Geschwindner S, Dekker N, Horsefield R, Tigerström A, Johansson P, Scott CW, Albert JS: Development of a plate-based optical biosensor fragment screening methodology to identify phophodiesterase 10A inhibitors. J Med Chem 2013;56:3228–3234.
Abstract: We describe the development of a novel fragment screening methodology employing a plate-based optical biosensor system that can operate in a 384-well format. The method is based on the “inhibition in solution assay” (ISA) approach using an immobilized target definition compound (TDC) that has been specifically designed for this purpose by making use of available structural information. We demonstrate that this method is robust and is sufficiently sensitive to detect fragment hits as weak as KD = 500 mM when confirmed in a conventional surface plasmon resonance approach. The application of the plate based screen, the identification of fragment inhibitors of PDE10, and their structural characterization are all discussed in a companion paper.
Commentary:In recent years, the use of fragment-based drug discovery (FBDD) to enhance libraries used in high-throughput screening (HTS) has grown in acceptance. A fragment typically refers to compounds with molecular weights less than 250, and libraries have been constructed to contain thousands of such fragments to cover many different scaffolds. Methods that have been applied for FBDD include, among others, X-ray crystallography, nuclear magnetic resonance, and surface plasmon resonance (SPR). Due to the general weak binding nature of small chemical fragments to biological targets, lack of assay sensitivity and/or throughput are some of the challenges FBDD faces. SPR, along with optical waveguide grating (OWG), are two major optical biosensor platforms. The availability of plate-based OWG platform permits screening at increased speed, but limitations remain associated with the detection limit. In the article highlighted here, Geschwindner and coworkers present an improved plate-based OWG system for fragment-based screening. In this work, instead of having a protein tethered onto a biosensor surface, a reverse immobilization method was developed—a tethered target definition compound (TDC; see
first figure
). Locating an attachment point in a small molecule for either immobilization or fluorescent labeling is often not a trivial matter. However, immobilization of a TDC allows the detection of fragment binding through a competitive mode (inhibition in solution assay [ISA]). The extent of fragment displacement reflects its binding potency, which correlated well with SPR binding affinity data in the present study of phosphodiesterase 10A (PDE10A; see
second figure
). Protein concentration and TDC immobilization density were shown to be key parameters in determining the assay signal window and detection limit. This ISA-based OWG assay in a plate format represents a promising generic screening platform for FBDD, with enhanced assay sensitivity at lower fragment concentrations. Contributed by Wendy Lea.
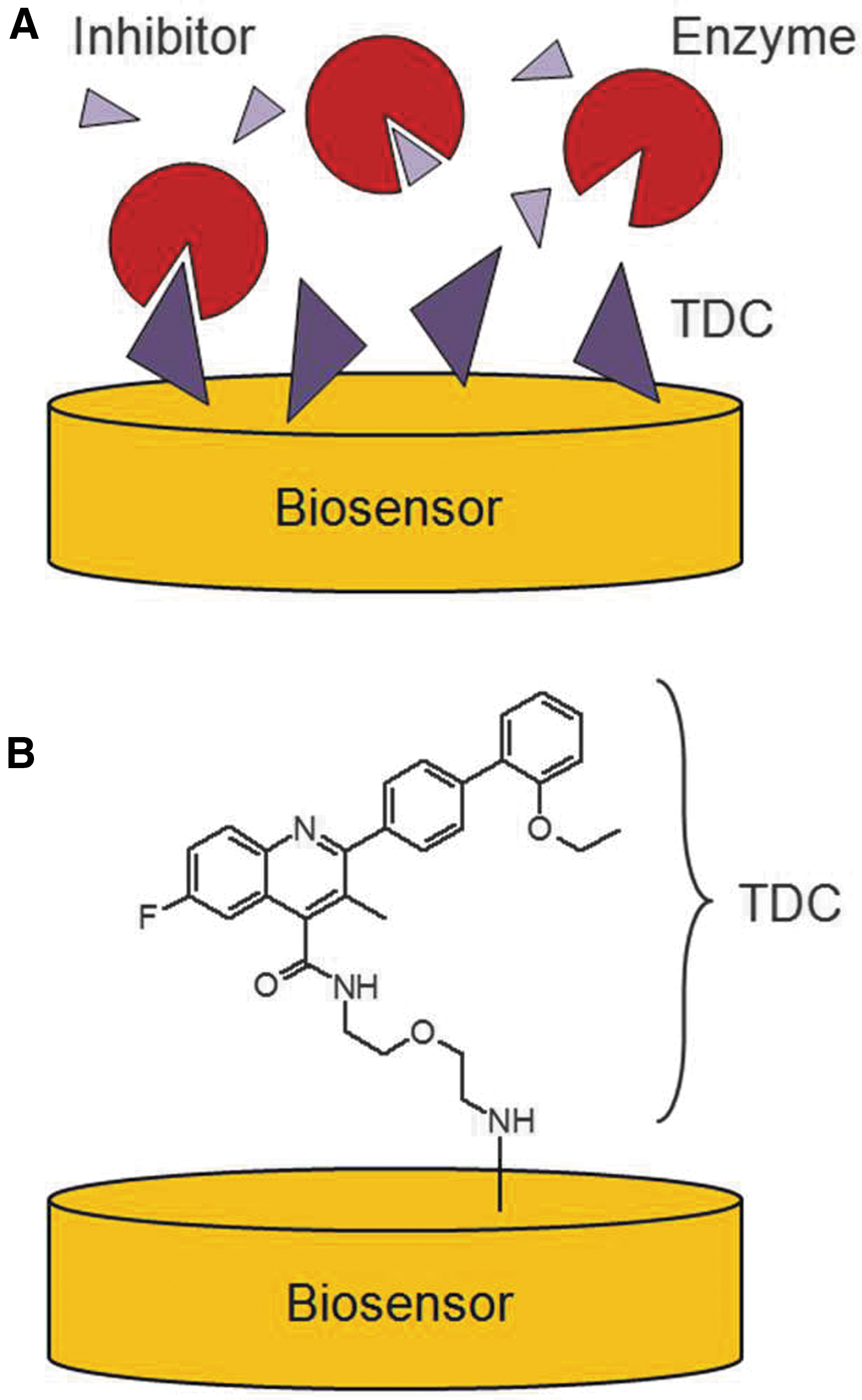
Schematic illustration of the ISA format used in SPR studies. (A) The ISA format employs an immobilized TDC that ideally displays high affinity to the enzyme. Competing inhibitors that are present in solution will subsequently lower the observable binding signal. (B) Illustration of the TDC (as employed in the present study) linked to the sensor surface. ISA, inhibition in solution assay; SPR, surface plasmon resonance; TDC, target definition compound.
Representative results from the OWG ISA. (A) Shown are typical sensorgrams (example of 14 compounds displaying different potencies at 20 μM and highlighted by different colors) illustrating the experimental setup of the OWG ISA. After buffer equilibration, the addition of PDE10A to TDC-containing wells at 7 min results in specific binding to the biosensor surface. Addition of protein to unmodified wells is used as control and to assess specificity. The addition of compounds after equilibration at 33 min results in a displacement of PDE10A from the biosensor surface due to specific competition with the TDC. The magnitude of the displacement reflects the potency of the compounds. (B) Correlation of compound activity in the OWG ISA and compound affinity determined via SPR ISA in the test set of 20 PDE10A inhibitors (details in supplemental information). In (A) and (B) the binding responses are shown as PDE10A binding %; i.e., the binding of PDE10A to the TDC-modified biosensor in the presence of fragment in relation to the controls containing only PDE10A protein. PDE10A binding percentage is defined by the OWG signal prior to addition of PDE10A (defined as 0% binding) in relation to the equilibrated OWG signal after addition of PDE10A at the given concentration (defined as 100% binding). The magnitude of the displacement correlates well with the affinity of the compounds (linear regression line depicted in black) and can thus be used for selection of compounds and preliminary affinity ranking. OWG, optical waveguide grating; PDE10A, phosphodiesterase 10A.
Probing Neurites with PKI Subsets
Al-Ali H, Schürer SC, Lemmon VP, Bixby JL: Chemical interrogation of the neuronal kinome using a primary cell-based screening assay. ACS Chem Biol 2013;8:1027–1036.
Abstract: A fundamental impediment to functional recovery from spinal cord injury (SCI) and traumatic brain injury is the lack of sufficient axonal regeneration in the adult central nervous system. There is thus a need to develop agents that can stimulate axon growth to re-establish severed connections. Given the critical role played by protein kinases in regulating axon growth and the potential for pharmacological intervention, small molecule protein kinase inhibitors present a promising therapeutic strategy. Here, we report a robust cell-based phenotypic assay, utilizing primary rat hippocampal neurons, for identifying small molecule kinase inhibitors that promote neurite growth. The assay is highly reliable and suitable for medium-throughput screening, as indicated by its Z′-factor of 0.73. A focused structurally diverse library of protein kinase inhibitors was screened, revealing several compound groups with the ability to strongly and consistently promote neurite growth. The best performing bioassay hit robustly and consistently promoted axon growth in a postnatal cortical slice culture assay. This study can serve as a jumping-off point for structure activity relationship (SAR) and other drug discovery approaches toward the development of drugs for treating SCI and related neurological pathologies.
Commentary:Protein kinases (PKs) are found universally among signaling pathways and are key targets for drug discovery. As such, there is a large amount of low molecular weight PK inhibitors (PKIs), and many assays are available for this class of enzymes. Focus libraries of PKIs have been developed for use in lead discovery and subsets of PKIs with well-validated targets are now available (see, for example, the GlaxoSmithKline PKI set; Dranchak et al.: PLoS One 2013;8:3.e57888). The article by Al-Ali et al. used a commercial PKI library to identify PKs operating in neurite outgrowth. Neurons of the central nervous system lack regenerative capacity, which presents a major complication in spinal cord injuries. A high content screen was developed using an immunostain for βIII-tubulin (to visualize cell bodies and neurites) and a Hoechst stain for nuclei. Following automated microscopy in 96-well plates the growth of neurites could be enumerated. Morphological parameters for neurite growth were captured, and compound activity was clustered based on four morphological parameters to define groups of PKIs that induced similar phenotypes (see
figure
). The PKI library was selected to cover a broad range of kinases. There were too few active compounds to develop robust structure activity relationships, but the inhibitors could be grouped into broad chemotypes. For example, isoquinolinyl sulfonamides were found to promote neurite outgrowth, and among these were potent Rho kinase (ROCK) inhibitors (showing a maximal effect at 20 μM). ROCK has been implicated in mediating neurite growth. Other PKIs included PKC, CDKs, and several tyrosine kinase inhibitors. Non-PKIs were also identified, including PI3K and a sphingosine kinase inhibitor. In all, 40 kinase inhibitors were identified in the high-content assay. To gauge which pathways were affected by the set of active PKIs, the authors examined Western blots using six downstream phosphoproteins as markers of different signaling pathways. Some PKIs showed similar activity profiles, which suggests that they affect overlapping pathways. This data provide a starting point for understanding the mode of action of these PKIs in neurite growth, but further work is needed to understand the critical pathways that mediate this biological process and to discern between on- and off-target activity. The most potent PKI, ML-7 (maximum effect at 160 nM) was tested in a cortical slice assay, which demonstrated that this compound could promote axon growth in an organotypic context. This article describes chemical probes toward PKs that should be useful tools for studying neurite growth as well as potential starting points for drug discovery. Contributed by Doug Auld.
Effects of hit compounds on distinct neurite parameters. (a) The Pearson correlation coefficient (r2) was calculated for each pair of neurite features using the data obtained for each hit KI at the concentration at which maximum neurite total length (NTL) was observed. (b) The effects (% change from control) of KI hits on NTL, NAL, NBA, and NC are shown at the concentrations at which maximal effect on NTL was observed for each compound. Clustering was performed in MATLAB using a Euclidean metric in the KI dimension and a correlation metric in the neurite feature dimension. Activity clustering shows that certain compounds, such as ML-7 and Flt-3 inhibitor III, increased multiple aspects of neurite growth, while others, such as VEGF receptor tyrosine kinase inhibitor II, appeared to have more isolated effects on neurite length. KI, kinase inhibitor; NAL, neurite average length; NBA, neurite branch-point average; NC, neurite count; VEGF, vascular endothelial growth factor.
Substrate-Pocket-Targeted Src Inhibitor
Naing A, Cohen R, Dy GK, Hong DS, Dyster L, Hangauer DG, Kwan R, Fetterly G, Kurzrock R, Adjei AA: A phase I trial of KX2-391, a novel non-ATP competitive substrate-pocket–directed SRC inhibitor, in patients with advanced malignancies. Invest New Drugs 2013 Jan 30 [Epub ahead of print]; DOI: 10.1007/s10637-013-9929-8.
Abstract:
Background. Src kinase is central to tumor cell proliferation, apoptosis, and metastasis. KX2-391 is a synthetic, orally bioavailable small molecule inhibitor of Src tyrosine kinase (TK) signaling and tubulin polymerization. This compound is distinct from other Src kinase inhibitors by targeting the peptide substrate rather than the ATP binding site; the binding site on hetero-dimeric tubulin is novel and distinct from the taxanes and other known tubulin inhibitors.
Methods. This multicenter Phase I trial utilized a 4+2 study design to determine the maximum tolerated dose (MTD), safety, and pharmacokinetics (PK) of KX2-391 in patients with refractory solid tumors.
Results. Forty-four (44) patients (18 M, 26 F; median age, 59) were enrolled in 9 dose cohorts. Dose-limiting toxicities, all reversible within 7 days, occurred in 7 patients and consisted of elevated AST (n = 4), ALT (n = 2), neutropenia (n = 1), thrombocytopenia (n = 1), failure to thrive (n = 1) and anorexia (n = 1). The MTD is 40 mg BID continuously. Eleven patients had stable disease for ≥4 months, including patients with ovarian, carcinoid, papillary thyroid, prostate, pancreas and head and neck cancer. Patients with prostate and pancreatic cancer also had significant biomarker decreases (PSA, 205 ng/mL to 39 ng/mL; CA19-9, 38,838 U/mL to 267 U/mL). The ovarian cancer patient has had stable disease >12 months. KX2-391 was orally available, rapidly absorbed, and exposure was proportional to dose across the range investigated.
Conclusions. KX2-391 has a favorable pharmacokinetic profile, is well tolerated, demonstrates preliminary evidence of biologic activity, and warrants further evaluation in Phase II trials.
Commentary:The nonreceptor tyrosine kinase SRC is highly expressed or activated in many cancer types, and inhibitors of its activity that bind to the ATP binding site are being evaluated for the treatment of solid tumors. An orally available compound, KX2-391, is described herein that inhibits SRC by targeting the peptide substrate rather than the ATP binding site. This compound is peptidomimetic. KX2-391 had a half-life of ∼4 hours. The PK profile was dose proportional, and there was no evidence of accumulation. The IC50 of KX2-391 inhibition of SRC substrate phosphorylation was ∼20 nM. The compound induces apoptosis by inducing p53 and stimulating caspase 3 and PARP cleavage. This report discusses the results from 44 patients who received KX2-391 in a phase I clinical trial. The maximum tolerated dose (MTD) was determined to be 40 mg. The types of dose-limiting toxicities (DLTs) observed at the higher doses are shown in the
Table
. All of these DLTs were reversible. There was preliminary evidence of biological activity based on biomarker changes; however, in this trial designed to determine the MTD, safety, and pharmacokinetics, no partial or complete responses were observed, although several patients remained on the drug for extended periods of time. The activity of KX2-391 in a mouse xenograft model with HT29 cells was about five times higher than for the ATP-binding site inhibitor dasatinib. Additionally, KX2-391 inhibited both primary tumor growth and lymph node metastases in an orthotopic mouse model with PC3-MM2 cells. An earlier article on this peptidomimetic series (Anbalagan et al., Breast Cancer Res Treat 2012;132:391–409) showed that combination treatment with tamoxifen lead to synergistic growth inhibition and apoptosis of estrogen receptor alpha (ERα) positive breast cancer cells in vitro. This earlier report showed that the compounds could induce G2/M arrest in a cell cycle assay. An additional effect of the compound that was highlighted in the earlier article and reiterated in this new article is that the compounds also interact with α and β tubulin and inhibit microtubule polymerization in colon cancer cells in a dose-dependent manner. It will be interesting to understand whether the efficacy observed to date is due to one or both of these mechanisms (i.e., SRC and/or tubulin inhibition). Contributed by Mindy I. Davis.
CTCAE, Common Terminology Criteria for Adverse Events; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Multitargeted Drug for Malignant Glioma
Albanese C, Alzani R, Amboldi N, Degrassi A, Festuccia C, Fiorentini F, Gravina GL, Mercurio C, Pastori W, Brasca MG, Pesenti E, Galvani A, Ciomei M: Antitumor efficacy on glioma models of PHA-848125, a multikinase inhibitor able to cross the blood brain barrier. Brit J Pharmacol 2013;169:156–166.
Abstract:
Background & Purpose. Malignant gliomas, the most common primary brain tumors, are highly invasive and neurologically destructive neoplasms with a very bad prognosis due to the difficulty to remove completely the mass by surgery and the limited activity of current therapeutic agents. PHA-848125 is a multikinase inhibitor with broad antitumor activity in preclinical studies and good tolerability in Phase 1 studies, that could affect two main pathways involved in glioma pathogenesis, the G1-S phase progression control pathway through the inhibition of CDKs and the signaling pathways mediated by tyrosine kinase growth factor receptors, such as tropomyosin receptors. For this reason we tested PHA-848125 in glioma models.
Experimental Approach. PHA-848125 was tested on a panel of glioma cell lines in vitro to evaluate inhibition of proliferation and mechanism of action. In vivo efficacy was evaluated on two glioma models both as single agent and in combination with standard therapy.
Key Results. When tested on a subset of representative glioma cell lines, PHA-848125 blocked cell proliferation, DNA synthesis and inhibited both cell cycle and signal transduction markers. Relevantly, PHA-848125 was also able to induce cell death through authophagy in all cell lines. Good antitumor efficacy was observed by oral route in different glioma models both with subcutaneous and intracranial implantation. Indeed, we demonstrate that the drug is able to cross the blood brain barrier. Moreover, the combination of PHA-848125 with temozolomide resulted in a synergistic effect and a clear therapeutic gain was also observed with a triple treatment adding PHA-848125 to radiotherapy and temozolomide.
Conclusions & Implications. All the preclinical data obtained so far suggest that PHA-848125 may become a useful agent in chemotherapy regimens for glioma patients and support its evaluation in phase 2 trials for this indication.
Commentary:There is a need to develop additional treatment options for malignant gliomas, the most common primary brain tumors. Glioblastoma is a complex disease, and it is thought that the simultaneous blocking of more than one pathway may be needed for an effective therapy. PHA-848125 is a multitargeted kinase inhibitor that can cross the intact blood–brain barrier and has shown good tolerability in phase 1 studies. In rat models the brain concentration exceeds the IC50 of the proliferation assay for at least 12 hours. The two main pathway types that PHA-848125 is thought to interact through include the CDK-mediated cell cycle control pathway and pathways mediated by tyrosine kinase growth factor receptors. This compound strongly inhibited c-Yes kinase activity, which is a target for cancer in its own right. In glioblastoma models c-Yes1 can mediate the activation of PI3K in response to extracellular signals. This compound was able to inhibit proliferation (IC50s 1.5–2.5 μM), DNA synthesis, cell cycle, and signal transduction. Four glioma lines were studied (SF539 Rb null, U251, U87MG, and SF268 with p16 deletion). In vivo efficacy was shown in various subcutaneously implanted models (see
figures
). Synergy was observed with temozolomide in a U87MG xenograft model, and there were a high number of tumor-free animals at the conclusion of the experiment. This synergy required dosing temozolomide prior to PHA-848125. Additionally, combining PHA-848125 with both radiation and temozolomide resulted in a clear therapeutic gain without additional toxicity. The polypharmacology of this compound may contribute to its apparent efficacy for treating gliomas. Contributed by Mindy I. Davis
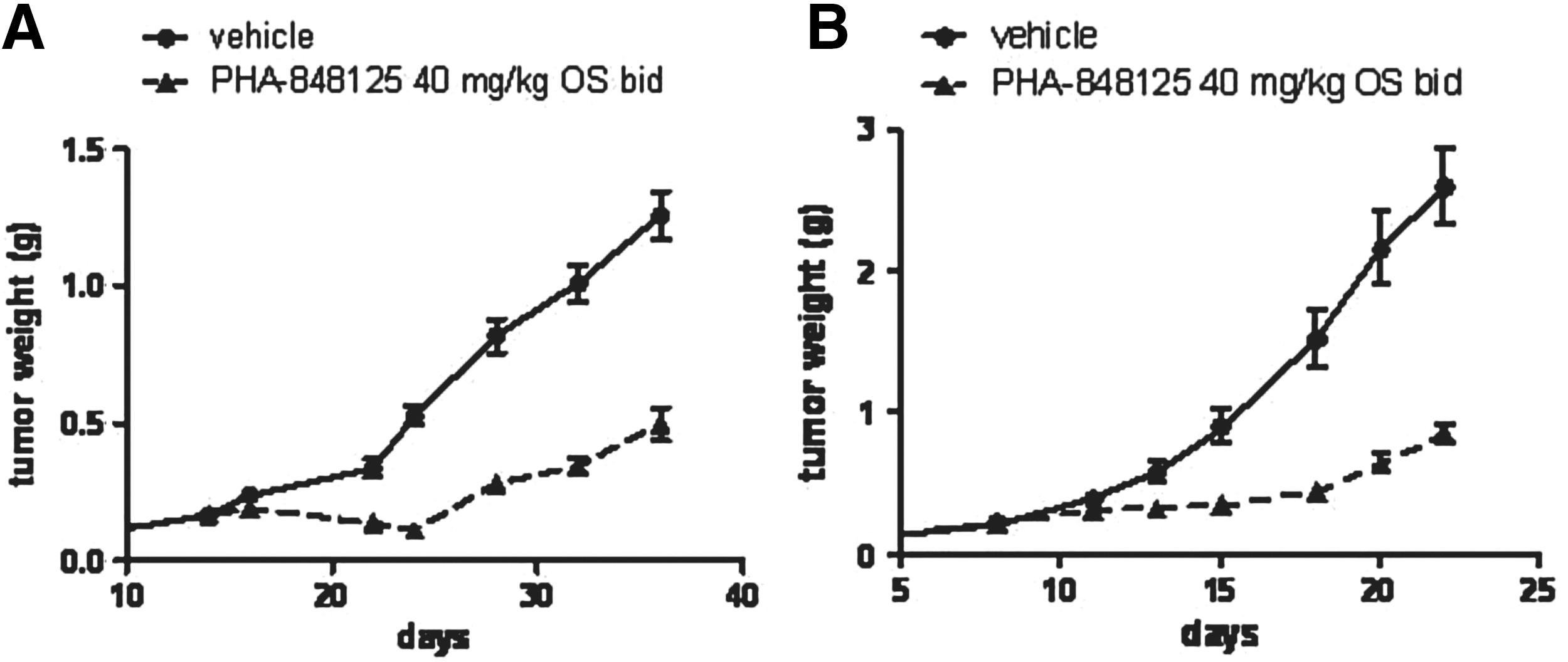
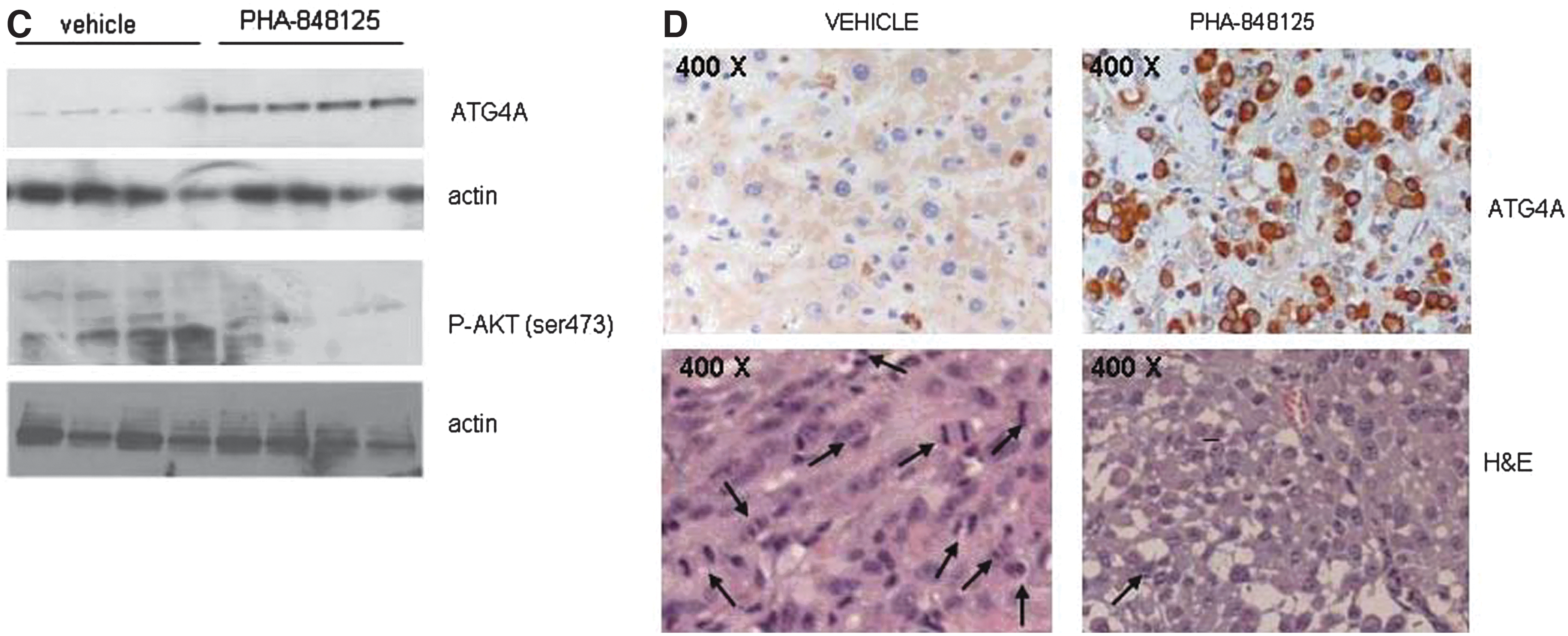
In vivo antitumor efficacy in subcutaneously implanted models. Animals were treated with vehicle or with PHA-848125 at 40 mg/kg twice a day for 10 consecutive days. (A) Tumor growth curves of U251 glioma model, treatment started on day 14; (B) tumor growth curves of U87MG glioma model, treatment started on day 8. For ex vivo analysis, 5 days after the last treatment, U87MG tumors from four control and four treated animals were excised. (C) Evaluation of ATG4A and P-AKT expression by Western blot analysis; (D) evaluation of ATG4A expression by immunohistochemistry. Images of the same tumors stained with H&E are reported. Morphological analysis allowed to count mitotic figures (indicated by arrows): CTR>100/field 200×; PHA 848125<10/field 200×. H&E, hematoxylin and eosin.
A Snapshot taken from the Inside
Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY: Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013;339:1328–1331.
Abstract: Microscopy and mass spectrometry (MS) are complementary techniques: The former provides spatiotemporal information in living cells, but only for a handful of recombinant proteins at a time, whereas the latter can detect thousands of endogenous proteins simultaneously, but only in lysed samples. Here, we introduce technology that combines these strengths by offering spatially and temporally resolved proteomic maps of endogenous proteins within living cells. Our method relies on a genetically targetable peroxidase enzyme that biotinylates nearby proteins, which are subsequently purified and identified by MS. We used this approach to identify 495 proteins within the human mitochondrial matrix, including 31 not previously linked to mitochondria. The labeling was exceptionally specific and distinguished between inner membrane proteins facing the matrix versus the intermembrane space (IMS). Several proteins previously thought to reside in the IMS or outer membrane, including protoporphyrinogen oxidase, were reassigned to the matrix by our proteomic data and confirmed by electron microscopy. The specificity of peroxidase-mediated proteomic mapping in live cells, combined with its ease of use, offers biologists a powerful tool for understanding the molecular composition of living cells.
Commentary:The approach reported by Rhee and colleagues promises to address a major deficiency of current proteomics platforms—their inability to report on protein distribution within the very small confines of discrete cell organelles and compartments. Here, the team uses a genetically targetable enzyme that catalyzes the generation of short-lived, highly reactive radicals in live cells; in turn, these radicals covalently react with electron-rich amino acids such as Tyr, Trp, His, and Cys. The enzyme selected was ascorbate peroxidase (APEX), chosen for its stability and activity in all cellular compartments. In initial studies, APEX was targeted to the mitochondrial matrix of human embryonic kidney (HEK) cells and labeling was started by adding biotin-phenol and H2O2 to the cell medium; the phenol portion of the reagent served as the source of radicals to tag the nearby proteins, while the biotin unit was intended for use in streptavidin-mediated microscopy analysis. Fixing the cells 1 min after labeling initiation served to also stop the APEX reaction (see
figure
). Analysis of the experiment revealed that the radicals produced did indeed have a very short range of action (few tens of nanometers), allowing for good spatial resolution of the labeling reaction, and that labeling occurred in a strict APEX- and H2O2-dependent manner. The proof-of-concept work was extended to include various cytosol-oriented APEX fusions, as well as a mitochondrial matrix facing variant. Analysis of the proteome labeled in the latter case revealed a number of proteins previously unknown to be associated with the mitochondria. The new technique appears to be well-validated through the present work and should be extendable to nearly all systems in which fusing APEX to an organelle- or compartment-targeting signal is available. Contributed by Anton Simeonov.
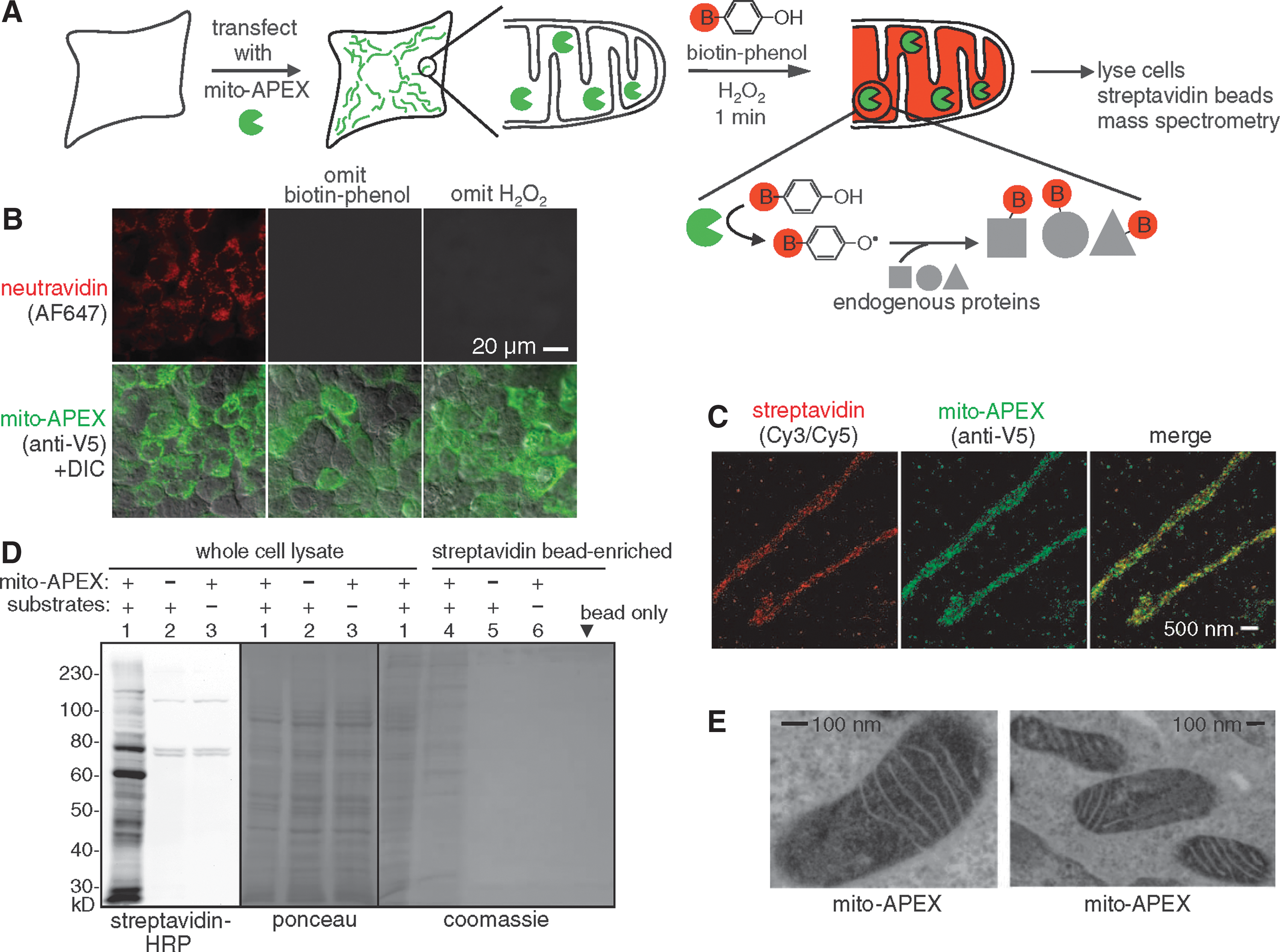
Labeling the mitochondrial matrix proteome in living cells. (A) Labeling scheme. The APEX peroxidase was genetically targeted to the mitochondrial matrix via fusion to a 24–amino acid targeting peptide (Martel et al., Nat Biotechnol 2012;30:1143–1148). Labeling was initiated by the addition of biotin-phenol and H2O2 to live cells for 1 min. Cells were then lysed, and biotinylated proteins were recovered with streptavidin-coated beads, eluted, separated on a gel, and identified by MS. The peroxidase-generated phenoxyl radical is short-lived and membrane-impermeant and, hence, covalently tags only neighboring and not distant endogenous proteins. (B) Confocal fluorescence imaging of biotinylated proteins (stained with neutravidin) after live labeling of HEK cells expressing mito-APEX as in (A). Controls were performed with either biotin-phenol or H2O2 omitted. (C) Superresolution stochastic optical reconstruction microscopy (STORM) images showing streptavidin and APEX (AF405/AF647) localization patterns at 22-nm resolution in U2OS cells. Samples were reacted as in (B). (D) Gel analysis of biotinylated mitochondrial matrix proteins, before (lanes 1 to 3) and after (lanes 4 to 6) streptavidin bead enrichment. Samples were labeled as in (B). Substrates are biotin-phenol and H2O2. Mammalian cells have four endogenously biotinylated proteins, three of which were observed in the negative control lanes (2 and 3) of the streptavidin blot. (E) Electron microscopy of HEK cells expressing mito-APEX. EM contrast was generated by treating fixed cells with H2O2 and diaminobenzidine. APEX catalyzes the polymerization of diaminobenzidine into a local precipitate, which is subsequently stained with electron dense OsO4 (Martel et al.). Dark contrast is apparent in the mitochondrial matrix, but not the intermembrane space. B, biotin; DIC, differential interference contrast.
Very Platonic
Zhu J, Larman HB, Gao G, Somwar R, Zhang Z, Laserson U, Ciccia A, Pavlova N, Church G, Zhang W, Kesari S, Elledge SJ: Protein interaction discovery using parallel analysis of translated ORFs (PLATO). Nat Biotechnol 2013;31:331–334.
Abstract: Identifying physical interactions between proteins and other molecules is a critical aspect of biological analysis. Here we describe PLATO, an in vitro method for mapping such interactions by affinity enrichment of a library of full-length open reading frames displayed on ribosomes, followed by massively parallel analysis using DNA sequencing. We demonstrate the broad utility of the method for human proteins by identifying known and previously unidentified interacting partners of LYN kinase, patient autoantibodies, and the small-molecules gefitinib and dasatinib.
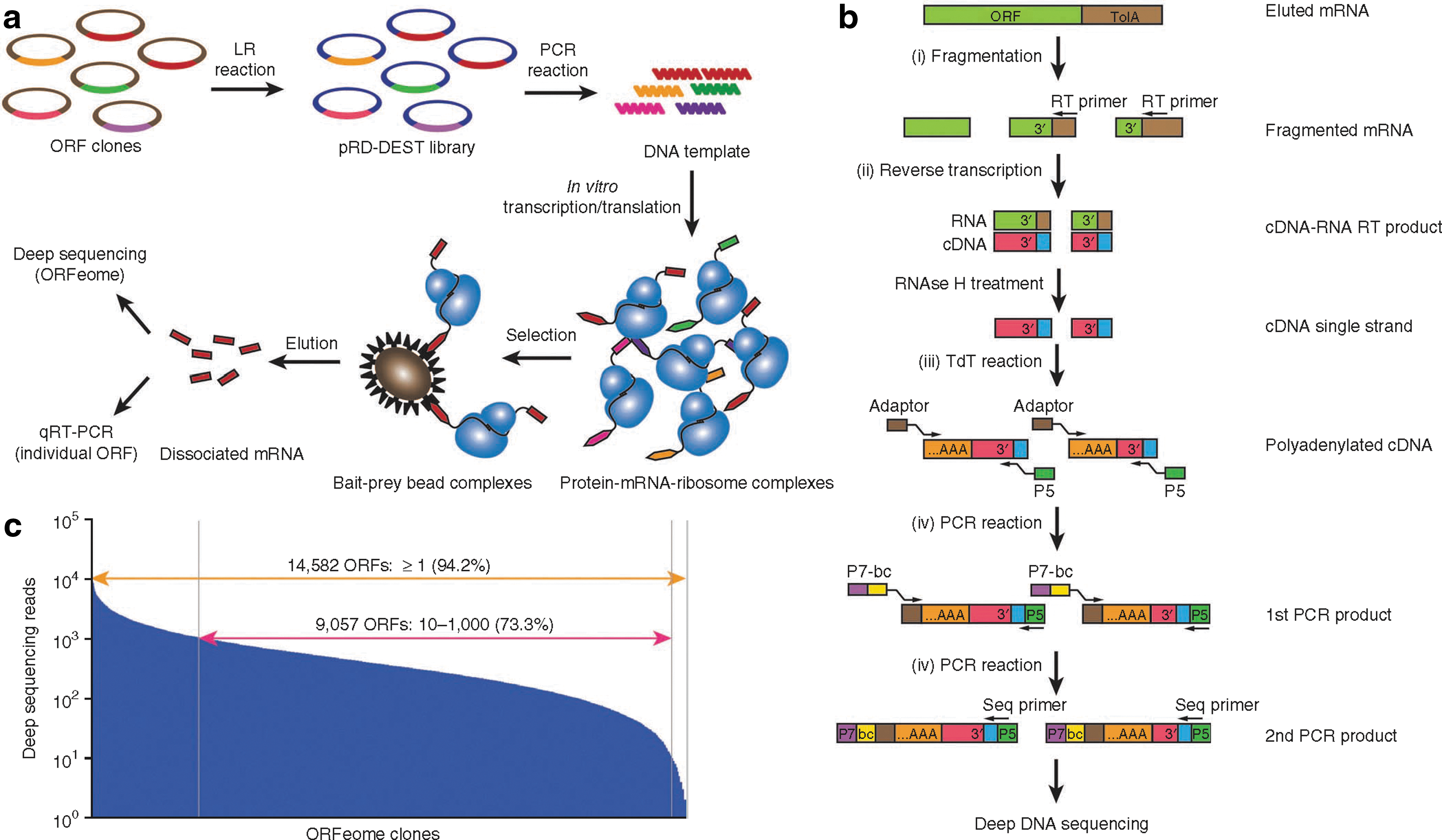
Commentary:High-throughput testing of the specificity of protein-binding ligands has typically involved the use of protein microarrays. However, these arrays typically require that the multiple proteins be purified and surface immobilized, which has made the overall technique difficult to expand to a wide range of targets and overall costly. The present approach, dubbed PLATO (parallel analysis of translated open reading frames [ORFs]), tackles the issue by binding analysis of in vitro displayed full-length proteins whose identity is determined through high-throughput DNA sequencing of the corresponding mRNA. Using ribosome display, the authors prepared a library of proteins based on the human ORFeome consisting of 15,483 cDNAs. Through the processes of in vitro transcription and translation, the proteins were produced, while the corresponding mRNAs remained tethered to them through the ribosomes. The ribosome-displayed library was screened for binding to surface-captured ligands of interest; after appropriate washing and mRNA elution steps, the top binders were elucidated through sequencing (see
figure
). The overall process of ribosome display, affinity selection, and sample preparation for sequencing could be automated for high-throughput application. The first proof-of-concept experiments involved mapping the interactome for LYN kinase, identifying protein targets for antibodies from autoimmune disease patients, and finally, profiling gefitinib, a small molecule epidermal growth factor tyrosine kinase inhibitor, for interactions with additional targets. While limitations of PLATO, such as inability to capture the entire proteome through the existing ORFeome libraries and potential complications associated with the spatial constraints posed by the ribosome display, are being discussed by the authors, it is expected that this new approach will have a following in the near future. Contributed by Anton Simeonov.
Parallel analysis of translated open reading frames (PLATO). (a) ORF display scheme. The pooled human open reading frame (ORF)eome v5.1 entry vector library is cloned by means of attL-attR (LR) recombination into the pRD-DEST expression vector. Expression plasmids are PCR amplified to generate the DNA templates for in vitro transcription. After in vitro translation, the protein–mRNA–ribosome complexes are incubated with protein, antibody, or small-molecule bait immobilized on beads. The enriched mRNA library is recovered from bait–prey bead complexes for further analysis. (b) Processing of mRNA samples for deep DNA sequencing. After fragmentation and reverse transcription (RT) using a universal primer to recover the 3′ end of ORFeome transcripts, cDNA is polyadenylated with terminal deoxynucleotide transferase (TdT) and amplified for multiplex deep sequencing using primers containing a sample barcode (bc) and the P5 and P7 Illumina sequencing adaptors. (c) Sequencing reads of the unenriched human pRD-ORFeome mRNA library (the “input” library). Most ORFs were sequenced at least once.
Predicting What Fits: GPCR Agonists
Weiss DR, Ahn S, Sassano MF, Kleist A, Zhu X, Strachan R, Roth BL, Lefkowitz RJ, Shoichet BK: Conformation guides molecular efficacy in docking screens of activated β-2 adrenergic G protein coupled receptor. ACS Chem Biol 2013;8:1018–1026.
Abstract: A prospective, large library virtual screen against an activated β2-adrenergic receptor (β2AR) structure returned potent agonists to the exclusion of inverse-agonists, providing the first complement to the previous virtual screening campaigns against inverse-agonist-bound G protein coupled receptor (GPCR) structures, which predicted only inverse-agonists. In addition, two hits recapitulated the signaling profile of the co-crystal ligand with respect to the G protein and arrestin mediated signaling. This functional fidelity has important implications in drug design, as the ability to predict ligands with predefined signaling properties is highly desirable. However, the agonist-bound state provides an uncertain template for modeling the activated conformation of other GPCRs, as a dopamine D2 receptor (DRD2) activated model templated on the activated β2AR structure returned few hits of only marginal potency.
Commentary:The recent availability of certain GPCR crystal structures has enabled virtual screens to identify new ligands and better understand the ligand-binding modes for this large class of cell surface receptors. These studies have found potent ligands, however, the GPCR crystal structures employed were all of the inactive receptor state, and all the compounds identified were found to be inverse agonists of the targeted receptor. Crystals structures of the inactive β2AR state and two activated high resolution structures of β2AR are available—one with a potent agonist bound (BI-167107) and another that used a G protein mimetic nanobody to stabilize the activated state. The preferential identification of inverse agonists in the previous virtual screening campaigns is surprising given the slight conformation changes found between activated and inactive states in the crystal structures of the β2-adrenergic receptor (β2AR). These past docking results could be due to a bias in the library or a genuine reflection of the different binding pockets. To address this question, the authors used the ZINC library containing approximately 3.4 million “lead-like” and “fragment-like” molecules in a virtual screen against both active and inactive structures of the β2AR. Docking was performed initially using 30 known agonists and 30 known inverse agonists along with similar “decoy” compounds to test the fidelity of the docking predictions. For these 60 known β2AR ligands, the activated structure was found to enrich for the 60 known ligands over the random computational decoys and to preferentially identify agonists. For example, 6 out of the 30 agonists were found in the top 1% of the data, while 22 of the 30 agonists were found in the top 10%. In contrast, docking to the inactive structure showed no agonists in the top 1% of the data and only 4 out of 30 agonists in the top 10%. However, the inactive structure identified 14 of 30 inverse-agonists in the top 10% of the data. The high frequency of inverse agonists found when virtual screens are performed with the inactive structure, and conversely the enrichment in agonists found when virtual screens are performed with the activated structure, support that the docking results reflect the different requirements of the binding site and not a bias in the library. As well, the agonists were found to dock in reasonable poses (see
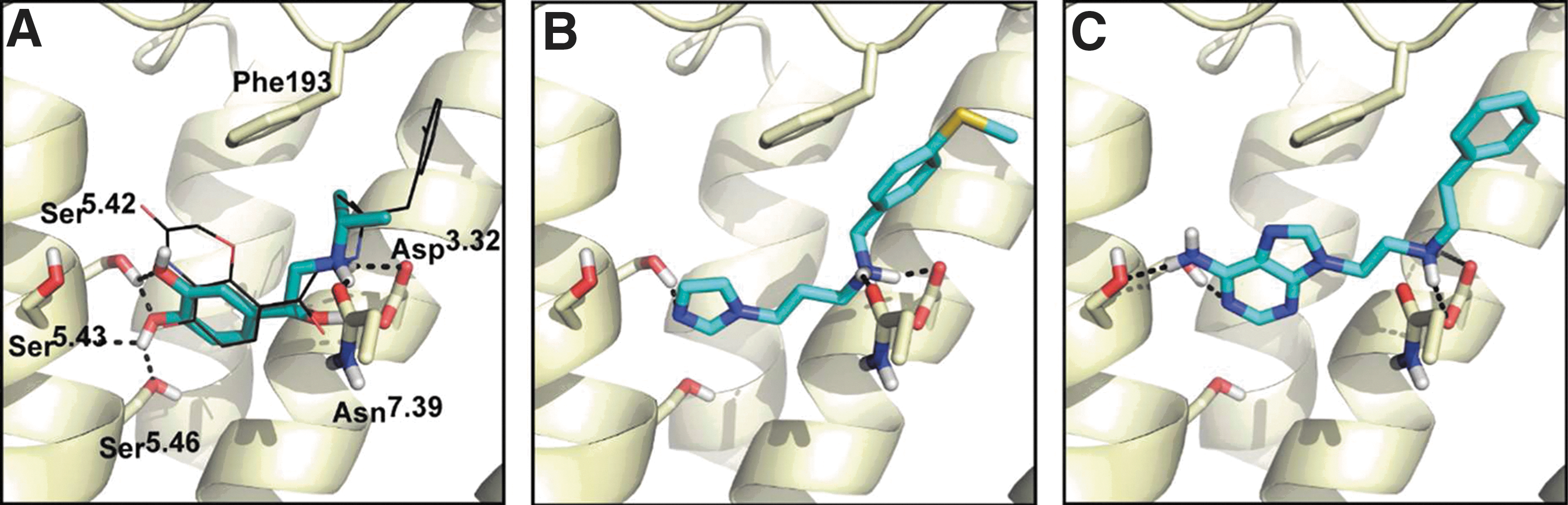
figure
). Next a virtual screen was performed against the 3.6 million member library, and 22 high-ranking compounds were tested for cAMP stimulation and β-arrestin recruitment. Increases in cAMP production were found for 6 of the 22 compounds, and four of these also resulted in β-arrestin recruitment. Two compounds showed some bias toward β-arrestin recruitment compared with cAMP stimulation. The authors then tried to use the β2AR activated structure as a template to construct a homology model for the dopamine D2 receptor (DRD2). Virtual screening against the modeled DRD2 structure showed much lower enrichments for agonists than the virtual screen for the β2AR, therefore suggesting that the activated structure of β2AR cannot serve as a general template to identify agonists for other GPCRs. This article illustrates the distinct, but subtle differences between GPCR binding pockets in inactive and active states. The virtual screening approach described here could enable the identification of new ligands as more GPCR crystal structures become available. Contributed by Doug Auld.
Two partial β2AR agonists with new activating chemotypes in their docked poses. (A) The docked pose of isoproterenol, with a salt-bridge between the amine group and key residue Asp1133.32 and hydrogen bonds to Ser2035.42, Ser2045.43, and Ser2075.46 in TM5. The co-crystal ligand BI-167107 is shown in black sticks. Previously unreported (B) imidazole, compound 10, and (C) amino-purine, compound 14, polar head groups make activating hydrogen bonds with TM5.