pathway. 12 Recently, the sumoylation pathway has been identified as an essential component in nononcogene addiction of myc-driven tumorigenesis. 13
Trichorhino-phalangeal syndrome type I (TRPS1), a disease characterized by sparse and slow growing scalp hair and craniofacial and skeletal abnormalities, occurs through the dominant inheritance of mutations in the TRPS1 gene. 14 Missense and nonsense mutations in the TRPS1 C-terminal GATA4-binding domain prevent its interaction with RunX2 and Osteocalcin promoters, thereby disrupting its function as a transcriptional repressor of these genes which are involved in cartilage and bone development, respectively. 15 –17 Ubc9 enhances TRPS1 repressive effects via sumoylation at two acceptor sites (K1192 and K1201) located within the minimal C-terminal repression domain (RD) of TRPS1, which binds promoter sequences. 18 Sumoylation at these two lysines results in enhanced GATA4 promoter-driven transcription, and mutation of these two lysines to arginine abrogates TRPS1-mediated repression in transfected cells.
Since Ubc9-dependent sumoylation of TRPS1 enhances the transcriptional repression of genes involved in bone and cartilage production, a high-throughput assay was developed to identify chemical inhibitors of Ubc9-directed sumoylation for potential therapeutic application in diseases associated with cartilage and bone loss. An optimized fluorescence polarization (FP) assay was developed that monitored the conjugation of SUMO-1 to a hexapeptide substrate bearing the Lys-1201-containing sumoylation site within TRPS1. This assay was run in a coupled-enzyme format with E1 (Aos1/Uba2), E2 (Ubc9), and SUMO-1 (Eqs. 1 –4) to screen the GlaxoSmithKline (GSK) high-throughput screening (HTS) chemical library. Compounds exhibiting adventitious reactivity toward thiols or which destabilized Ubc9 accounted for most of the validated inhibitors that were found. Other than compounds acting through these undesirable mechanisms, only one type of specific SUMO-Ubc9 inhibitor was identified, which belonged to a class of L-ornithine–containing diaminopyrimidines that acted as alternative substrates which were functional to produce adducts of SUMO-1.
Materials and Methods
The sumoylation control kit was obtained from LAE Biotech International (kit K007). pDonor221, pDEST8, BP/LR clonase, horseradish peroxidase (HRP) streptavidin (SA), and BL21 (DE3) star competent cells were purchased from Life Technologies. Ni-NTA agarose, DNA purification, and agarose gel extraction kits were obtained from Qiagen. QuikChange Site-directed mutagenesis kit was from Agilent. Bovine serum albumin (BSA; fatty-acid–free) was from Boehringer Mannheim laboratories (100 069). Isopropyl β-D-1-thiogalactopyranoside (IPTG) was from Roche. Enhanced chemiluminescent Western blotting substrate was from Thermo Scientific. X-OMAT film was from Kodak. Synthetic genes were purchased from GenScript, Inc. Custom oligonucleotides were purchased from Integrated DNA Technologies, Inc. Custom syntheses of N-terminally tetramethylrhodamine-labeled (TMR), N-terminally biotin labeled, and unmodified peptides were purchased from 21st Century Biochemicals. Ultrapure ATP (10 mM solution) and the AMP-Glo assay kit was from Promega. Tween-20 (10% solution) was from Teknova. 0.5 M Tris[2-carboxyethyl] phosphine (TCEP) buffer neutralized solution was purchased from Thermo Scientific. Formic acid and acetonitrile (ACN) were purchased from Burdick and Jackson. 7-Diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (CPM) was purchased from Molecular Probes. Dapoxyl sulfonic acid was purchased from Life Technologies. N-acetyl cysteine was from Sigma Aldrich. All other reagents were obtained from Sigma.
Expression Construct Generation
The DNA sequences encoding Aos1/SAE1 (NP_005491), Uba2/SAE2 (NP_005490), Ubc9 (NP_003336), SUMO-1 (NP_003343), RAN BP IR1-M (from NP_006258), and TRPS1 RD (from NP_054831) were synthesized with optimized codon preferences for Escherichia coli expression (Supplementary Figs. S1—S7; Supplementary Data are available online at
The coding sequence of Aos1 (M1-K346) was synthesized with 5′ NcoI and 3′ NotI restriction sites in which a 3′ sequence encoding a Strep tag and Tobacco Etch Virus protease (TEV) cleavage site (WSHPQFEKENLYFQG) was fused directly to the Aos1 sequence. The resulting Nco1/Not1 cassette was isolated from a pUC57 holding vector, and subcloned into the corresponding sites in pAC28. 19
Uba2 (A2-D640), Ubc9 (S2-S158), SUMO-1 (S2-G97), and RANBP-IR1-M (L2633-I2711) coding sequences were synthesized in the same format with flanking attB1/attB2 Gateway™ recombination sites. A 5′-sequence encoding a TEV cleavage site (ENLYFQ/G) was fused directly to the second codon of each gene. The resulting constructs were subsequently transferred by attB x attP recombination to pDonor221 and by attL x attR recombination to N-terminal tag vectors derived from pET22b, in which the pelB leader sequence is replaced with in-frame tag modules. Uba2 was transferred to the pDESTT7His vector to generate an in frame fusion of a hexahistidine tag through the N-terminal Gateway sequence (MGHHHHHHTS TSLYKKAGFENLYFQG). Ubc9 was transferred to pDESTT7FlagHis to generate an in-frame fusion encoding a tandem Flag, hexahistidine tag followed by a TEV cleavage site (MGDYKDDDDKGHHHHHHTSTS LYKKAGFENLYFQG). SUMO-1 and RanBP IR1-M were both transferred to pDESTT7HisGST to generate an in frame fusion encoding a tandem hexahistidine-GST tag.
The sequence encoding codon-optimized Ubc9, preceded by an in-frame TEV tag, was mutated using the QuikChange Site-Directed Mutagenesis Kit within the pDonor 221 vector to create a single base change to provide the C93S point mutation. The following primers were used for this reaction: Forward, 5′-CAA ATG TAT ACC CGA GTG GGA CTG Taa GCC TGT CTA TTC TTG AAG AAG ATA AG-3′; Reverse, 5′-CTT ATC TTC TTC AAG AAT AGA CAG GCt TAC AGT CCC ACT CGG GTA TAC ATT TG-3′.
The resulting clone was subsequently transferred to expression backbones pDESTT7FlagHis and pFNcmvHA using LR Clonase as per manufacturer's directions. All clones were sequenced on both strands to confirm the full coding sequence, including inserted mutation.
TRPS1 RD (G1192- E1294) was synthesized with a 3′-extension encoding a C-terminal tandem Flag and hexahistidine dual tag (GDYKDDDDKGHHHHHH) fused directly to the E1294. The resulting cassette was transferred by attB x attP recombination to pDonor221 and by attL x attR recombination to pDEST8 baculovirus expression vector.
Protein Expression and Purification
Aos1/Uba2, Ubc9, SUMO-1, and RANBP IR1-M were individually expressed in E. coli BL21 star™ (DE3) cells in Superbroth plus 1× Vogel-Bonner salts. Overnight cultures (1 L) grown at 37°C in Luria-Bertani Broth (LB) plus 1% (w/v) glucose were pelleted and used as inoculum for 20-L scale production. Cells were induced mid-log phase with 0.5 mM IPTG. Fermentations were carried out in a 30 L Applikon Stirred Tank Reactor (20 L working volume, Applikon Biotechnology) stainless steel vessel with water-jacketed temperature control. Dissolved oxygen was monitored at a set point of 40%. Cell paste was recovered through centrifugation using a CARR Powerfuge (20,000 g).
Aos1/Uba2 heterodimer
pAC28Strep-Aos1 and pDESTT7His-Uba2 vectors were cotransformed for coexpression of Aos1 and Uba2 at 28°C overnight (pH 6.5) under antibiotic selection (100 μg/mL carbenicillin, 50 μg/mL kanamycin). The StrepAOS1/HisUba2 heterodimer was purified by nickel NTA Superflow followed by Superdex 200 size exclusion chromatography (SEC). Cell paste (160 g) from the equivalent of 6.8 L cell culture was lysed in 1.2 L lysis buffer (50 mM Tris-HCl, pH8.0, 0.5 M NaCl, 20% glycerol, 1 mM TCEP, 15 mM Imidazole, 1% Triton X-100, and complete proteinase inhibitor (Roche), 50 mL/tab at 7.5 mL/g of cells) using a microfluidizer (Avestin Emulsiflex-C50 at 100,000 kPa for three passes). Protein was purified by chromatography on Ni-NTA Superflow (50 mL) and eluted with 300 mM imidazole followed by SEC (1,800 mL, 5 cm×92 cm Superdex 200 column) at 1.5 mL/min. The elution volume was 132 mL. Fractions were pooled and stored at 2.2 mg/mL in storage buffer A (20 mM Tris-HCl pH8.0, 20% v/v glycerol, 75 mM NaCl, and 1 mM TCEP). Protein concentration was measured spectrophotometrically at A280 nm using an average extinction coefficient of 79,300 M−1·cm−1. The final yield was 0.5 mg/g of cell paste, 38.8 mg/L of cell culture at 70% purity (Supplementary Fig. S8).
Ubc9
pDEST T7FlagHis-Ubc9 was transformed for expression at 28°C for 20 h (pH 6.5) under antibiotic selection (100 μg/mL carbenicillin). FlagHisUbc9 was purified by nickel NTA and SEC. 395 g FlagHisUbc9 E. coli cell paste from 17.7 L cell culture was lysed in 3 L lysis buffer using microfluidizer (three passes at 100,000 kPa). Protein was bound to a 120 mL Ni-NTA Superflow column (XK50; 5 cm×6 cm), washed with lysis buffer (20 column volumes), and on- column tag cleavage was carried out by incubating with 1:7 molar ratio TEV protease at 4°C for 18 h. Detagged protein isolated from the Ni-unbound fraction was further purified by SEC (1,000 mL Superdex 200) into storage buffer A. Final concentration was 5.3 mg/mL with 95% purity. Yield was 2.5 mg/g of cell, 55 mg/L of cell culture (Supplementary Figs. S9 and S10).
To biotinylate Ubc9, protein was desalted into 20 mM HEPES, pH7.5, 20% (v/v) glycerol, 0.5 mM TCEP via chromatography on sephadex G25, and biotinylated at a 20:1 molar ratio of biotin:protein at 4°C overnight using EZ-Link Sulfo-NHS-LC-LC-Biotin (Pierce #21338), followed by a desalting step into the final storage buffer. An average of 5–8 lysine residues out of 14 total lysines were biotinylated as determined by liquid chromatography/mass spectroscopy (LC/MS; data not shown).
Mature SUMO-1
pDESTT7GST-Sumo1GlyGly was transformed for expression at 18°C for 20 h (pH 6.5) under antibiotic selection (100 μg/mL carbenicillin). HisGSTSUMO-1 (mature form) was purified by successive chromatography using nickel NTA Superflow followed by Superdex 200 size exclusion. 138 g HisGSTSumo-1 (mature form) E. coli cell paste from 5 L cell culture was lysed in 1 L lysis buffer at 7.5 mL/g using a microfluidizer (three passes at 100,000 kPa). Protein was purified by chromatography using Ni-NTA Superflow (100 mL) eluting with 300 mM imidazole, and followed by SEC (1,800 mL). Fractions were pooled and stored at 10 mg/mL in storage buffer B (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 20% v/v glycerol, 0.5 mM TCEP). The yield was 22 mg/g of cell, 600 mg/L of cell culture at 70% purity (Supplementary Fig. S11).
To prepare detagged SUMO-1, the HisGST tag was cleaved with 1:10 molar ratio (TEV protease: HisGSTSUMO-1), for 18 h at 4°C during dialysis into lysis buffer. The TEV treated sample was subsequently bound to a column of Ni-NTA, and the detagged SUMO-1 was eluted in the unbound fraction and further purified by SEC into the same final storage used for HisGSTSUMO-1 (Supplementary Fig. S12).
Preparation of RANBP IR1-M
The pDESTT7 HisGSTRanBP IR1-M construct was coexpressed with DNAK, DNAJ, and GroEL/GroES chaperone proteins at 15°C for 18 h under antibiotic selection (carbenicillin (100 μg/mL), chloramphenicol (50 μg/mL) at pH 7.0. Before induction, cells were grown to an OD600 of 1.5 at 37°C. Temperature was lowered to 30°C, and 0.2% arabinose was added to induce DnaK/DnaJ chaperone protein for 1 h. Temperature was then lowered to 15°C, and samples were treated with 0.5 mM IPTG for induction of RANBP IRM1 and 80 ng/mL tetracycline for induction of GroES and GroEL chaperone proteins.
HisGSTRanBP IR1-M was purified by nickel NTA and SEC. 70 g HisGSTRanBP IR1-M E. coli cell paste from a 4.5-L cell culture was lysed in 500 mL lysis buffer using sonication (Branson Sonifier, level 6, output 40%, 50% duty cycle, 30 s on/30 s off for 8 min in total). Protein was purified by chromatography using Ni- NTA (15 mL) and 1 L SEC into storage buffer C (20 mM Tris-HCl, pH8.0, 0.15 M NaCl, 20% v/v glycerol, 1 mM TCEP). The final yield was 1.18 mg/g of cell paste, 18.4 mg/L of cell culture at 50% purity (Supplementary Fig. S13).
TRPS1 was expressed in baculovirus-infected Spodoptera frugiperda (Sf9) cells using the bac-to-bac system (Life Technologies). Baculovirus-infected insect cells (BIIC) were harvested and stored frozen in liquid nitrogen as described. 20 Sf9 cells were seeded into the shake flask at a density ∼0.6×106cells/mL at 110 rpm. After 16 h, cells were infected with 1×106 BIIC cells for each liter of cell culture. The infected cells were cultured at 27°C and harvested after 66 h. C-terminally FlagHis-tagged TRPS1 RD was purified by chromatography using nickel NTA Superflow followed by Superdex 200 SEC. 16 g C-terminally FlagHis tagged TRPS1 RD from 2L-baculovirus-infected Sf9 cell culture was lysed in 80 mL lysis buffer using sonication (Branson Sonifier at level 6, output 40%, 50% duty cycle, 30 s on/30 s off for total 8 min). Protein was purified by chromatography using Ni- NTA Superflow followed by Superdex 75 SEC into storage buffer C. Final yield was 3.75 mg/g of cell paste, 30 mg/L of cell culture at 95% purity (Supplementary Fig. S14).
Assays
In-gel detection of SUMO-1 conjugation to biotinylated peptides
Sumoylation reactions (2 h at ambient temperature) carried out in reaction buffer containing 20 mM HEPES pH 7.5, 5 mM MgCl2, 2 mM ATP in the presence of 1 μM biotinylated peptides were resolved by 4–12% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) gels, 1× MES buffer (50 mM MES, 50 mM Tris, 0.1% SDS 1 mM EDTA, pH 7.3. Gels were transferred to 2 μm nitrocellulose (1 h at 100 volts), and Western detection was performed with HRP-SA. Blots were blocked for 1 h in PBS containing 5% BSA, 0.05% Tween-20, and then probed with HRP-SA at 1:1500 in PBS plus 0.5% BSA, 0.05% Tween-20. Blots were washed in PBS plus 0.05% Tween-20, and treated with enhanced chemiluminescent Western blotting substrate for detection on x-ray film.
FP assay using untagged and GST-tagged SUMO-1
Development and optimization of assay formats measuring sumoylation were evaluated in 384-well black low volume assay plates (Greiner, 784076); HTS reactions were evaluated in 1,536-well black low volume assay plates (Greiner 782076). All reactions were evaluated at ambient temperature (22°C±2°C) in assay buffer solution containing 50 mM HEPES (pH 7.5), 10 mM MgCl2, 0.1 mM TCEP, 0.01% Tween-20. The Molecular Devices Analyst GT fluorimeter was used for primary assay development, characterization of reagents, and reaction kinetics. FP was monitored with excitation/emission filters 530/580 nm, and a 561 nm dichroic mirror. High-throughput screening and profiling was monitored as endpoint reactions on the Perkin Elmer Envision, utilizing a BODIPY TMR FP 531 nm excitation filter (barcode 105), Bodipy TMR FP P-pol 595 nm/BODIPY TMR FP S-pol 595 nm emission filters (barcodes 211 and 210, respectively), and a BODIPY TMP FP Dual dichroic mirror (barcode 482).
For assay development and kinetic evaluations, 5 μL of a 2× substrate-containing reaction mixture comprised of combinations of ATP, SUMO-1, peptide, and other additives, was dispensed to reaction wells. Sumoylation reactions were initiated with a 5 μL addition of a 2× enzyme mixture of E1 and Ubc9. In general, reaction plates were centrifuged at 500 rpm for 30 s, after each reagent addition. Kinetic reactions were monitored at 2 min intervals for up to 2 h. Reaction rates, assessed as the change in FP as a function of time, were obtained from the linear portion of reaction progress profiles.
HTS assay and data analysis
Assay robustness and validation studies were conducted to miniaturize the Ubc9 sumoylation assay, to ensure compatibility with 1,536-well assay formatting and tolerance to production-scale liquid handling (Multidrop Combi, Thermo Scientific). HTS assay-ready plates were prepared by dispensing 50 nL of either DMSO or 1 mM compound solution in DMSO using acoustic dispensing technology (Echo model 555, Labcyte). Equal volumes (2.5 μL) of stock reagents were dispensed for negative-control samples (-ATP), protein (E1, GST-SUMO-1, Ubc9, TMR-peptide), and ATP solutions with a Multi-drop Combi liquid handler, to provide a 100-fold dilution of compound into a 5 μL assay reaction mixture (10 μM final compound concentration). Final reaction conditions included 162 nM E1, 400 nM Ubc9, 5 μM detagged SUMO-1, 1 μM TMR-VVKTEK peptide, 100 μM ATP, 50 mM Hepes (pH 7.5), 10 mM MgCl2, 0.01% Tween-20, 0.1 mM TCEP. Reactions were run to ∼80% completion to obtain a larger S/B and improved assay robustness, and were quenched by the addition of TFA to a final concentration of 1% v/v (1 μL addition to 384 well reaction plates, 0.5 μL addition to 1536 well HTS plates). The FP signal was stable for 5 h after reactions were quenched. See Table 1 for additional details. An average Z′ factor was calculated using Equation 5 from 229,888 wells from 1,796 plates with an average S/B of 3.5.
Ubc9 Fluorescent Polarization High-Throughput Screening Assay Protocol
1. Compound or DMSO dispensed using acoustic liquid handling (Labcyte Echo 555).
2. Assay buffer=50 mM HEPES (pH 7.5), 10 mM MgCl2, 0.01% Tween-20, and 0.1 mM TCEP. 2× concentrations are: [E1]=324 nM, [Ubc9]=800 nM, [SUMO-1]=10 μM, [TMR-VVKTEK]=2 μM.
3. 2× [ATP]=200 μM.
4. Using extreme caution, dilute TFA to a final concentration of 10% (v/v) in water.
5–7. All reagents are dispensed using the multidrop combi (Thermo) equipped with a small tube metal tip cassette. Low control signal is generated by leaving ATP out of the reaction (columns 35 and 36). High control signal is generated by leaving compound out of the reaction (columns 11 and 12).
10. The assay signal was stable for at least 5 h after quench was added.
11. FP signal was read on an Envision (Perkin Elmer) using a BODIPY TMR FP 531 nm excitation filter (barcode 105), BODIPY TMR FP P-pol 595 nm/BODIPY TMR FP S-pol 595 nm emission filters (barcodes 210 and 211), and a BODIPY TMR FP Dual dichroic mirror (barcode 482).
Ubc9, ubiquitin carrier protein 9; HTS, high-throughput screening; TMR, tetramethylrhodamine; TCEP, Tris[2-carboxyethyl] phosphine; FP, fluorescence polarization.
Michaelis-Menton kinetics of the E1-catalyzed reaction with variable MgATP were determined by fitting initial velocity data to Equation 6. Inhibition of ATP analogs or unlabeled peptides was obtained by fitting initial velocity data from inhibitor titrations to an IC50 template (GraFit; Erithacus Software Limited; Version 5.0.12). IC50 evaluations for HTS hits were obtained using AbaseXE's full curve analysis bundle (Eq. 7). Hit population analysis and visualization, activity weighted diversity selection
21
and inhibition frequency index (IFI; defined as the relative frequency with which a compound has scored >50% inhibition in multiple HTS assays)
22
were conducted using Spotfire® DecisionSite™ (Tibco Software).
AMP-Glo assay
AMP formation from the E1-catalyzed reaction was quantified by the use of the Amp-Glo assay, using instructions provided by the assay kit. Optimized reaction conditions for sumoylation (162 nM E1, 400 nM Ubc9, 5 μM GST-SUMO-1, 1 μM TMR-VVKTEK, 50 mM HEPES [pH 7.5], 10 mM MgCl2, 100 μM ATP, 0.01% Tween, 0.1 mM TCEP, containing either DMSO or compound suspended in DMSO [1% final]) were evaluated in 5 μL reaction mixtures over 2 h at ambient temperatures. Reactions were quenched with a 5 μL addition of Reagent 1 and allowed to incubate for 1 h. The luminescence signal was developed for 1 h after a 10 μL addition of the AMP detection reagent (Reagent 2) The AMP-Glo kit Reagent 1 removes endogenously added ATP; addition of Reagent 2 provides the luminescence glo signal via proprietary technology through the recycling of AMP produced in the primary reaction back to ATP, which is utilized by a luciferase: luciferin coupled reaction. The resulting luminescence signal was detected with a Perkin Elmer Viewlux plate imager, using a 613/55 nm emission filter.
Compound susceptibility to reaction with thiols
Compounds from the HTS screen were evaluated for their reactivity toward the free thiol on N-acetyl cysteine, as assessed by a simple high-throughput fluorescence-based assay (Supplementary Fig. S15). Briefly, compounds were allowed to react for 15 min with N-acetyl cysteine, after which the remaining unreacted free thiol was quantified using CPM, a pro-fluorescent thiol reactive dye. Assay plates (Greiner 784076) were prepared containing 100 nL of serially diluted compounds in DMSO (200 μM maximal concentration, 11-point curve, 1:3 dilution scheme). Two solutions were prepared for the assay: (1) 10 μM N-acetyl cysteine mixed in reaction buffer (50 mM Hepes pH 7.5, 10 mM MgCl2, 0.01% Tween-20), and (2) 10 μM CPM mixed in reaction buffer (protected from light). First, 5 μL of N-acetyl cysteine was dispensed to the plate using a Multidrop Combi (Thermo Scientific). The reaction with compound was allowed to proceed for 15 min at ambient temperature. Next, 5 μL CPM was dispensed to the plate and allowed to react with the remaining free thiol for 15 min at room temp. Finally, fluorescence was quantified using an Acquest plate reader (Molecular Devices) at an excitation wavelength of 360 nm and an emission wavelength of 460 nm, using a 400 nm dichroic mirror. High signal control wells contained 100 nL of DMSO, N-acetyl cysteine, and CPM; whereas low signal control wells contained 100 nL DMSO, buffer in place of N-acetyl cysteine, and CPM. High and low control samples were used to normalize the data between 0% and 100% thiol consumed. A parallel compound interference reaction was run whereby the N-acetyl cysteine was premixed with CPM for 15 min at room temp (in the absence of any compounds) followed by dispensing the mixture to the plate and recording the resultant fluorescent signal. Data for the test and interference assays was superimposed and only those compounds which led to consumption of free thiol and did not optically interfere with the detection were deemed thiol reactive.
Thermal shift assay
The thermal stability of protein in the presence or absence of titrated compounds was analyzed as a thermal shift assay (TSA) using a LightScanner instrument (Idaho Technology, Inc.). The readout from a TSA is the melting temperature of the protein (T m), which is defined as the midpoint of the transition from the folded to the unfolded state of the protein as measured by changes in dapoxyl fluorescence. Assay plates (HSP3865 from Bio-Rad) were prepared containing 100 nL of serially diluted compounds in DMSO (333 μM final top concentration, 11-point curve, 1:3 dilution scheme). A solution containing 5 μM Ubc9 and 15 μM dapoxyl sulfonic acid mixed in TSA buffer (50 mM HEPES pH 7.5, 10 mM MgCl2, 0.01% Tween-20, and 100 μM TCEP [77720 from Pierce]) was prepared, and 3 μL of the solution was added across the plate using a multichannel pipette. The assay solution was then overlaid with 1 μL of silicone oil DC200 to prevent evaporation and immediately analyzed. The change in fluorescence due to protein unfolding was monitored with the excitation wavelength set to 380 nm and the emission wavelength at >435 nm over a temperature range of 35°C–85°C at a ramp rate of 1°C/min. Secondary plots (T m vs. [compound]) were created and qualitatively examined for deviations in protein T m in response to increasing compound concentrations. Compounds were then binned according to their effect on T m: either no effect, increase (protein stabilizer), or decrease (protein destabilizer).
LC/MS to assess the sumoylation small molecules
An LC/MS-based assay was developed to assess the substrate potential of small molecules which would exhibit competitive behavior with the TRPS1 peptide (VVKTEK). Compounds were incubated in sumoylation reaction mixtures in the absence of the TRPS1 peptide substrate, and resulting covalent SUMO-1 adducts were detected by LC/MS. Briefly, 5 μM detagged SUMO-1 was mixed with 0.15 μM E1, 0.5 μM Ubc9, and 2.5 μM small molecule in assay buffer (50 mM HEPES pH 7.5, 10 mM MgCl2, 0.01% Tween-20, and 100 μM TCEP). Assays were initiated with the addition of 150 μM ATP followed by incubation at 37°C for 3 h and overnight incubation at 4°C, before analysis by LC/MS. For each compound, an ATP-negative control was included to demonstrate dependence on the enzymatic pathway. For LC/MS detection, a 40 μL reaction mixture prepared as described above was mixed with 60 μL of LC/MS buffer (0.1% TFA in H2O) and a 50 μL sample was analyzed. A Poros R2/10 2.1 mm×30 mm column was used with a flow rate 1.25 mL/min. After the column, the flow stream was split 1:1 to lower the amount and rate of material entering the mass spectrometer, optimizing the ionization spray characteristics. The APoros column was equilibrated in 95% (0.1% formic acid in ACN:H20) for 1 min, followed by a gradient of 5–95% of these solvents over the subsequent 2 min and followed by further elution for 0.5 min. The column was then equilibrated to 5%:95% of 0.1% formic acid in ACN:H20.
Prediction of the binding modes of the diaminopyrimidines
The conformations of the diaminopyrimidine compounds were aligned to the crystallographic conformation of L523-KS-E526 of RanGAP1 (the recognition motif on RanGAP1 by Ubc9) which is shown in the crystal structure of the SUMO-RanGAP1-Ubc9-RanBP2 complex (PDB code: 1Z5S). The Flexible Alignment module in MOE (Molecular Operating Environment software package version 2011, Chemical Computing Group), was used to carry out the alignment calculations. MMFF94 was selected as the force field. The conformation of L523-KS-E526 was held fixed during the calculations, and the diaminopyrimidines were flexibly aligned onto the conformation of L523-KS-E526. Default settings of the Flexible Alignment module were used.
Results
Validation of Sumoylation Substrates
We wished to develop a high-throughput assay for the identification of inhibitors of the Ubc9-dependent sumoylation of target proteins, such as TRPS1. Four peptides comprising the sequences of known sumoylation sites in p53, TRPS1, and TCF4 transcription factors were evaluated as substrates for assay development. 12,18,23 For ease of detection, each of the peptides was synthesized with an N-terminal biotin group, designated Btn-peptides 1 to 4 for p53 K536, TRPS1 K1201, TRPS1 K1192, and TCF4 K297, respectively (Table 2). Initial characterization of these biotinylated peptide substrates was performed by analyzing the products of coupled enzyme reactions containing purified E1 (Aos1/Uba2), E2 (Ubc9) and SUMO-1 (Fig. 1A). The covalent attachment of SUMO-1 or GST-SUMO-1 to the biotin peptide substrates resulted in 14 and 36 kDa products, respectively, that were resolved by SDS-PAGE and detected by SA-HRP binding to the sumoylated biotin-peptide products after Western blot transfer to a nitrocellulose membrane. Unreacted peptides (each <2 kDa) were detectable near the leading edge of the gel blot in both the presence or absence of SUMO-1 addition indicating that substrate was still available in the reactions at the completion of the 2 h reactions, although the extent of conversion could not be accurately quantified (data not shown). As shown in Figure 1A, each of the four peptide substrates functioned as SUMO-1 acceptors in the assay. As expected, no biotin peptide conjugation was observed in the absence of SUMO-1 (Fig. 1A-i). Conjugation occurred using either untagged or N-terminally GST-tagged SUMO-1 (Fig. 1A-ii, iii, respectively). A 98 kDa product was also observed with each of the peptide substrates using GST-SUMO-1, the size of which is consistent with product conjugated with two molecules of GST-SUMO-1 (Fig. 1A-iii, B). The sensitivity of this format enabled peptide sumoylation detection with as little as 0.2 μM peptide (Fig. 1B). Although a slight preference for TRPS1 peptide 2 was observed using untagged SUMO-1 as evidenced by increased band intensity (Fig. 1A-ii), all four peptides served as suitable substrates in the assay.

Gel-based demonstration of E3-independent sumoylation of peptide substrates by the coupled E1:Ubc9 assay format. Synthetic N-acetyl biotin peptide substrates based on known sumoylation sites in p53 (K356), TRPS1 (K1192 and K1201), and TCF4 (K297) were evaluated in an in vitro sumoylation assay (see
Materials and Methods
, Assays; see Table 2 for peptide sequence and identification).
Synthetic Peptide Substrates of Sumoylation
Target lysines of sumoylation are underlined and bolded within each peptide sequence.
FP Assay Configuration
Several formats were considered for the development of a high-throughput assay. Initial attempts to develop a time resolved Forster resonance energy transfer (FRET)–based assay using Cy5 labeled substrate in conjunction with Europium–anti-GST labeled GST-SUMO-1 failed to yield a quantifiable signal using each of the four Cy5-labeled peptides. Subsequently, an FP assay was configured using TMR-labeled peptides for detection of peptide substrate sumoylation with designated TMR-peptides 1 to 4 for p53 K536, TRPS1 K1201, TRPS1 K1192, and TCF4 K297, respectively (Table 2). This assay was configured in a coupled-enzyme format with E1 (Aos1/Uba2) and E2 (Ubc9) in the absence of E3 (Fig. 2A). The Ubc9-catalyzed transfer of the SUMO-1 protein from SUMO-Ubc9 onto the fluorescent peptide substrate resulted in a FP signal increase due to the greatly increased mass of the fluorescent SUMO-peptide conjugate (Fig. 2B). Using this format, only TMR-peptide 2 (Table 2) was found to yield a signal window suitable for assay development. Assay optimization was, thus, carried out with the TMR-labeled 14mer peptide 2, which is based on the sequence of the sumoylation site of TRPS1 S5 which includes Lys-1201. 18
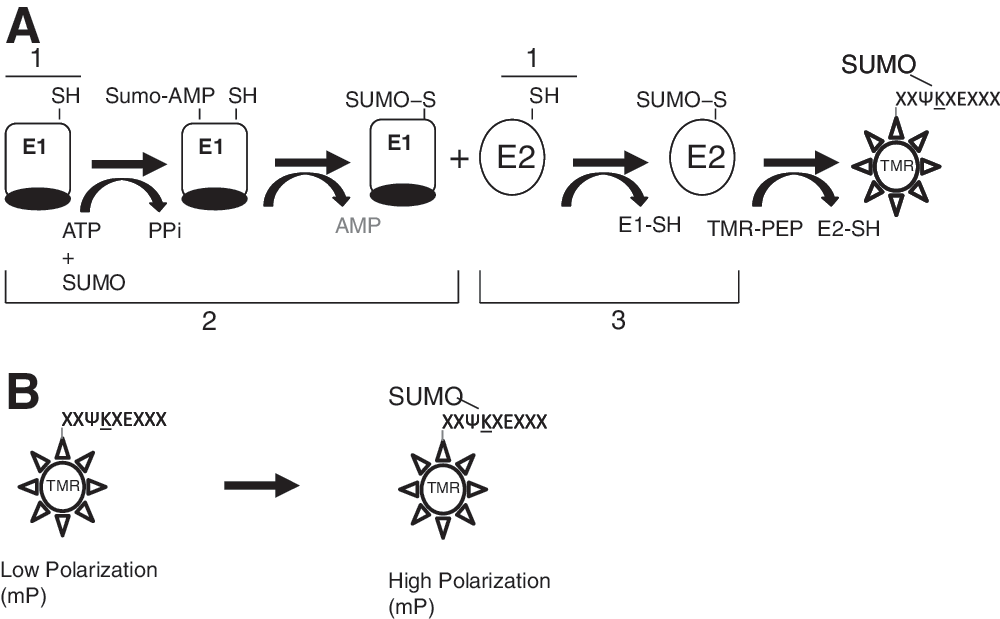
Schematic of an E2:Ubc9 coupled fluorescence polarization assay. Depiction of the coupled sumoylation, reaction, including purified E1 (Aos1/Uba2), E2 (Ubc9), SUMO-1, and TAMRA-labeled peptide designed to monitor the formation of sumoylated peptide.
Assay conditions were initially optimized by varying the concentrations of ATP, E1 (Aos1/Uba2), and E2 (Ubc9) using a fixed concentration of TMR-peptide 2 (1 μM). Reaction component concentrations were plotted against reaction rates calculated for each concentration (Fig. 3A–C). The nonlinearity of rate curves for both E1 and ATP reflects saturation kinetics in contrast to the linear Ubc9 rate curve which reflects nonsaturating kinetics up to 1.6 μM Ubc9. Concentrations of 2 μM MgATP, 162 nM E1, and 400 nM Ubc9 were determined to yield an optimal signal which was linear over a time course of 2 h. Under these conditions, the Ubc9 transfer of SUMO onto the peptide acceptor represents the rate limiting step of this reaction pathway in the presence of saturating E1 concentrations.

E1:Ubc9 coupled FP reagent optimization.
To further establish that the FP signal was dependent on functional Ubc9, the active site Cys-93 of Ubc9, the residue which forms a thioester bond to the SUMO-1 protein, was mutated to serine, and the mutant was evaluated in comparison to wild-type Ubc9 (Fig. 3D). As expected, no activity was observed using the mutant Ubc9, indicating that trans-sumoylation of the peptide was specifically dependent upon SUMO-1 thioester conjugation to Cys-93. The use of GST-SUMO-1 instead of SUMO-1 resulted in a 1.6-fold increase in FP (Fig. 3E). The reaction rates were intentionally accelerated with the inclusion of 1.2 μM Ubc9 (∼3× the assay optimized concentration) to best visualize the ΔmP differential due to the molecular weight differences between the two SUMO species. Although the rate of peptide sumoylation was greater for GST-SUMO-1 compared with untagged SUMO-1 (3.37 and 2.51 mP units/min, respectively), the reaction end points and total mP change observed differed considerably (ΔmP of 175 and 130 for GST-SUMO-1 and untagged SUMO-1, respectively), which reflects on the effect of the larger hydrodynamic radius of GST on molecular tumbling, yielding larger polarization. The GST tag itself had no effect on the binding affinity for Ubc9 as determined by isothermal titration calorimetry (K d=2 μM for both GST-SUMO-1 and detagged SUMO-1; data not shown). The 5 min lag in the FP signal observed in Figure 3E is likely due to the need to build up steady state levels of E1-SUMO followed by SUMO-Ubc9.
Optimization of Substrate Specificity
The observed substrate preference for the TRPS1 K1201 target sequence in TMR-peptide 2 (V
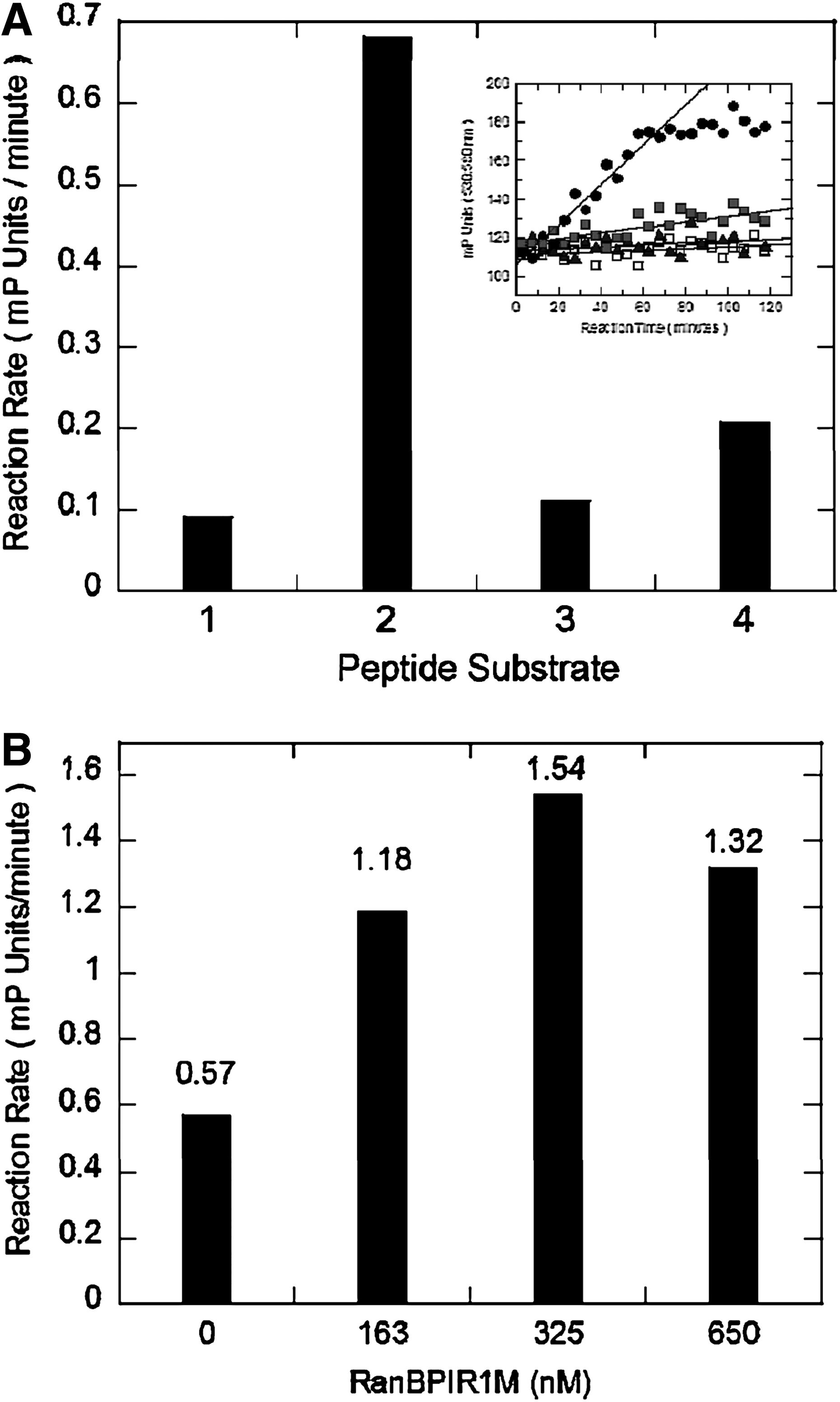
initial assessment of peptide substrate sumoylation preferences.
Substrate selectivity was further explored by competition experiments with each of the four TMR-labeled peptides in the FP assay under optimized conditions. Btn-peptides 1–4 used in the gel-based assay in Figure 1 and corresponding N-terminally acetylated peptides, designated Ac-peptides 1–4 (Table 2), were titrated into the assay to a top concentration of 2 mM (Supplementary Fig. S16). As summarized in Table 3, Btn-TRPS1 peptide 2 and Ac-TRPS1 peptide 2 were able to compete in the assay with apparent IC50s of 14 and 16 μM, respectively. Btn- and Ac-peptides 1, 3, and 4 were also able to compete in this assay but to a much lesser extent (∼10–100-fold weaker). These results indicated a relative substrate preference of peptides based on TRPS1 K1201 > TCF4 K297 > P53 K536 > TRPS1 K1192.
Inhibition of TMR-Peptide 2 Sumoylation in the Coupled-Enzyme Fluorescence Polarization Assay
Error limits refer to the standard error of the mean (n=4). Biotinylated (Btn) and N-acetyl peptides were added to the FP assay over a dose range, competing directly with 5-TMR-Ahx-VVKTEK-OH as substrates for Ubc9 trans-sumoylation.
n.d., could not be accurately determined due to poor solubility of peptide.
The ability of a surrogate E3 ligase to catalyze the transfer of SUMO-1 from SUMO-Ubc9 to a cognate lysine residue on a peptide substrate was also assessed (Fig. 4B). An E3 ligase specific for the sumoylation of TRPS1 has yet to be identified, and accordingly we utilized the internal repeat domain (IR1-M) from the E3 RANBP which has broad substrate specificity. 6,24 The IR1-M domain has previously been shown to promote constitutive Ubc9-dependent trans-sumoylation of substrates, including P53, enhancing the reaction rate of sumoylation by 18-fold or more. 6 FP was measured using TMR-peptides 1–4 in the coupled kinetic FP reaction (Fig. 2) in the presence of GST-RAN BP IR1-M; however, the results failed to show rate enhancement using P53, TCF4, or TRPS11194 peptides as substrates (data not shown). A modest 2.5-fold enhancement in reaction rate compared to identical coupled reactions run in the absence of added GST-RAN-BP IR1-M was observed only with TMR-peptide 2 based on TRP11201 (Fig. 4B). These results reaffirmed TMR-peptide 2 as the preferred substrate for further development of the FP assay. However, E3 RAN BP IRM1 did not appreciably improve assay robustness and was therefore omitted from the final screening assay.
To further characterize the specificity of sumoylation of peptides based on the sequence surrounding K1201 of TRPS1 (GPLNVVKTE KVDRS), a series of smaller peptides was assessed as substrates in the FP assay (Table 4). The results show that a minimal hexapeptide TMR-VVKTEK is adequate to serve as a substrate in the assay with comparable kinetic values to the larger peptides tested. The smaller tetrapeptide was poorly utilized as a substrate in the assay. Specificity toward the active internal lysine residue was tested by replacing lysine 1201 to arginine in an octapeptide (NVV
TRPS-Peptide 2 Substrate Optimization
TMR-peptides were evaluated as substrates in the FP kinetic assay (1 μM Peptide, 100 μM ATP, 160 nM E1, 1.63 μM Ubc9, and varying concentrations (0.4–50 μM of GST-SUMO-1). Reaction rates were obtained from the linear portion of reaction rate profiles monitored for 2 h; values for kinetic parameters were obtained by fitting rate data to Equation 5. Vmapp, Kmapp, and V/Km are expressed in terms of mP units/min, μM, and (mP units·μM)/min, respectively, and are represented as the mean±standard error (n=2).
n.a.o., no activity observed.
Inhibition of the FP Assay Using Nonhydrolyzable Analogues of ATP
Inhibition was evaluated using nonhydrolyzable ATP analogs as tool inhibitors to block E1 adenylylation of SUMO-1 which requires ATP hydrolysis. Titration of AMP-PNP, AMP-CPP, and AMP-PCP into the assay resulted in a dose-dependent inhibition, each with similar potency between (IC50s of 1.9, 6.4, and 3.4 mM, respectively; Fig. 5). These results demonstrated that inhibitors are detectable in this assay through a signal decrease. Ginkgolic acid (GA) which is reported to inhibit RanGAP1 sumoylation with an IC50 of 3 μM in gel-based assays, 25 was evaluated; however, only 11% and 33% inhibition of peptide sumoylation was observed at 2.5 mM and 5 mM GA, respectively (data not shown).
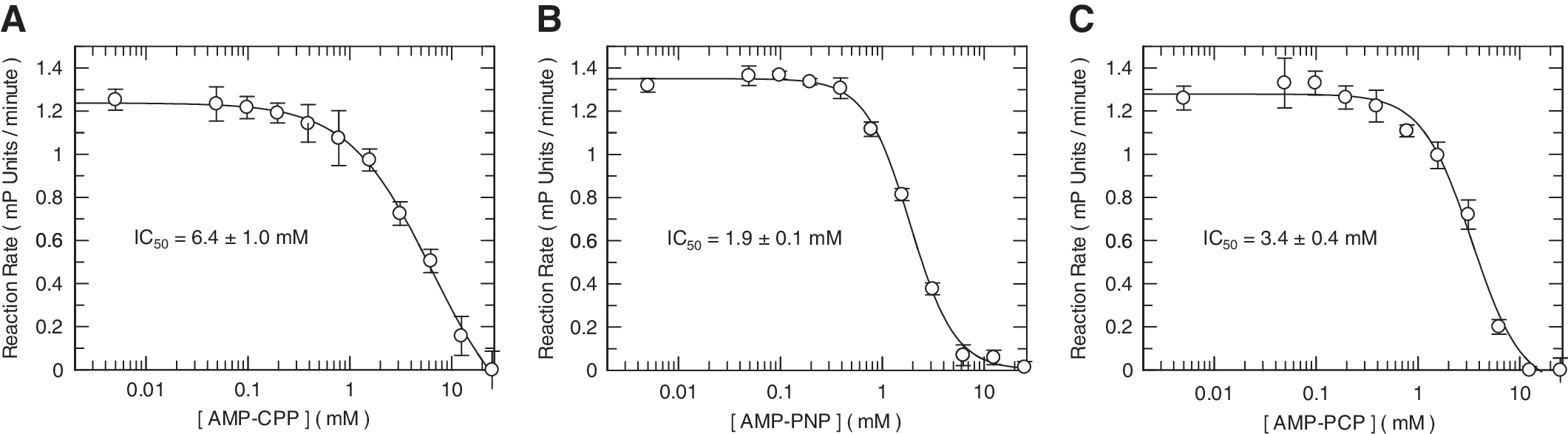
Inhibition of E1-dependent sumoylation with ATP analogue inhibitors. Inhibition of peptide sumoylation reactions, detected as a decrease in the observe rate (ΔmP/min), were evaluated in the presence of varying concentrations of nonhydrolyzable ATP analogues. Reactions containing 160 nM E1, 1.6 μM Ubc9, 10 μM GST-SUMO-1, 100 μM ATP, 1 μM TMR-VVKTEK, were evaluated in the presence of a titration of AMP-CPP
Detection of Substrate Sumoylation
Additional sumoylation of nonspecific peptide substrate lysyl residues (or other amino acids) would not be directly detectable in the assay in which the signal is derived exclusively from the trans-sumoylation from GST-SUMO-1 onto TMR-peptides, but these extraneous sumoylations could still adversely affect assay robustness. For example, unspecific sumoylation could deplete the available pools of GST-SUMO-1 formed in situ or aberrantly could abrogate the activity of (E1 or E2) in the reaction. Therefore, direct analytical characterization was performed to assess this possibility. LC/MS analysis confirmed the mass of sumoylated TMR-peptide 2 using GST-tagged and detagged SUMO-1 at 40,731 and 12,269 Da, respectively (Fig. 6A, B). When GST-SUMO-1 was used, higher molecular weight species were observed indicating that additional GST-SUMO-1 adducts were formed at lysines within the GST sequence in addition to the sumoylation of the cognate lysine (Lys-1201) found in the TRPS1 peptide 2 (Fig. 6A). These higher molecular adducts were not observed when detagged SUMO-1 was used (Fig. 6B).
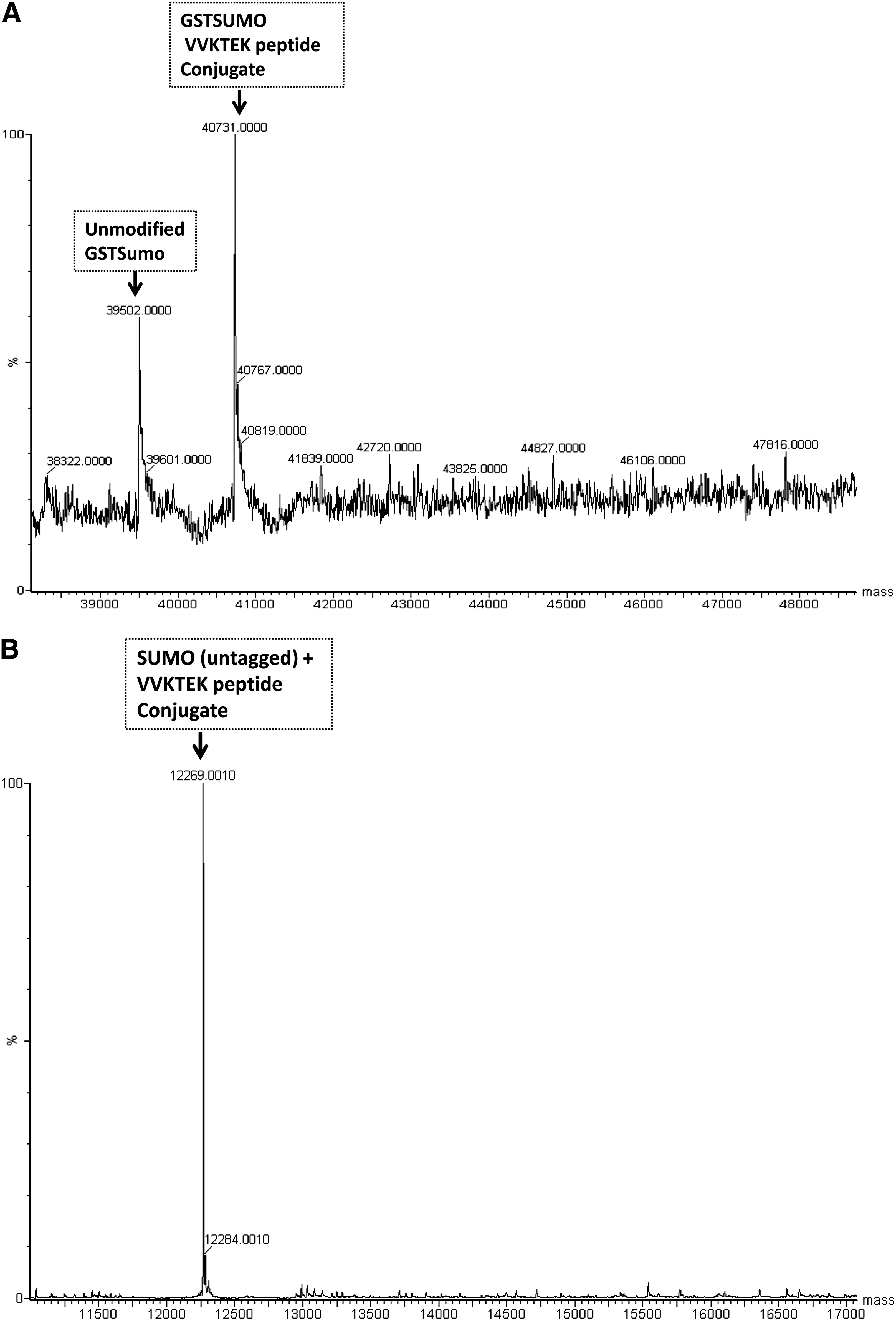
Optimization of TRPS1 peptide and TRPS1 repression domain (RD) substrate sumoylation.
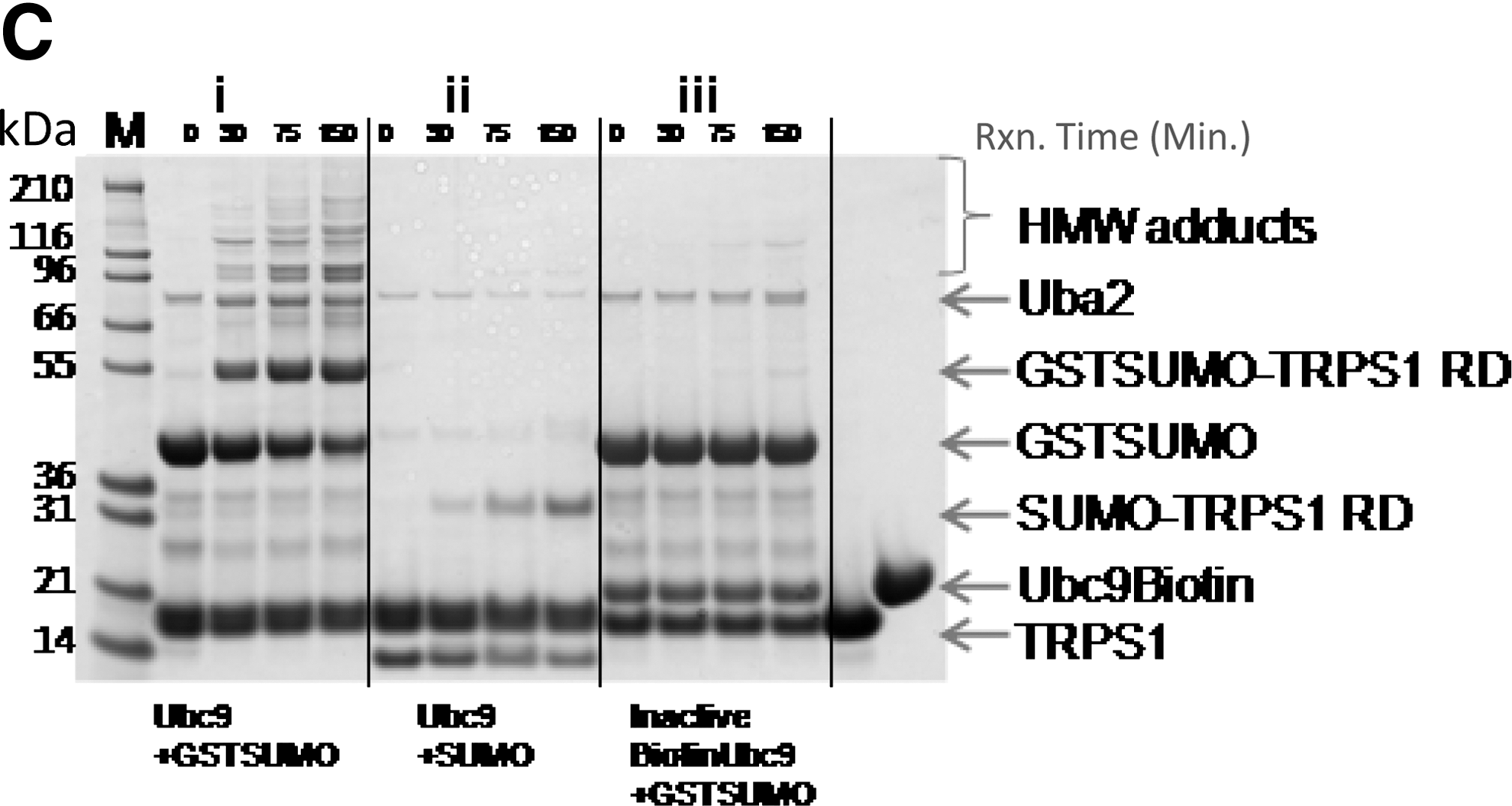
To better visualize this difference, coupled-enzyme reactions were also carried out using purified TRPS1 protein comprised of the RD (TRPS1 1192–1294) containing both K1192 and K1201 sumoylation sites 18 to analyze sumoylation by SDS PAGE. Reactions were conducted out over a time course of 150 min using either GST-SUMO-1 or detagged SUMO-1. The results indicated that although the major GST-SUMO-1 adduct corresponded to GST-SUMO-TRPS1 as expected, extraneous sumoylation was also apparent (Fig. 6C-i). This extraneous sumoylation appeared as laddering, the masses of which were consistent with sequential GST-SUMO additions to itself (39.5+39.5 kDa) or to TRPS1-GST SUMO (53+39.5+39.5 kDa). These high molecular weight (HMW) adducts likely represent adventitious GST tag sumoylation. Reactions with detagged SUMO-1 showed minimal HMW adduct formation (Fig. 6C-ii). Biotinylated Ubc9 prepared through extensive biotinylation of Lys residues (see Materials and Methods ) was empirically found to be inactive when tested in the fluorescent polarization assay (1 μM peptide, 100 μM ATP, 130 nM E1, 1.4 μM biotinylated Ubc9, and 10 μM GST-SUMO-1; data not shown). Inactivated Ubc9 prepared through biotinylation in this way, was unable to promote either on-target or off-target sumoylation; thus, demonstrating its dependence on Ubc9 trans-sumoylation (Fig. 6C-iii). Catalytically inactivated Ubc9 C93S, as expected, was also incompetent to promote transacylation from SUMO-Ubc9C93S onto TRPS1 RD as was evident from the absence of species of TRPS1 of higher molecular weight than the unlabeled protein (data not shown).
Independently, evaluation by both LC/MS and SDS PAGE showed that TRPS1 is the optimal target of the sumoylation reaction under our assay conditions (Fig. 6A–C). Although more off-target sumoylation was observed using GST-tagged SUMO-1, the larger polarization effect resulting as a consequence of the larger mass of the GST tag yielded a net positive signal enhancement over the use of detagged SUMO-1.
Additionally, in coupled-enzyme assay formats using Flag-tagged Ubc9 resulted in additional HMW species. We surmised that this may be negatively affecting the signal in the assay by depleting the available GST-SUMO-1 through adventitious sumoylation to sites other than that of TMR-peptide 2. Indeed, the use of detagged Ubc9, purified after TEV protease cleavage of the N-terminal FlagHis6 tag, enhanced the signal by 1.6-fold and resulted in a concomitant reduction in higher molecular weight adducts (data not shown). Analysis of the Flag tag sequence revealed a potential sumoylation site, VKVD, upon which sumoylation could be occurring. Although specific sumoylation at this site was not confirmed, detagged Ubc9 was used for subsequent HTS assay development.
To further optimize the assay signal, TCEP was evaluated as a general reducing agent as a replacement for DTT which could de-acylate SUMO-E1 and SUMO-Ubc9 by the formation of adventitious thioester adducts. Inclusion of 100 μM TCEP provided a better signal to background ratio and improved values of Z′ in the final assay format in 1,536-well plates (see Materials and Methods ).
High-Throughput Screening and Triage of Ubc9 Inhibitors
The primary high-throughput screen against 2,268,941 compounds (n=1) yielded a hit rate of 0.47% at a cutoff of 24.15% (3 standard deviations from the sample mean, 3SD). The quality of the assay was monitored by the statistical factor Z′, which was calculated from 64 high control wells and 64 low control wells on every HTS plate. This calculation accounts for the distance between the high and low signals as well as the error on these measurements. 26 Typically, a Z′ value≥0.4 is considered acceptable. The average Z′ of 0.61 for this HTS fell well within the range of acceptability. The screen resulted in 10,626 statistically robust hits identified at 3SD, and in a follow up single concentration experiment, 1,518 of these hits were confirmed to be inhibitors of sumoylation (14.29% of the total hits; Supplementary Figure S17). This set of compounds was evaluated over a range of concentrations (0.2–200 μM), resulting in a list of 728 compounds that demonstrated reproducible inhibition (pIC50>4) and did not optically interfere with the FP detection method (Fig. 7).
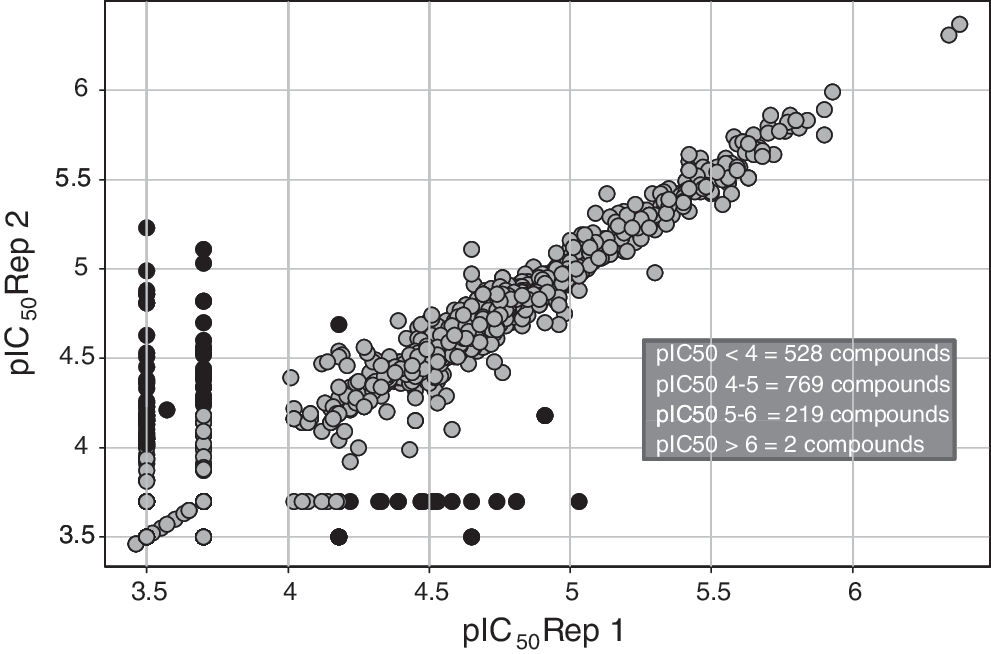
Replicate pIC50 pivot of compounds selected from HTS for dose-response testing. The 1,518 confirmed HTS hits were tested in duplicate in an 11-point dose response experiment (0.2–200 μM). Replicate set pIC50 values were pivoted to visualize assay reproducibility. pIC50 for compounds that differ by >0.5 logs between reps are colored black and pIC50 for compounds that differ by <0.5 logs between reps are colored gray.
The panel of 728 inhibitors identified from the HTS screen was triaged to remove compounds that either inhibited the E1 enzyme or reacted with thiols, as exemplified by reaction with N-acetyl-cysteine, both of which were not desired mechanisms of action (Fig. 2A). E1 inhibitors were identified in an AMP-Glo assay format, which measures the AMP released from the hydrolysis of ATP in the E1-catalyzed adenylylation and acylation of E1 SUMO-1; only 12 E1 inhibitors were found from the HTS triage. In contrast, a much larger subset of 242 inhibitors was identified as thiol reactive compounds evaluated in a high-throughput N-acetyl Cys reactivity assay (see Materials and Methods ). An additional 216 compounds were eliminated from further evaluation due to untoward structural properties to arrive at a final list of 258 compounds of interest derived from the total 728 confirmed hits (pIC50>4; cLogP<5, MW<500, and IFI<0.05).
In addition, a low-throughput fluorescence-based TSA was used to assess compound binding to free Ubc9 (Supplementary Fig. S18). Stabilization of Ubc9 by compounds would be consistent with the desired mechanism involving binding and inhibition of Ubc9 trans-sumoylation. In the presence of compound, an increase or decrease in T m reveals binding to the native or unfolded state, respectively. 26 Of the 258 compounds tested in dose response in the Ubc9 TSA, none exhibited an increase in T m, 247 exhibited a decrease in T m (usually occurring at >10 μM), and 11 had no effect on the protein T m. The results from this assay suggested that none of the identified compounds bound to the native state and stabilized Ubc9. In an extension of this experiment, 21 of the 247 compounds were tested in an orthogonal TSA with an unrelated protein, and it was observed that 18 compounds (>80%) appeared to behave as general protein destabilizers (data not shown).
Interestingly, 21 of the 258 triaged hits contained primary amines. We hypothesized that although these compounds appeared to destabilize Ubc9 at concentrations >10 μM, it was possible that they could act as alternative substrates for Ubc9 SUMO-1 thioester formation at lower concentrations (<10 μM). To test this possibility, compounds were added in place of the TMR-peptide 2 in the functional assay at comparable concentrations (2 μM); LC/MS analysis was performed to assess the sumoylation of confirmed inhibitor compounds containing primary amines. Surprisingly, several diaminopyrimidine compounds, which contained ornithine moieties in the middle of a linear peptide-like scaffold, were found to be sumoylated. The representative compound, GSK145A (shown in Fig. 9) was found to form a 455 Da adduct of SUMO-1 (Fig. 8). The two related compounds, GSK149A and GSK705A (Fig. 9) also were identified to be sumoylated. All three of these compounds were identified as inhibitors, each with a pIC50 value of 4.9. As these compounds were competent substrates for sumoylation at the ornithine substituents, the pIC50 values likely represent the Michaelis constants of these alternative substrates. Altogether, these results demonstrate that this coupled HTS assay is configured to identify multiple mechanisms of inhibition, including the discovery of alternate substrates of sumoylation that present as competitors. We were nonetheless unable to find inhibitors by any other specific mechanism which bound reversibly to the native state of Ubc9 and prevented sumoylation of TRPS1.
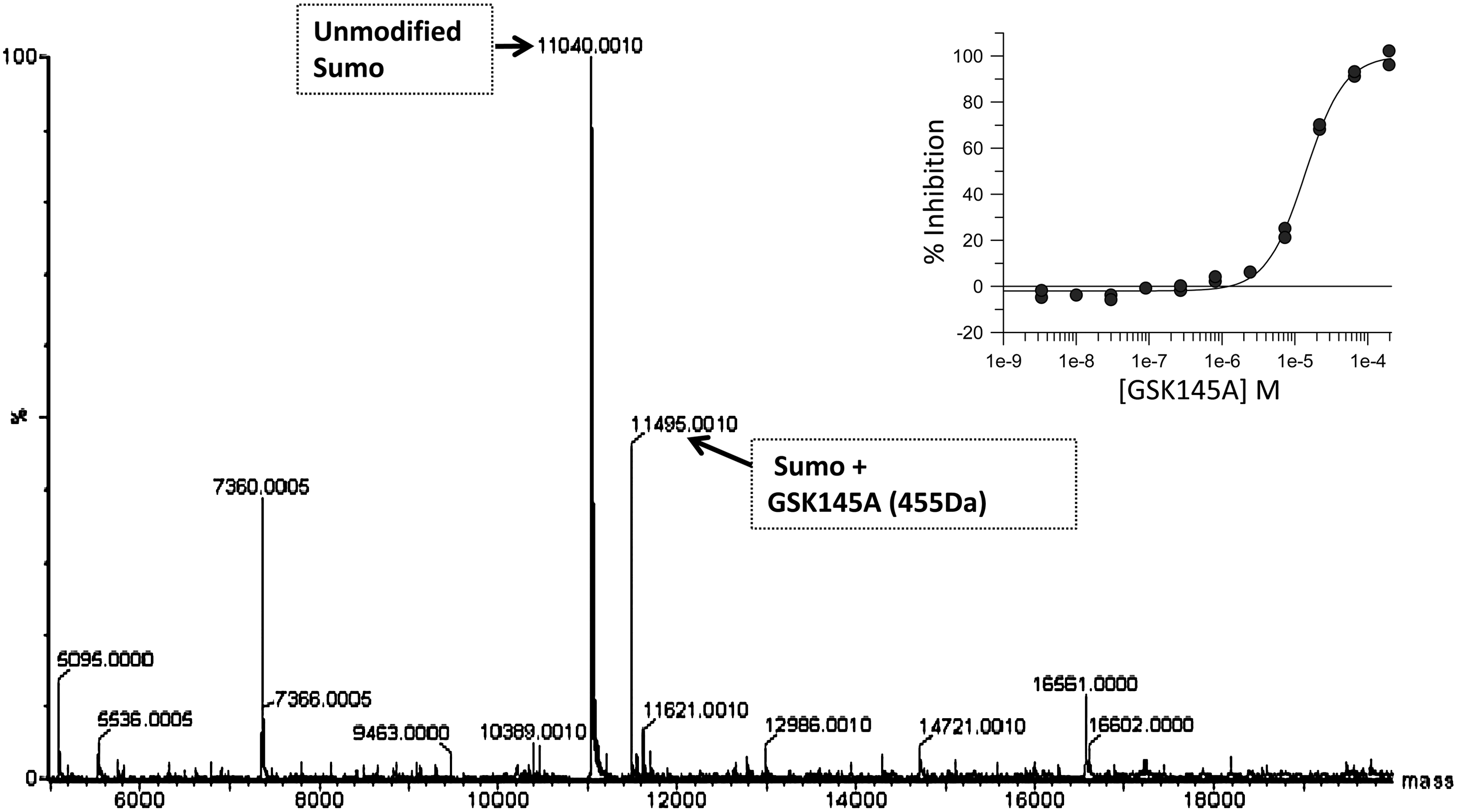
Detection of sumoylated adducts of L-ornithine–containing diaminopyrimidines. Reactions were carried out in the presence of detagged SUMO-1 (5 μM), E1 (0.15 μM), Ubc9 (2.5 μM), plus 2 μM compound in the absence of the TRPS1 hexapeptide substrate. Reactions were analyzed by LC/MS. Sumoylated GSK145A is detected at 11,495 Da. Unreacted SUMO-1 is observed at 11,040 Da. The inset plot shows the original IC50 curve obtained for GSK145A (pIC50=4.85 and Hill slope=1.6) during the dose response phase of the HTS.
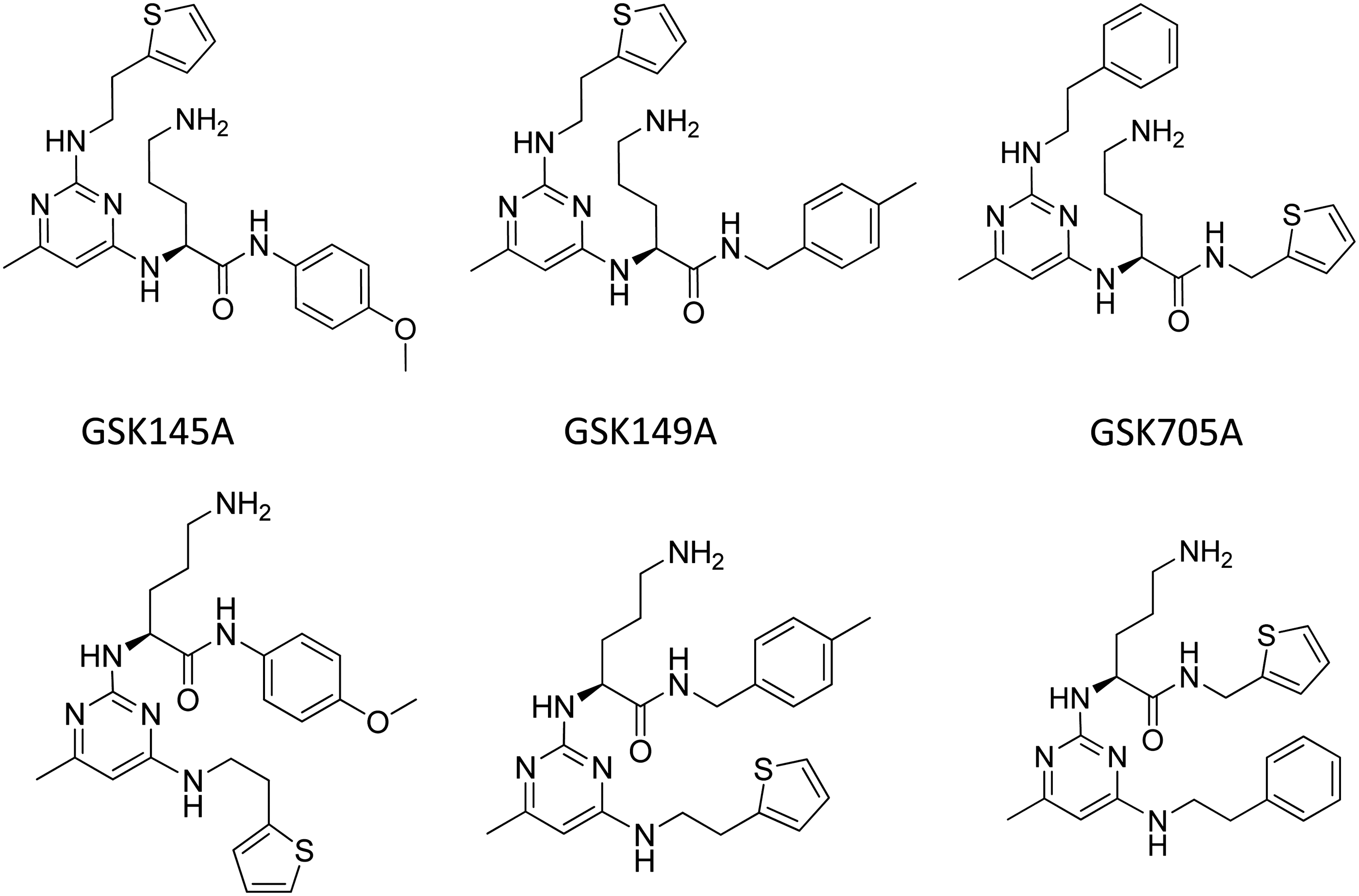
Identified structures of representative L-ornithine-containing diaminopyrimidines sumoylated in the coupled assay. Each of these compounds (top) was synthesized using solid phase array technology. QC showed a second peak of identical mass representing regioisomers (bottom). No attempt was made to separate these mixtures.
Discussion
To establish a high-throughput assay to screen for inhibitors of Ubc9 trans-sumoylation activity, peptide substrates were synthesized based on conserved, lysine-containing consensus sequences of known sumoylation sites in p53, TRPS1, and TCF4. 12,18,23 Consistent with previous reports, we were able to show that all of these substrates contained suitable acceptor sites of sumoylation in the protein products of the coupled-enzyme assay format as detected by Western analysis of SDS PAGE gels (Fig. 1). However, only one of these substrates, derived from the RD of the transcriptional repressor protein TRPS1 (peptide 2), could be configured into a robust high-throughput assay format using fluorescent polarization detection. In this format, Ubc9-dependent sumoylation was remarkably specific towards this substrate and K1201 contained within a minimal hexapeptide derived from this consensus sequence. Therefore, reaction conditions were optimized using TMR-VVKTEK to develop a high-throughput assay which was used to screen the GSK HTS compound collection. Although confirmed inhibitors and alternative nonpeptide substrates of sumoylation were found, no specific noncovalent inhibitors of Ubc9 trans-sumoylation were identified. Many compounds either destabilized Ubc9 or were cysteine reactive, indicative of undesirable properties and nuisance mechanisms. One class of compounds identified, representing L-ornithine-containing peptide mimetics, acted as surrogate substrates. This result correlates with the findings using competing peptides (Table 3) and demonstrates that this class of L-ornithine-containing diaminopyrimidines can compete for sumoylation of TRPS1 peptides in the assay. Development of alternative substrates could be further evaluated as potent disruptors of the cellular sumoylation machinery due to their depletion of available SUMO-Ubc9 through unproductive sumoylation.
We were surprised to find substrate preferences using the FP assay format compared to Western analysis of SUMO-1 conjugation on SDS PAGE gels. Although sensitive, the detection of SUMO-1 conjugated biotin-peptides by Western analysis could not readily or quantitatively discern differences in substrate specificity. This may be due to the high concentrations of peptide-SUMO-1 conjugates formed, saturating the detection signal and giving the false appearance that all substrates were equivalent. In contrast, the FP assay was more sensitive to discriminate substrate preferences. Although we cannot rule out the effects that specific sequences may have on their suitability as substrates in the FP assay, the original peptides chosen for our assays spanned equivalent distances around the target sumoylation site which should have minimized these effects. Additionally, we were able to show that similarly-sized biontinylated peptides were substrates in a gel-based format, and that each competed with TMR-VVKTEK with varying efficiencies in the FP assay (Table 3). Therefore, it is possible that the peptide substrate preferences observed in the FP assay may reflect actual target site preferences. The FP assay developed here may be useful as a platform to further characterize Ubc9 substrate preferences and sequence specificity.
The consensus motif (VKTE) of TMR-peptide 2 is unusual in that it contains a second lysine located proximal to the target lysine. Mutation of Lys-1201 to Arg in the TRPS1 peptide 2 completely abolished the substrate activity of this peptide indicating that the retained lysine at amino acid residue 1204 in TRPS1 is not a conjugation site, and that the fidelity of sumoylation from SUMO-Ubc9 requires the target Lys-1201 (Table 4). A peptide based on the second sumoylation site located within nine residues of this site (Lys-1192) appears to be a much poorer substrate based on the competition results shown in Table 4. Similarly, peptides with sequences KLMF
The peptide which spans residues 380–393 of p53 had been reported to have a sumoylation rate of 5 pM/sec in the absence of E3 and 330 pM/sec in the presence of the constitutively active E3 RANBP–IR1-M as determined using gel-based detection of sumoylation. 6 This dramatic enhancement in sumoylation efficiency was not observed using this p53 peptide (KLMFKTEGPPSD) in the FP assay suggesting that the affinity of IR1-M for P53 may have been compromised in the assay conditions used here. The hexapeptide spanning TRPS1 K1201 exhibited a 2.5-fold enhancement in reaction rate in the presence of RANBP-IR1-M compared to the rate in its absence. This rate enhancement is quite small compared to that reported for other substrates, but indicates that RAN BP IR1-M ligase activity provides nominal catalysis of trans-sumoylation in the FP assay.
TMR-peptides 1, 3, and 4, were all poorer substrates than TMR-peptide 2 in the FP assay suggesting that SUMO-Ubc9 is able to utilize other sequence motifs in addition to the conserved tetrapeptide sequence ΨKxE/D. The preference for TRPS1 peptide 2 over TRPS1 peptide 3 is consistent with the finding that the peptide consensus sequence as found proximal to Lys-1201 in TRPS1 is the optimal sequence for sumoylation within the RD as previously reported. 18 However, the effects resulting from the sequential mutation of these two sites on GATA4 transcription appear to be additive in cell culture indicating that modification of both sites is still biologically important. 18 This observation reinforces the concept that substrate preferences of SUMO- Ubc9 are influenced by interacting proteins, possibly, including E3 ligases which contribute to substrate recognition in vivo. 2,28
Although SUMO E3 ligases appear to play a critical role in defining substrate preferences in cells, the observed preferences for TMR-peptide 2 over TMR peptides 1, 3, and 4 in the coupled-enzyme assay without an E3 enzyme indicates that SUMO-Ubc9 trans-sumoylation can be directly affected by substrate preferences. However, the E3-independent mechanisms which have been described do not adequately explain how this can be achieved. In one mechanism, the noncovalent binding of SUMO at Lys-14 creates a second binding interface which enhances substrate interactions through SUMO-Interaction motifs (SIMs) located on target proteins. 29 Although it is possible that the optimized TRPS1 hexapeptide VVKTEK substrate serves both as a minimal acceptor and minimal SIM, this seems unlikely due to its small size and lack of potential exosites for SUMO-1 binding. In a second mechanism, downstream negatively charged amino acid-dependent sumoylation motifs (NSDM's), such as the one defined for E twenty-six–like transcription factor 1 (ΨKXEXXEEEE), can further influence substrate binding affinity. 6,30 The minimal TRPS1 hexapeptide does not include an NSDM. Furthermore, the theoretical isoelectric point for peptide 2 which predicts a net positive charge is inconsistent with negative charge preferences. Our results infer that a different, as yet undefined E3-independent mechanism may contribute to the observed TMR-peptide 2 preference in the coupled-enzyme assay format, possibly involving the local charge interactions at or directly proximal to the sumoylation consensus motif (VKTE).
The coupled-fluorescent polarization assay used for this study was developed to detect inhibitors acting through diverse mechanisms (Fig. 2), including E1 inhibition, thiol reactivity, and interception of activated intermediates (SUMO-adenylate/thioester). Since we were specifically interested in identifying inhibitors of Ubc9-dependent trans-sumoylation of TRPS1, the triage assays were chosen to identify compounds able to reversibly bind to the native state of Ubc9. We reasoned that compounds acting through inhibition of E1, through covalent modification of cysteinyl residues on E1 or Ubc9, or through the interception of activated intermediates (SUMO-adenylate/thioester), were likely to lack specificity for TRPS1, the desired sumoylation target. While binding to free TRPS1 would also represent available, specific mechanisms of the inhibition of trans-sumoylation, it is not likely that our peptide-based assay would identify compounds with this mechanism. Unfortunately, no tool inhibitors of Ubc9-dependent trans-sumoylation are available to validate this mechanism in the coupled reaction. However, through the use of nonhydrolyzable ATP analogues we demonstrated that it is feasible to inhibit the first step of SUMO-1 adenylylation and E1 charging with SUMO-1 (Table 3). The high concentration needed to elicit an effect (IC50s of 3.4 and 6.4 mM for AMP-PCP and AMP-CPP, respectively) also demonstrates that this mechanism of inhibition would not likely have been detected in the final screening format. In fact, the inclusion of high ATP concentrations (100 μM) in the coupled assay, which is 40-fold greater than the K m for ATP (2.5 μM), was intended to specifically reduce the likelihood of detecting inhibitors which acted via such a mechanism. Overall, 12 inhibitors of E1 activity were identified (data not shown). Half of these were also cysteine-reactive compounds, likely acting as nonspecific inhibitors to intercept the active-site cysteine of E1, which is found in the Uba2 subunit (Fig. 2). Recently, adenosine sulfamate inhibitors of Neddylation have been identified, which form covalent adducts to NEDD, and thereby inhibit Neddylation by tightly binding at the active site of NAE1. 31,32 A similar mechanism of inhibition is also possible for sumoylation. However, amongst the six hits which inhibited the activity of E1 but were not cysteine-reactive, no adenosine sulfamate analogues were identified. TSA of non—cysteine-reactive compounds failed to reveal any Ubc9 compounds that stabilized the protein. Instead, most compounds appeared to destabilize Ubc9 at high concentration (>10 μM) indicative of a higher affinity to the unfolded state of Ubc9.
Other HTS configurations which have been reported for identifying inhibitors of sumoylation are distinct from the FP method used in this study and have been applied with varying success. Time-resolved FRET (TR-FRET) in vitro biochemical and cellular assay methods which measure the binding of Ubc9 and SUMO-1 have been used to screen small compound library sets. 33 –35 Although no inhibitors were identified from a 10,000 inhibitor set in a biochemical format, 39 validated hits were identified from a set of 2,000 compounds evaluated in a cell-based format. 35 This equates to a 2% hit rate after filtering out auto fluorescent compounds, but no follow-up studies to identify mechanism of inhibition were described. Assays employing AlphaScreen 36 and Lanthascreen 36,37 technologies have also been described which establish reproducible and highly sensitive in vitro coupled screening formats that are optimized to monitor E1:Ubc9 dependent sumoylation of RanGap1; however, neither assay was assessed with compound sets designed to identify inhibitors of protein sumoylation.
The HTS assay used in this study, which was configured to run in a high-throughput 1,536-well format, exhibited very low assay variability (Z′=0.6), in spite of observed off-target sumoylation (known as GST-SUMO-1 laddering) associated with the use of GST-SUMO-1 compared to detagged SUMO-1 (Fig. 6). The 728 validated hits (pIC50>4) represents a true hit rate of <0.05% (pIC50>4). The identification of diaminopyrimidine L-ornithine–containing peptide mimetics (Figs. 2 and 10) is surprising, given the specificity of the reaction for the TRPS1 peptide.

Model of diaminopyrimidine GSK705A alignment with Ubc9. Structural alignment of GSK705A with the conformation of L523-KS-E526 (the recognition motif on RanGap1 by Ubc9) observed in the crystal structure of the SUMO-RanGap1-Ubc9-RanBP2 complex (PDB 1Z5S). 6 GSK705A and L523-KS-E526 of RanGAP1 are shown in green and cyan ball-and-stick representations, respectively. The molecular surface of P128, A131 and Y134 of Ubc9, L523 of RanGAP1 and methyl-substituted pyrimidine moiety of GSK705A is shown in brown mesh, cyan mesh and green mesh, respectively. The structural isomer, GSK705A, fits the model in a similar way except that the distal phenyl rather than pyrimidine ring overlays Leu523 (data not shown).
However, structural alignment of these mimetics with the RanGAP1 recognition motif L523-KS-E526 reveals several structural similarities. A model derived from this alignment indicates that the ornithine substituent of the diaminopyrimidines can adopt a conformation that is similar to that of K524 in RanGAP1, which suggests that the ornithine moiety of the peptide mimetics can covalently link with G97 of SUMO-1, as observed (Fig. 10). For the regioisomers shown in the top row of Figure 9, the alignment result suggests that the methyl-substituted pyrimidine moiety mimics L523 of RanGAP1 and may have similar hydrophobic contact with a hydrophobic patch on the surface of Ubc9 consisting of P128, A131, and Y134. In contrast, for the regioisomer in the bottom row of Figure 9, it is the aromatic functional group connected to the pyrimidine via an ethylene linker that was found to overlay with the side chain of L523 of RanGAP1. For Ubc9-directed sumoylation to occur, a large hydrophobic amino acid residue is required at this position on Ubc9 substrates (for example, L523 of RanGAP1 and V1200 of TRPS1). It is possible that the methyl-substituted pyrimidine moiety or the distal aromatic ring connected to the pyrimidine of these peptide mimetics is able to mimic this conserved hydrophobic residue needed to be an Ubc9 substrate. These structural similarities between the diaminopyrimidines and L523-KS-E526 of RanGAP1 provide some structural rationalization of how the diaminopyrimidines can be substrates of Ubc9 and thus, compete with the TRPS1 K1201.
Given the apparent difficulties in finding specific inhibitors of SUMO-Ubc9 by any mechanism, achieving the identification of substrate mimetics provides a possible path toward developing irreversible suicide substrates for the specific inhibition of target-specific sumoylation. The inhibition of TRPS1's transcriptional repression in this way would have potential benefits in the treatment of bone and cartilage diseases. This would be preferable to inhibition of Ubc9 alone which is the sole E2 enzyme responsible for regulating intracellular sumoylation. Recently, conditional Ubc9 knockouts have been shown to be highly deleterious, leading to the depletion of intestinal crypt stem cells. 38 Tool inhibitors of Ubc9 will be important toward understanding the biological effects of inhibiting Ubc9-dependent sumoylation compared to the disruption of sumoylation-independent Ubc9 functions in transcriptional repression and nuclear translocation. 39 –41
Footnotes
Acknowledgments
We thank our colleagues (current and former) from GlaxoSmithKline's Molecular Discovery Research Unit: Quinn Lu, Jessica Schneck, Scott Pesiridis, and Kristin Brown for critical reading of this manuscript and for their contributions toward improving the content; Sean McLarney and John Feild for molecular biology support; Stephanie van Horn, Elizabeth Thomas, and Christopher Traini for DNA sequencing; and Michael Schaber for isothermal titration calorimetry determinations. This work is dedicated in memoriam to Dr. Siegfried B. Christensen, IV, PhD, late of the VPoC DPU.
Author Disclosure Statement
All authors were employed by GlaxoSmithKline during the work described in this article. No competing financial interests exist.