
Abstract
Maintenance of genome integrity by preventing and overcoming DNA damage is critical for cell survival. Deficiency or aberrancy in the DNA damage response, for example, through ataxia telangiectasia mutated (ATM) signaling, lead to pathophysiological perturbations in organs throughout the body. Therefore, control of DNA damage is of major interest for development of therapeutic agents. Such efforts will greatly benefit from convenient and simple diagnostic and/or drug development tools to demonstrate whether ATM and related genes have been activated and to then determine whether these have been returned to normal levels of activity because pathway members sense and also repair DNA damage. To overcome difficulties in analyzing differences in multitudinous ATM pathway members following DNA damage, we measured ATM promoter activity with a fluorescent td-Tomato reporter gene to interrogate the global effects of ATM signaling pathways. In cultured HuH-7 cell line derived from human hepatocellular carcinoma, cis-platinum, acetaminophen, or hydrogen peroxide caused DNA strand breaks and ATM pathway activation as shown by γH2AX expression, which in turn, led to rapid and sustained increases in ATM promoter activity. This assay of ATM promoter activity identified biological agents capable of controlling cellular DNA damage in toxin-treated HuH-7 cells and in mice after onset of drug-induced acute liver failure. Therefore, the proposed assay of ATM promoter activity in HuH-7 cells was appropriately informative for treating DNA damage. High-throughput screens using ATM promoter activation will be helpful for therapeutic development in DNA damage-associated abnormal ATM signaling in various cell types and organs.
Introduction
Numerous insults, including physiological processes, genetic perturbations, acquired disorders, environmental agents, chemicals, drugs, ionizing radiation, and so on cause DNA damage. To maintain DNA integrity, which is critical for cell survival and proliferation, elaborate and conserved responses to detect and repair DNA damage evolved. 1,2 The DNA damage response (DDR) contributes in cell fate decisions, that is, whether cells would remain viable and replicate or undergo cell cycle arrest and death. Therefore, controlling DDR to treat organ damage is highly significant. 3 Also, mitigating DNA damage, for example, during therapeutic use of alkylating agents or ionizing radiation, 4,5 is significant. Approaches to control DDR will greatly benefit from availability of simple, convenient, and effective screens to identify drugs, chemicals, or biological agents.
In DDR, seminal roles were identified many years ago of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and rad3-related (ATR) kinases and these remain of much interest. 2 Studies in several biological systems established ATM/ATR kinases preserve genomic stability by recruiting complex and as yet incompletely defined cascade of interacting partners. 6 In concert with sophisticated signaling mechanisms, ATM/ATR kinases are rapidly activated during DDR and through interlinked protein–DNA, protein–protein, and protein–RNA interactions, recognize DNA damage, followed by initiation of DNA repair mechanisms and/or cell cycle arrest. 1 –3 For instance, the complex of Mre11-Rad50-Nbs1 (MRN) proteins is important in recruiting ATM/ATR to DNA damage sites. Subsequently, dozens of genes play roles in intracellular signaling. Use of standard biochemical and molecular biology approaches with study at a time of one or more genes and proteins has been helpful in unraveling components of ATM signaling cascade. Typically, assays of ATM/ATR signaling concern study of DNA strand breaks, differences in gene expression with cytostaining methods, assays of RNA or protein levels, functional analyses with protein–protein interactions, kinase activities, and so on, and flow cytometry or other assays of cell cycling, apoptosis, cell death, and others. 6 –15 These require substrates ranging from cell extracts, prokaryotic organisms, for example, xenopus laevis, and mammalian cells. 6
Cell-based screens utilized activation of ATM/ATR pathway members or events to identify potential drug candidates. 10,16,17 However, demonstrating differences in ATM/ATR signaling at a global level in whole cells has been difficult. For instance, issues are posed by potential redundancies in interactions among ATM/ATR signaling genes, 3 release of toxic cytokines by cells with ATM-related DDR, 17 transcriptional regulation of ATM and other genes by extracellular molecules, such as growth factors, 18 transcriptional or posttranscriptional regulation of individual genes by intracellular molecules, such as microRNAs, 19 alterations in ATM protein itself after oxidative damage, 20 and unknown nature of feedback loops driving expression of ATM and related genes during DDR.
We considered that after DDR, if feedback loops demanded replenishment of ATM kinase via greater gene transcription in cells, this could offer suitable assays for interrogating the sum of ATM signaling. This possibility was suggested by rapid increases in ATM promoter activity after DNA damage in intact mice. 21 For proof-of-principle, we used HuH-7 cell line, which was derived from human hepatocellular carcinoma, 22 and these cells exhibit gene expression profiles associated with mature hepatocytes. 23 We permanently modified these cells with a lentiviral vector (LV) to express under human ATM promoter a fluorescent tdTomato (tdT) reporter gene. 24 Transduced HuH-7 cells showed DDR with anti-neoplastic drug, cis-Platinum (cis-P), antipyrectic drug, acetaminophen (APAP), and pro-oxidant chemical, hydrogen peroxide (H2O2), which permitted screening of candidate agents to protect cells from DNA damage.
Materials and Methods
Drugs, Chemicals, and Growth Factors
Stock solutions of 3 mM cis-P and 5 mM APAP (Sigma Chemical Co., St. Louis, MO) were prepared in normal saline or dimethyl sulfoxide, respectively. H2O2 (Sigma Chemical Co.), fibroblast growth factors (FGFs), hepatocyte growth factor (HGF), insulin-like growth factor (IGF)-1, vascular endothelial growth factor (VEGF) (Peprotech, Rocky Hill, NJ), and filgrastim (analog of granulocyte-colony stimulating factor, GCSF) (Amgen, Inc., Thousand Oaks, CA) were diluted in cell culture medium.
Lentiviral Vector
Human peripheral blood mononuclear cells were used to extract genomic DNA with polymerase chain reaction (PCR) amplification of ATM promoter sequence (Tm: 55°C, 35 cycles) and flanking of ClaI restriction site in forward primer, 5′-TCCATCGATGGAGATCAA AACCACAGCAGG-3′, and XbaI restriction site in reverse primer, 5′-CCCTCTAGAGGGCGTTCTCTCGCCTCCTCCC GTG-3′. 18 Amplified locus spanned 108,093,264 to 108,093,849 positions on chromosome 11 (UCSC Genome Bioinformatics, Santa Cruz, CA). After digestion with ClaI and XbaI, PCR product was ligated upstream of tdT reporter gene in third generation LV. Positive clones were confirmed by commercial sequencing. LV with cytomegalovirus (CMV) promoter upstream of tdT reporter gene was used as nonspecific control. LV with tdT reporter under ATM or CMV promoters was prepared as previously described. 25 Cells were transduced with LV at 10 multiplicity of infection. Fluorescence of tdT was analyzed by microscopy and flow cytometry.
Reporter HuH-7 Cell Lines
Key steps of the protocol are described in Table 1. Cells were cultured in RPMI 1640 medium 10% fetal bovine serum, 2 mM L-glutamine, and 1% penicillin-streptomycin (10,000 U/mL, Cellgro; Mediatech, Inc.) under 5% CO2 at 37°C. For cell viability and promoter activity assays, 8×104 cells were plated per well in 48-well dishes and cultured overnight with or without cis-P, APAP, or H2O2. After 16–18 h, cells were washed twice with phosphate-buffered saline (PBS), pH 7.4, and incubated for 30 min with 6 μg/mL Hoechst 33428 (Sigma Chemical Co.). After 2 more washes with PBS fluorescences were measured by a FLUOstar plate reader (BMG Lab Technologies, Life Technologies, Carlsbad, CA) at 520 nm (excitation) and 590 nm (emission) for Hoechst 33,428 and 355 nm (excitation) and 460 nm (emission) for tdT.
Protocol for ATM Promoter Expression Analysis
1. Using cells transduced from organs or tissues of interest with lentiviral constructs to express ATM promoter-driven reporter, for example, tdTomato or another.
2. Negative controls containing empty vector and also no treatment.
3. Examples of DNA damage-inducing agents are cisplatinum, hydrogen peroxide, and others. Stock solution in normal saline of cisplatinum is 3.3 mM and of hydrogen peroxide is 10 mM. The final dose range will require tittering according to given cell types. Additional compounds may be tested one at a time or together to elicit cytotoxicity. Other compounds may be added to elicit cytoprotection.
4. Cell culture plates are incubated under gas exchange conditions for desired period.
5. Hoechst 33428 dye–stock, 6 mg/mL in normal saline, PBS or culture medium, final concentration, 6 μg/mL. May use another assay, for example, calcein AM incorporation.
6. Reading in spectrofluorometer, with necessary optical filters for example, FLUOstar plate reader (BMG Lab Technologies, Life Technologies, Carlsbad, CA). ATM promoter activity is corrected for viable cells in individual measurements=(tdTomato fluorescence)/(Hoechst 33428 fluorescence). Each condition is assayed in 3–6 replicates. P values are obtained for assigning significance to differences and should be at least <0.05 with established statistical tests.
Comet Assay for DNA Strand Breaks
Previous procedures were used with minor modifications for this well-established test of DNA breaks. 26,27 Slides were dipped in methanol overnight, air-dried, dipped in 0.5% normal melting-point agarose (NMPA) (Sigma Chemical Co.), and redried. Then, 80 μL of 0.5% NMPA was rapidly covered with coverslip and gelled for 5 min at 4°C. To 500 μL of 0.5% low melting-point agarose (LMPA) (Cambrex, East Rutherford, NJ), 50 μL of 1×105 cells/mL PBS was added, with pipetting of 50 μL mixture beneath coverslip of NMPA slide and cooling at 4°C for 5 min. Next, slides were incubated in cold lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris [pH 10], 1% sodium lauryl sarcosine, plus freshly added 1% Triton X-100), in dark at 4°C for 1 h followed by wash in water and incubation in alkali unwinding solution (300 mM NaOH and 1 mM EDTA, pH 13) in dark at 4°C for 1 h. After another wash in water, slides were immersed in 1×TAE buffer with application for 1 h of 25 V at 300 mA. Slides were neutralized for 5 min in 0.4 M Tris, pH 7.5, immersed in 70% ethanol for 5 min, air-dried, and stained with 0.02 mg/mL ethidium bromide (Promega, Madison, WI). Comets were examined under 200× under epifluorescence (Observer Z.1 Zeiss) and 50 randomly selected cells were analyzed per sample in triplicate conditions. Comet tail length was measured with imaging software (Comet Score; TriTek Systems).
Gene Expression
RNA was extracted by TRIzol Reagent (Life Technologies) and cleaned by RNeasy (Qiagen Sciences). Human DNA Damage Signaling Pathway (PAHS-029) RT 2 Profiler PCR Array, RT 2 Real-Time SyBR Green PCR Mix, and RT 2 First Strand Kit were used, with cDNA synthesis and PCR according to manufacturers' instructions (SABiosciences, Life Technologies). Data were analyzed by 2−ΔΔCt method. Gene expression was normalized against housekeeping genes in individual samples. Fold-changes in gene expression were determined by log-normalized ratios of treated/untreated cells and ≥2-fold differences were considered significant.
Cell Immunostaining
Cells were fixed in 4% paraformaldehyde in PBS, blocked/permeabilized in 5% goat serum, 0.2% Triton X-100 (Sigma Chemical Co.) in PBS for 1 h, and incubated overnight at 4°C with rabbit anti-phosphoS139 H2AX (1:300, ab2893; Abcam) or mouse anti-phosphoS1981 ATM (1:100, 56007; BD Pharmingen, followed by anti-rabbit Alexa Fluor 546 IgG (1:500) or anti-mouse Alexa Fluor 488 IgG (1:300) (Molecular Probes, Life Technologies), respectively, for detection.
Animal Studies
Protocols were approved by Institutional Animal Care and Use Committee at Einstein. Healthy male C57BL/6 mice of 8–10 week age and weight of 25–30 g were from Special Animal Core of Liver Research Center. Mice were maintained under standard light/dark conditions. To induce liver injury, 500 mg/kg i.p. APAP in normal saline was given. To some animals APAP was followed by 100 μg GCSF i.p. Animals were sacrificed if moribund.
Statistical Methods
Data were expressed as mean±SEM. Differences was analyzed by t-tests, Chi-square test, or ANOVA as appropriate. Linear regression and Pearson correlation was also performed. These analyses used SigmaStat 3.0 (SysStat). P values<0.05 were considered significant.
Results
Generation of HuH-7 Cells Expressing tdT Under ATM Promoter
HuH-7 cells were efficiently transduced and expressed tdT under ATM and CMV promoter, whereas transduction of cells with empty LV produced no signals (Fig. 1a–c). The viability and proliferation capacity of cells transduced with empty LV was unchanged (not shown). In unselected populations, 37% and 53% HuH-7 cells expressed tdT under ATM or CMV promoter, respectively. We designated these LV-transduced lines as HuH-7-ATM Prom-tdT and HuH-7-CMV Prom-tdT cells. To quantitate differences in the levels of ATM and CMV promoter activity, we utilized FACS, which showed mean fluorescent intensity (MFI) of tdT reporter was greater in HuH-7-CMV Prom-tdT cells, 53.8 versus 22.8 arbitrary units, respectively, indicating basal ATM promoter activity was approximately 2-fold less than CMV promoter activity, P<0.05.

HuH-7-ATM-Prom-tdT and HuH-7-CMV-Prom-tdT cells.
DNA Damage and ATM Signaling in HuH-7 Cells
After cis-P, APAP, or H2O2, phase contrast microscopy revealed morphological evidences of cytotoxicity with rounding, cytoplasmic vacuolation or nuclear fragmentation in HuH-7 cells (Fig. 2a–d). Cell viability assays with Hoechst 33342 dye staining of nuclear DNA showed 50% inhibitory concentrations (IC50) were approximately 15 μM, 10 mM, and 100 μM for cis-P, APAP, and H2O2, respectively, which were then used for further studies. Under these conditions, >80% cells expressed γH2AX histone, indicating onset of DDR. Cells showed pan-nuclear and punctate nuclear staining for γH2AX, which confirmed single-stranded and double-stranded DNA breaks, respectively. 15 Comet assays showed the extent of DNA strand breaks was generally similar after cells were exposed to toxins.
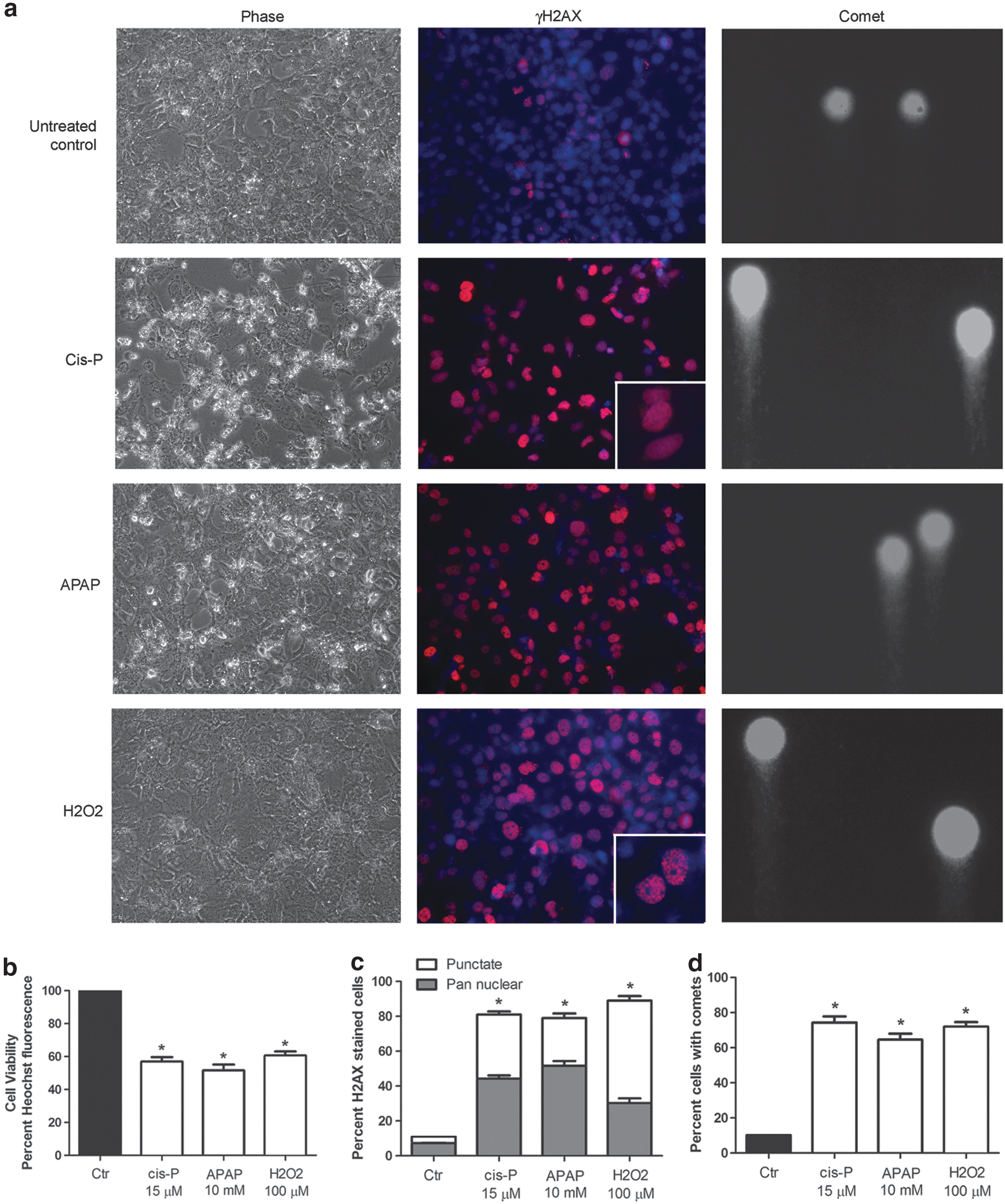
ATM-related DDR in HuH-7 cells.
DDR and ATM Promoter Activity in HuH-7 Cells
We anticipated onset of DDR will rapidly induce ATM promoter activity if feedback loops in ATM signaling demanded ATM gene transcription. After cis-P, APAP, or H2O2 for 24 h, ATM promoter activity increased by 4–8-fold in HuH-7-ATM Prom-tdT cells, compared with toxin-untreated control cells, P<0.05 (Fig. 3a). However, HuH-7-CMV Prom-tdT cells exposed to cis-P, APAP, or H2O2 exhibited decreases in viability but without alterations in CMV promoter activity, indicating tdT expression was unchanged (Fig. 3b). When HuH-7 cells were cultured with cis-P in a series of low (5 μM) and high (25 μM) concentrations, we observed dose-dependent cytotoxicity, which was as expected based on the IC50 concentration of 15 μM (Fig. 3c). We observed ATM promoter activity was significantly increased even when cells were cultured in the lowest drug concentration of 5 μM with limited step-up in higher drug concentrations indicating this assay was quite sensitive (Fig. 3d). Similarly, the assay was sensitive for toxicity induced by APAP. Moreover, cytostaining of cells verified that greater tdT expression in drug-treated cells was associated with increased expression of endogenous phosphorylated ATM protein (Fig. 3e). Therefore, greater ATM promoter activity in HuH-7-ATM Prom-tdT cells after cis-P, APAP, or H2O2 was due to DDR and ATM signaling. The ATM promoter response in HuH-7-ATM Prom-tdT cells was evident within 2–4 h after exposure of cells to toxins prior to loss of cell viability (not shown). Taken together, we surmised that ATM signaling and feedback loop-driven demand for ATM transcription in cells was rapid and persistent under this setting.

Promoter responses in HuH-7 cells treated overnight with cis-P, APAP, and H2O2.
To determine whether ATM promoter activity corresponded to DDR in other ways, for example, at gene expression level, we used quantitative reverse-transcription real-time PCR with a commercial array of 84 relevant genes in DNA Damage Signaling Pathway Array (PAHS-029, SABiosciences). Gene expression in vehicle-treated and toxin-treated HuH-7-ATM Prom-tdT cells was markedly different (Table 2). Annotated gene lists indicated cis-P toxicity was responsible for largest differences in DDR, for example, multiple genes other than ATM, ATR, and MRN complex (MRE11A, RAD50, and NBS1), were downregulated (Supplementary Table S1; Supplementary Data are available online at
Differences in Numbers of DNA Damage Response Genes Expressed in Toxin-Treated Cells Versus Untreated Control HuH-7 Cells
ATM Promoter Activity as Screen to Identify Drugs Capable of Mitigating DDR
Previous studies suggested ATM signaling could be regulated by cellular growth factors, for example, GCSF, HGF, and others. 18 Similarly, IGF-1 was found to attenuate cis-P-induced activation of ATM, ATR, and their targets CHK2, CHK1, and TP53 along with other aspects of DDR. 28 Therefore, we performed a “mini-screen” with ATM promoter activity in HuH-7-ATM Prom-tdT cells to examine whether several growth factors could attenuate toxin-induced DDR and ATM signaling. If so, we should have observed higher cell viability and lower DNA damage.
After culture of HuH-7-ATM Prom-tdT cells for 16–18 h with cis-P, APAP, or H2O2 with and without FGF-1/2, GCSF, IGF-1, or VEGF, ATM promoter activity significantly declined, P<0.05 (Fig. 4). In mock-treated cells with empty LV we observed no differences related to background fluorescence. In cases of GCSF and IGF-1, ATM promoter activity declined by up to 75% against toxin-treated cells. In case of FGF-2, ATM promoter activity declined to 34%–63% of promoter activity in control cells with cis-P, APAP, or H2O2 alone. VEGF and FGF-1 less effectively restored ATM promoter activity, which was 31%–50% of promoter activity in cells with cis-P, APAP or H2O2 alone. HGF was ineffective. Restoration of ATM promoter activity was reflected in improved cell viability after culture of cells with cis-P, APAP, or H2O2 plus GCSF, IGF-1, FGF-2, VEGF, and FGF-1.

ATM promoter activity to screen for drugs to protection against DNA damage. Differences in cell viability and ATM promoter activity after HuH-7-ATM Prom-tdT cells were cultured with only cis-P
Changes in ATM promoter activity and cell viability after treatment of HuH-7-ATM Prom-tdT cells with growth factors, that is, GCSF, IGF-1, FGF-2, VEGF, and FGF-1, correlated with one another (Fig. 5). To determine whether improvement in cell viability and ATM promoter activity were related to amelioration of DDR in growth factor-treated cells, we performed Comet assays for DNA strand breaks. In cis-P-, APAP-, or H2O2-treated cells, the number of cells with Comet formation and comet tail-lengths, which represents the extent of DNA strand breaks, decreased after GCSF (Fig. 6).
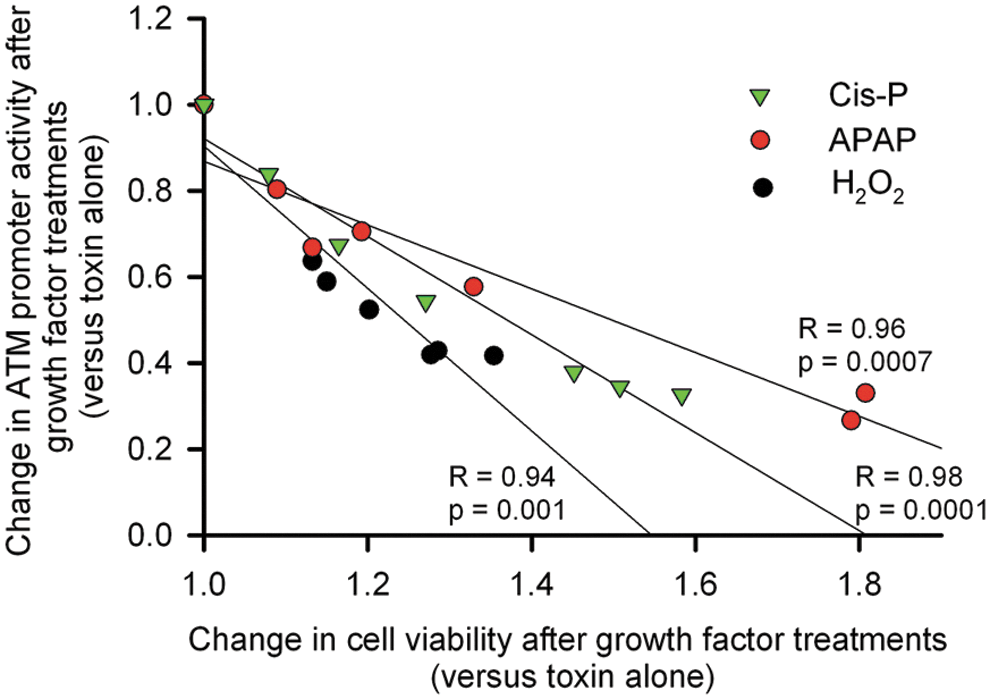
Correlations between changes in ATM promoter activity and cell viability. Linear regression analysis of cells treated with cis-P, APAP, or H2O2 alone and with toxins plus GCSF, IGF-1, FGF-1, and −2, and VEGF. Correlation coefficients calculated by Pearson method and p-values are indicated. The extent by which ATM promoter activity induced by toxins was decreased correlated highly significantly with cell survival with protective factors.
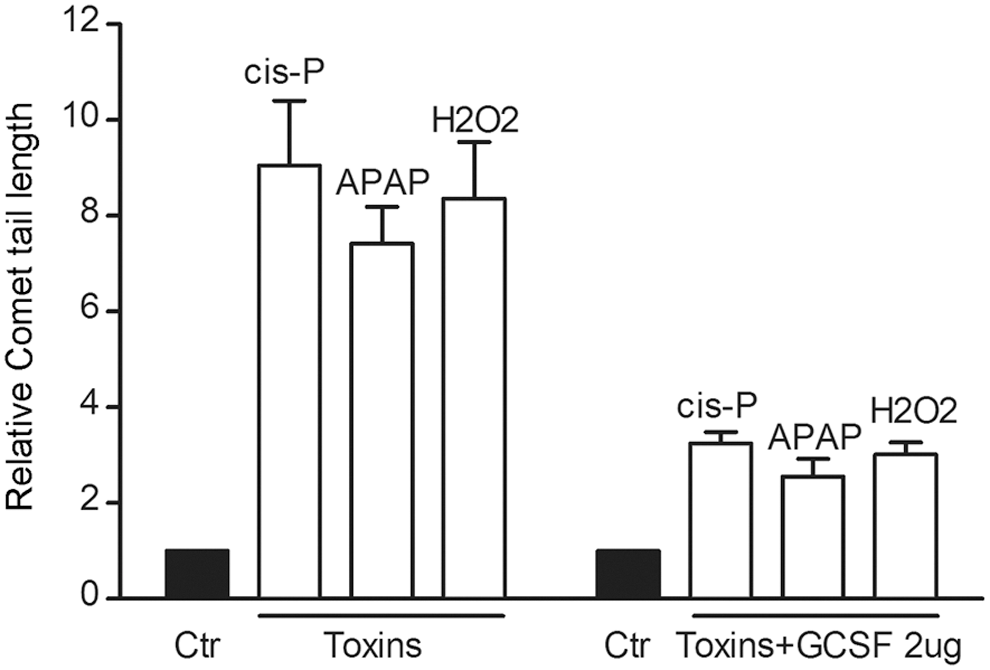
Amelioration of toxin-induced DNA damage in HuH-7 cells. Comet assays and comet tail length measurements showed significant amelioration of cis-P, APAP, or H2O2-induced DNA strand breaks after GSCF. Comet tail lengths decreased in GCSF-treated cells by 3–5-fold, P<0.05.
To determine whether restoration of ATM signaling and cytoprotection from toxins could be realized in vivo, we studied APAP-induced acute liver failure (ALF) in C57BL/6 mice. After 500 mg/kg APAP i.p. (n=12), 50% (n=6) of mice died over 30 h. By contrast, when 1 h after 500 mg/kg APAP, 100 μg/kg GCSF i.p. was given, 83% (n=10) mice survived (n=12) over 14 d duration of study, P<0.001. After APAP, mice showed perivenous necrosis and widespread γH2AX expression by immunostaining, whereas after APAP plus GCSF, liver histology improved and γH2AX expression substantially declined (not shown).
Discussion
These results indicated successful interrogation by tdT reporter of ATM signaling and feedback loop demanding ATM transcription and the ability to identify candidates to restore cell viability. Despite improving insights into DDR and ATM signaling in DNA damage repair, cell cycling, and others, major uncertainties remain in mechanisms by which individual pathway members interact and contribute in DDR. 1,2 Typically, analysis of ATM-related changes requires multiple and often cumbersome assays of DDR. 6 The processes include post-translational modifications, for example, kinase activities, of ATM and other members of ATM and additional pathways. Our studies were restricted to DNA toxicity although ATM pathways subserve roles in many pathophysiological or disease states. Therefore, effective approaches to generate global readouts of ATM signaling should be very attractive for a broad range of investigations but further analysis will be appropriate in given conditions to establish the utility of proposed ATM promoter assay.
The tdT reporter in intact cells showed ATM promoter activity increased after cytotoxicity with well-established toxins. For instance, Cis-P is widely used for chemotherapy, APAP is the major cause of ALF in the western world, 29 and H2O2 is routinely used as an inducer of oxidative stress. 20 In case of all 3 toxins, we verified onset of DDR in HuH-7 cells with conventional assays of DNA strand breaks, that is, expression of γH2AX and Comet assays. The latter technique has been widely adopted to verify presence of double-stranded DNA breaks followed by quantitation of the extent of such changes by Comet tail-length analysis with commercial software, as in our studies here, and extending to not only cells but whole tissues too. 30 However, gene expression was far more different in cis-P-treated cells versus APAP- or H2O2-treated cells, despite substantial cytotoxicity and DNA strand breaks in all 3 instances. This likely reflected differences in the mechanisms of cytotoxicity. While cis-P is thought to produce damage by DNA adducts, 4 H2O2 or APAP produce cytotoxicity through oxidative stress, including biochemical alterations in ATM protein itself. 20,28 These changes are difficult to identify without sophisticated methodologies. By contrast, analysis of ATM promoter activity was simple and rapidly demonstrated onset of DDR after cis-P, APAP, or H2O2. The difference in ATM promoter activity was also much simpler than gene expression analysis of multiple ATM signaling pathway members.
The ATM promoter regulation in cells exposed to toxins served as effective screen for candidate cytoprotective agents. We used a mini-screen with growth factors with the ability to regulate ATM promoter, for example, GCSF, HGF, 18 or to alter intracellular signaling after cis-P-induced cytotoxicity. 27 We chose growth factor concentrations known to be within the range of physiological responses in cell culture studies. This screen established GCSF was particularly helpful for cytoprotection against cis-P, APAP, or H2O2. Moreover, a connection between restoration of ATM promoter activity and DDR was confirmed by Comet assays. Remarkably, GCSF was effective in APAP-induced hepatic injury in vivo. While GCSF was shown to protect organs in vivo, 31 –33 it was presumed to recruit hematopoietic cells to injured liver, whereas our results established that GCSF regulated intracellular ATM signaling. Although growth factors contribute to oncogenetic processes and ATM pathway may be dysregulated in cancer cells, 34 our purpose was to simply establish proof-of-principle in short-term culture conditions. Since HuH-7 cells were derived from hepatocellular carcinoma, transient growth factor exposures should not have introduced confounding due to perturbations in additional oncogenetic events or processes. Moreover, our results were verified in intact animals under a noncancer setting.
The rapidity by which DDR increased ATM promoter activity in HuH-7 cells was similar to regulation of murine ATM promoter in animals exposed to ionizing radiation. 21 This feedback loop driven regulation of ATM promoter activity in DDR offers possibilities to develop high-throughput assays for screening biological agents, small molecules, and drugs to ameliorate cytotoxicity. One could envision cell panels representing multiple tissues with ATM promoter reporter as diagnostic and drug discovery tools for DDR in multi-well format. As a variety of insults affecting organs throughout the body, including genetic and acquired disorders, induce ATM signaling, opportunities for therapeutic development will be broad. On the one hand, this will concern prospective organ protection, such as from chemotherapy or radiotherapy, and on the other hand, treatment of organ damage, degenerative diseases, or aging, where DDR and ATM signaling serve pathophysiologic roles, will also be relevant. Nonetheless, the underlying mechanisms driving and/or mitigating DDR under various settings are full of complexities, including involvement of DNA repair genes, cell cycle suppressor genes, and other genes, including as yet undiscovered genes. For instance, the MKK stress-activated protein kinase and ARF have recently been recognized as sensors of oxidative stress and participants in ATM/ATR-mediated DDR, leading to implications in cell repair, apoptosis, cycling, or oncogenesis, 35 although evaluation of the proposed ATM promoter approach could be helpful for these or other prospectively defined conditions.
Footnotes
Acknowledgments
Supported in part by NIH grants R01 DK071111 and R01 DK088561 and by Core facilities supported by P30-DK41296 and P30 CA13330.
Disclosure Statement
The authors declare no conflicts of interest.