
Abstract
Recent progress in embryonic stem cell (ESC) and induced pluripotent stem cell (iPSC) research led to high-purity preparations of human cardiomyocytes (CMs) differentiated from these two sources—suitable for tissue regeneration, in vitro models of disease, and cardiac safety pharmacology screening. We performed a detailed characterization of the effects of nifedipine, cisapride, and tetrodotoxin (TTX) on Cor.4U® human iPSC-CM, using automated whole-cell patch-clamp recordings with the CytoPatch™ 2 equipment, within a complex assay combining multiple voltage-clamp and current-clamp protocols in a well-defined sequence, and quantitative analysis of several action potential (AP) parameters. We retrieved three electrical phenotypes based on AP shape: ventricular, atrial/nodal, and S-type (with ventricular-like depolarization and lack of plateau). To suppress spontaneous firing, present in many cells, we injected continuously faint hyperpolarizing currents of −10 or −20 pA. We defined quality criteria (both seal and membrane resistance over 1 GΩ), and focused our study on cells with ventricular-like AP. Nifedipine induced marked decreases in AP duration (APD): APD90 (49.8% and 40.8% of control values at 1 and 10 μM, respectively), APD50 (16.1% and 12%); cisapride 0.1 μM increased APD90 to 176.2%; and tetrodotoxin 10 μM decreased maximum slope of phase to 33.3% of control, peak depolarization potential to 76.3% of control, and shortened APD90 on average to 80.4%. These results prove feasibility of automated voltage- and current-clamp recordings on human iPSC-CM and their potential use for in-depth drug evaluation and proarrhythmic liability assessment, as well as for diagnosis and pharmacology tests for cardiac channelopathy patients.
Introduction
The last 60 years represented a glamorous period in cardiac cellular electrophysiology and mathematical modeling of cardiac action potentials (APs), 1,2 marked by successive identification of individual ion current components that interplay to generate its well-known four phases. Cellular preparations evolved steadily from Purkinje network fragments (“false tendons”), used in 1951 by Draper and Weidmann 3 in microelectrode impalement experiments, and then in voltage-clamp experiments, allowing biophysical characterization of multiple K+ current components—the inward rectifier I X1 (renamed I K1) and delayed (outward) rectifier I X2 (comprising a fast component and a slow component, named I Kr and I Ks), 4 the transient outward currents I to1 5 –8 and I to2 9,10 (contributed by K+ and Cl− ions, respectively), the “funny” or hyperpolarization-activated current I f, 11 I Na, the “slow inward” calcium current (via L-type Ca2+ channels, I CaL), 12,13 and others. Isolated cardiomyocytes (CMs) obtained via enzyme dissociation and suitable for patch-clamp experiments were introduced later, and stem-cell-derived, cultured CMs are the most recent development in the field. These technology breakthroughs resulted in improved in vitro assays of arrhythmogenic or antiarrhythmic effect of drug candidates. Beyond cardiac risk assessment of drug effects at whole organ or tissue level, accomplished with Langendorff-type or myocardial wedge preparations, a highly successful approach during the last decade was the testing of inhibitory effects of compounds on human Ether-á-go-go related gene (hERG) channels expressed in different cell lines using automated patch-clamp methods. The rationale of this approach is a peculiar structural feature of these channels, underlying I Kr. The lack of two proline residues (the “Pro-X-Pro” sequence) in the C-terminal region of transmembrane helix S6, lining the permeation pathway, and their replacement with a more flexible motif (the Ile-Phe-Gly sequence for residues 655–657 in hERG), 14 renders them “pharmacologically promiscuous” due to a larger-than-normal inner vestibule that can be easily occupied by a variety of chemical compounds. This results in open-channel block, impaired repolarization, appearance of early after depolarizations (EADs) in the cardiac AP that may synchronize and lead to severe arrhythmias like torsades-des-pointes or polymorphic ventricular tachycardia. 15 According to current estimates, ∼40% of potential drug candidate lead compounds have to be withdrawn from the research pipelines due to arrhythmia risks via hERG blockade, not to mention the innumerable complications and major financial losses subsequent to withdrawal from the market of an already approved and commercially available drug. 16 However, a new paradigm, based on experimental evidence of the last 10 years, is challenging the already classical high-throughput hERG screening. It considers that many drugs discarded according to hERG-blocking criteria alone do not bear in fact a clinically manifest “torsadogenic” risk. In an outstanding study, 17 Kramer et al. defined the MICE (Multiple Ion Channel Effects) cardiac safety assay to have a better predictor for clinical outcome. Based on screening of drug effects on multiple channels, including at least hERG (I Kr), Cav1.2 (I CaL), and Nav1.5 (I Na), and correlating these data with the known “torsadogenic” risks via multiple logistic regression models, a model could be chosen with a much better predictive value than hERG screening alone. However, there were still a significant number of false-positive and false-negative compounds, where the arrhythmogenic risk was incorrectly predicted by the model. These findings have been endorsed by a number of regulatory agencies and high-profile research institutes, in the CiPA (Comprehensive in vitro Pro-Arrhythmia Assay) 18 initiative. The aim of this initiative consists in assessment of proarrhythmia scores based on a combination of voltage-clamp experiments on multiple cardiac ion channels, computer modeling of pharmacological effects on cardiac AP using advanced cardiac cell electrophysiology models, 19 and—as a final control—AP recordings in human stem-cell-derived CMs.
Accomplishment of this last approach (i.e., human stem-cell-derived CM AP recordings) has benefited substantially from recent progress in the field of CM differentiation starting from embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs), 20 obtained in turn from adult differentiated cells via transfection with pluripotency factor genes using the Yamanaka method 21 or later variants derived from it. Use of antibiotic resistance tags both for induction of pluripotency and for redifferentiation resulted in high-purity (over 99% CMs) cellular preparations, suitable for automated electrophysiology experiments. 22 Commercial availability of human iPSC (and ESC)–derived cardiomyocytes (hiPSC-CMs) allowed in recent years development of a variety of high-throughput cardiac safety pharmacology assays, based on optical AP and intracellular calcium transient recordings, 23 field potential recordings of AP propagation and activation maps with multielectrode array devices, 24 or impedance measurements on CM monolayers with interdigitated electrodes. 25 Although each of these methods presents advantages as well as shortcomings, in view of the CiPA guidelines, we consider that automated patch-clamp technology offers the most complete and accurate assessment of the proarrhythmic liability of a drug candidate, and in-depth exploration of its arrhythmogenic mechanisms at cellular level. Therefore, the aim of our study was to prove and explore the feasibility of pharmacology studies using the CytoPatch™ 2 automated patch-clamp equipment on commercial hiPSC-CM preparations, including in-depth AP analysis, and to develop improved assay protocols and quantitative analysis methods suitable to become standard approaches for automated pharmacology screening.
Materials and Methods
Cell Culture and Dissociation
Cor.4U® hiPSC-CM cells, obtained from human fibroblasts (defined donors with complete medical history, including genetic family history) upon retroviral transfection with the four pluripotency-inducing Yamanaka factors, and further differentiated into CMs using proprietary protocols, were kindly provided by Axiogenesis (Cologne, DE) as monolayers in 12.5- or 25-cm2 fibronectin-coated flasks. They were further cultured for up to 10 days in fibronectin-coated flasks, using a special medium provided by the cell supplier, replaced twice weekly. For detachment, the monolayers were rinsed twice with calcium- and magnesium-free phosphate-buffered saline, incubated for 10 min at 4°C in the same solution and then treated with TrypLE™ Express (Life Technologies) for 2 min at 37°C, and resuspended in a 1:1 mixture of culture medium and extracellular solution.
Automated Electrophysiology Assays
Experiments were performed in parallel on four CytoPatch 2 devices (Cytocentrics Bioscience GmbH, Rostock, DE). Silicon dioxide microfluidic chips were used with two embedded quartz pipette tips (2 μm in diameter) for simultaneous seal formation and recordings in two cells, each pipette surrounded by a Cytocentering channel for capture and coordination of cells out of the cell suspension to the pipettes. 26 The sequence to whole-cell configuration was fully automated, including chip filling, cell capture, gigaseal formation, and patch rupture. Capacitive transient cancellation was also automated, occurring in multiple stages: after gigaseal formation for fast capacitive transients, and after whole-cell achievement for slow capacitive transients. Subsequently, current densities were determined using a well-defined sequence of voltage-clamp protocols (Table 1). For I Na: from a holding potential of −90 mV, a 20-ms hyperpolarizing step at −110 mV was used for monitoring electrical equivalent circuit parameters, followed after a 100-ms pause by a 40-ms depolarizing step at −10 mV. For I CaL: from a holding potential of −80 mV, a 200-ms depolarizing step at −20 mV, used to activate and then inactivate voltage-dependent Na+ channels, was followed by a 100-ms step at −40 mV, during which L-type Ca2+ channels, but not Na+ channels, were removed from inactivation, and a second 200-ms test pulse at voltages between −50 and +50 mV, in 10-mV increments, was used to specifically activate I CaL. For I Kr: from a holding potential of −70 mV, a first 2-s depolarizing step at 0 mV was applied to activate hERG channels, followed by a second test pulse of equal duration at −50 mV, within the activation window, to reopen the channels that inactivated during the first pulse, and the peak value during this pulse was used to assess hERG current density.
Voltage-Clamp and Current-Clamp Protocols Applied for iPSC-Derived Cardiomyocyte Automated Electrophysiology and Pharmacology Assay
After these voltage-clamp recordings, the amplifier was switched to current-clamp mode, the fast capacitive transient compensation was readjusted if necessary, and a well-defined sequence of protocols was applied (summarized in Table 1). For excitability assessment, CMs were paced with a series of injected current stimuli of different durations and amplitudes, at 3-s intervals: 50 pA for 20 ms, 70 and 100 pA for the same duration, and 2 nA for 0.1, 0.5, and 1 ms. Afterward, a series of three 10-s steps with hyperpolarizing injected current values of 0, −10, and −20 pA was applied to induce cessation of spontaneous occurrence of APs; the injected current that stopped spontaneous APs was applied subsequently in all protocols as holding current. For AP-shape characterization we applied a unique stimulus of 400 pA and 10-ms duration. Based on the results of these first stimulation protocols, it was decided for which of the two cells to continue the assay sequence. For assessment of AP duration (APD) adaptation, a series of 5 sweeps, each containing 11 stimuli (2 nA, 0.5 ms), at progressively increasing interstimulus intervals—300, 650, 1,000, 1,350, and 1,700 ms—was applied to study adaptation of APD; this sequence was repeated after each concentration of pharmacological compound. To facilitate the effect of voltage- or use-dependent compounds, a series of sweeps containing 3 stimuli of 2-nA amplitude and 0.5-ms duration at 3-s intervals was repeatedly applied initially and during dispensing of pharmacological compounds.
The last two protocols were iterated for each tested drug and concentration, and stability plots were automatically generated for the resting potential (RP) and APD90 (APD from maximum upstroke velocity to 90% recovery from peak depolarization to RP) of each AP in a sweep during the pacing protocol with 3 stimuli at 3-s intervals.
Solutions and Chemicals
The extracellular solution had the following composition (in mM): 140 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, 10 glucose, and 15 sucrose. The pH was adjusted to 7.40 with 1 M NaOH and the osmolality to 320 (±5) mOsmol/kg with 1 M sucrose, and the storage temperature was 4°C. The intracellular solution contained (in mM): 100 K gluconate, 20 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 11 EGTA-KOH, 4 ATP-Mg2+, 3 phosphocreatine-Na2-H2O, and 9 sucrose. The pH was adjusted to 7.20 with 1 M KOH and the osmolality to 295 (±5) mOsmol/L and then it was stored in 10-mL aliquots at −20°C, thawed before usage, and applied for up to 4 h. The pharmacological compounds used for experiments were nifedipine (N7634; Sigma, St. Louis, MO), cisapride (Sigma C4740), and tetrodotoxin (TTX; BN0518; Biotrend, Zurich, CH). Working solutions were prepared from stock solutions in dimethyl sulfoxide (for nifedipine and cisapride) or aqueous stock solution (for TTX), at dilutions of 0.1% or higher.
Data Analysis
For each assessed cell, the automated protocols yielded values of seal resistance (R
seal), values for the components of the equivalent electrical circuit in whole-cell configuration (cell capacitance, C
m; membrane resistance, R
m; and series resistance, R
s), as well as for the fast capacitive transient cancellation (C
fast). The offset potential for each channel was determined prior to cell capture and used subsequently to correct current-clamp recordings. For voltage-clamp protocols, the peak and baseline current values were automatically measured. For current-clamp recordings, we developed software routines (available in the Supplementary Materials; Supplementary Data are available online at
Results
Quality Criteria and Assay Validation
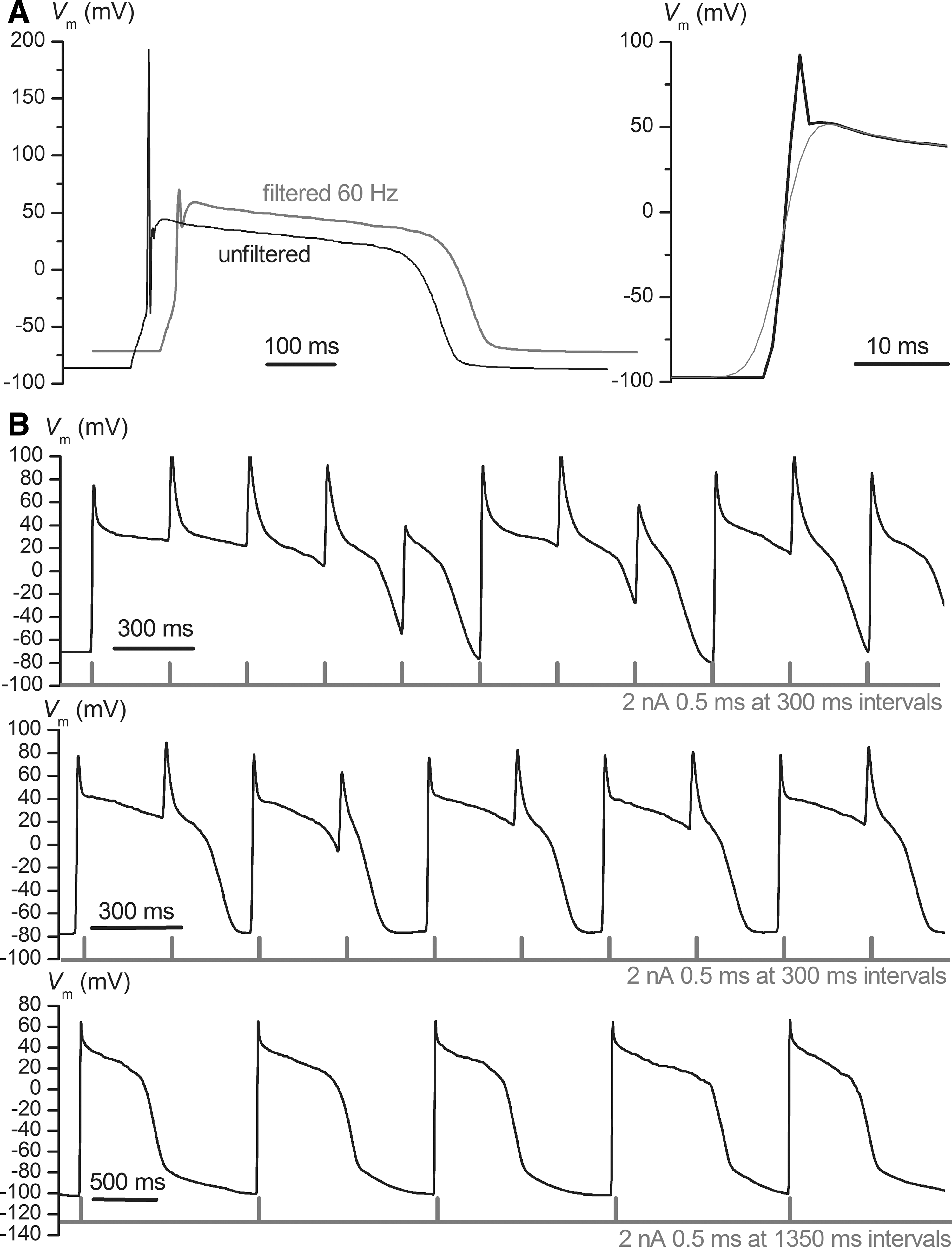
In the afore-mentioned conditions we consistently obtained high-quality seals with all hiPSC-CM preparations. Beyond the automated validation of cell capture, gigaseal cell-attached configuration, and whole-cell configuration, yielded by the CytoPatch 2 software during an experiment, we established additional quality criteria for the seal (both R seal and R m larger than 1 GΩ) and the cell type as inferred from the AP (RP<−50 mV, APD90 larger than 200 ms, ventricular AP shape with APD50/APD90≥0.7). We identified three main AP morphologies, as shown in Figure 1A—ventricular, atrial, or nodal—and a special type, called S-type (S for short). Ventricular CMs featured generally more negative RP compared with the other two types, a fast phase 0, and a consistent plateau; atrial/nodal cells had more depolarized RP, slower phase 0, and quite often sustained spontaneous firing, with a slow diastolic depolarization during RP, while S-type cells presented a fast, typical ventricular phase 0, but completely lacked a plateau. In cells with spontaneous rhythmic firing, we attempted to drive the RP to more negative values and suppress endogenous pacing, via continuous injection of a faint hyperpolarizing current of −10 or −20 pA, a maneuver that proved successful in most cases, as shown in Figure 1B. In these cells the minimum hyperpolarizing current that suppressed spontaneous pacing was applied in all subsequent current-clamp protocols, to study stimulus-triggered AP. In Table 2 we show a comparison of several quality parameters for iPSC-CMs at different ages in culture. Generally there was a tendency of decrease in propensity of seal formation with advance in culture age, as well as an increase in the average and median APD90; the best results were obtained before 2 weeks in vitro. In Figure 2 we show voltage-clamp recordings using three distinct voltage protocols, and in Table 2 average current densities of I Na, the inactivatable component of I CaL, I K DR measured at the end of plateau at +40 mV, and I to at the beginning of this voltage step.

Types of action potential (AP) and firing activity in human induced pluripotent stem cell–cardiomyocytes (hiPSC-CMs).

Voltage-clamp recordings in hiPSC-CMs.
Quality Criteria and Assay Validation Parameters for hiPSC-Derived Cardiomyocytes at Different Time Intervals In Vitro
a R seal≥1 GΩ.
b R seal≥1 GΩ, R m≥1 GΩ.
cRP<−50 mV, APD90>200 ms, ventricular AP shape (APD50/APD90≥0.7).
Response to External Pacing
In most recordings we encountered the problem of overshooting and damped oscillations of transmembrane potential (V
m) during phase 0—rapid depolarization, with the automatically established settings for C
fast, and the time constant of the compensation feed-back circuit (K
RM), that in this study were not readjusted when switching to current-clamp mode. We found a practical solution to this problem via subsequent off-line numeric filtering of the voltage traces with the Gaussian filter of Clampfit (Axon Instruments, part of Molecular Devices, Sunnyvale, CA) after conversion of the raw data to Axon Text File (atf) format. In Figure 3A we show an overlay of an unfiltered AP and the same signal after Gaussian filtering at a 60-Hz cutting frequency (fc
). Generally the AP waveform was not distorted significantly, except for phase 0, where the rising slope was diminished. We could quantify this effect using a formula for the 10% to 90% risetime (trise
10–90) in response to a step stimulus of a Gaussian filter
27
:
AP shape and response to external pacing.
where σ g is the standard deviation of the Gaussian function of the filter, and erf −1 the inverse error function. For the chosen fc value of 60 Hz we obtained a trise 10–90 of 5.66 ms, which corresponds, in the case of a maximal voltage change of 150 mV, to an upper limit in depolarization slope dV m/dt of 26.5 V/s, which is low compared with typical values in ventricular CMs. Therefore we have measured dV m/dt max in unfiltered AP traces, and the other quantitative AP parameters (RP, peak depolarizing potential V peak, APD10, APD50, and APD90) in filtered recordings, using self-written software routines.
In Figure 3B we show various patterns of response to repeated external current stimuli (rectangular pulses with duration of 0.5 ms and amplitude of 2 nA), applied at different pacing frequencies, to characterize APD adaptation. In the upper trace there is an incomplete repolarization after each stimulus, due to a fast pacing rate (300-ms interstimulus interval) in a cell with longer APD, similar to a high-degree block for atrio-ventricular conduction, while in the middle trace we have the more common finding of a 2:1 block (each second stimulus elicits an AP). In the lower trace there is a phenomenon of APD alternans: long AP alternating with shorter ones. The main explanation of this phenomenon, already retrieved in the 1962 model of Denis Noble for a Purkinje fiber, 1 is a pronounced activation of delayed rectifier K+ currents during a longer pulse, and therefore a larger K+ conductance during the subsequent AP, rendering it shorter than the previous one.
Effects of Nifedipine, Cisapride, and TTX
After defining the succession of pacing protocols and their timing for the current-clamp assay, and after checking the stability of AP parameters in control conditions, we performed pharmacology trials for three prototype compounds: nifedipine, a typical L-type Ca2+ channel blocker; cisapride, a medium-potency hERG blocker with relatively quick onset of effects and recovery during wash-out; and tetrodotoxin (TTX), able to block the relatively TTX-resistant Nav1.5 cardiac-voltage-dependent Na+ channels at concentrations in the medium micromolar range. In Figure 4A we show representative AP recordings with these three compounds at different concentrations. For nifedipine, the main effect is a progressive shortening of the AP plateau and triangularization of its shape; for cisapride a marked prolongation of the plateau, sometimes without return to RP until the next stimulus, and in some instances EAD during phase 3; and for TTX a marked slowdown of phase 0 and decrease in V peak. The effects of nifedipine at 1 and 10 μM on APD90, APD50, and APD10, at different pacing frequencies, are shown in Figure 4B. Both concentrations of nifedipine exert saturating effects, resulting in marked decreases in APD90 and APD50. A two-way ANOVA was performed for APD90 and APD50 values with 1 μM nifedipine, taking interstimulus interval-specific values. For APD90 both variables exerted significant effects: F=213 (83.23% of total variance), P=0.0001 for the presence of drug vs. control; F=9.73 (15.21% of total variance), P=0.0244 for pacing frequency. For APD50 only the presence/absence of drug produced significant results: F=181.98 (94.08% of total variance), P=0.0002. The quantitation of pharmacological effects of these three compounds on different AP characteristics is presented in Table 3, where significant changes, from a mechanistic point of view, expressed as relative percentage values in the presence of drug versus control, are typed in bold. Thus, the main effect of nifedipine is a reduction in APD50 (to 16% and 12% of control values for 1 and 10 μM, respectively), a less-pronounced reduction in APD90, and decrease of APD50/APD90 ratios from 0.8 to roughly 0.2, corresponding to the afore-mentioned AP triangularization. For cisapride, the main effect is a pronounced increase in APD90, while for TTX it is the decrease in maximum slope of phase 0 to one-third of control values, together with reduction in V peak and a slight APD shortening. The same effects are presented graphically in Figure 5, where individual average values recorded in the same cell in control conditions and then after drug application are connected with lines.
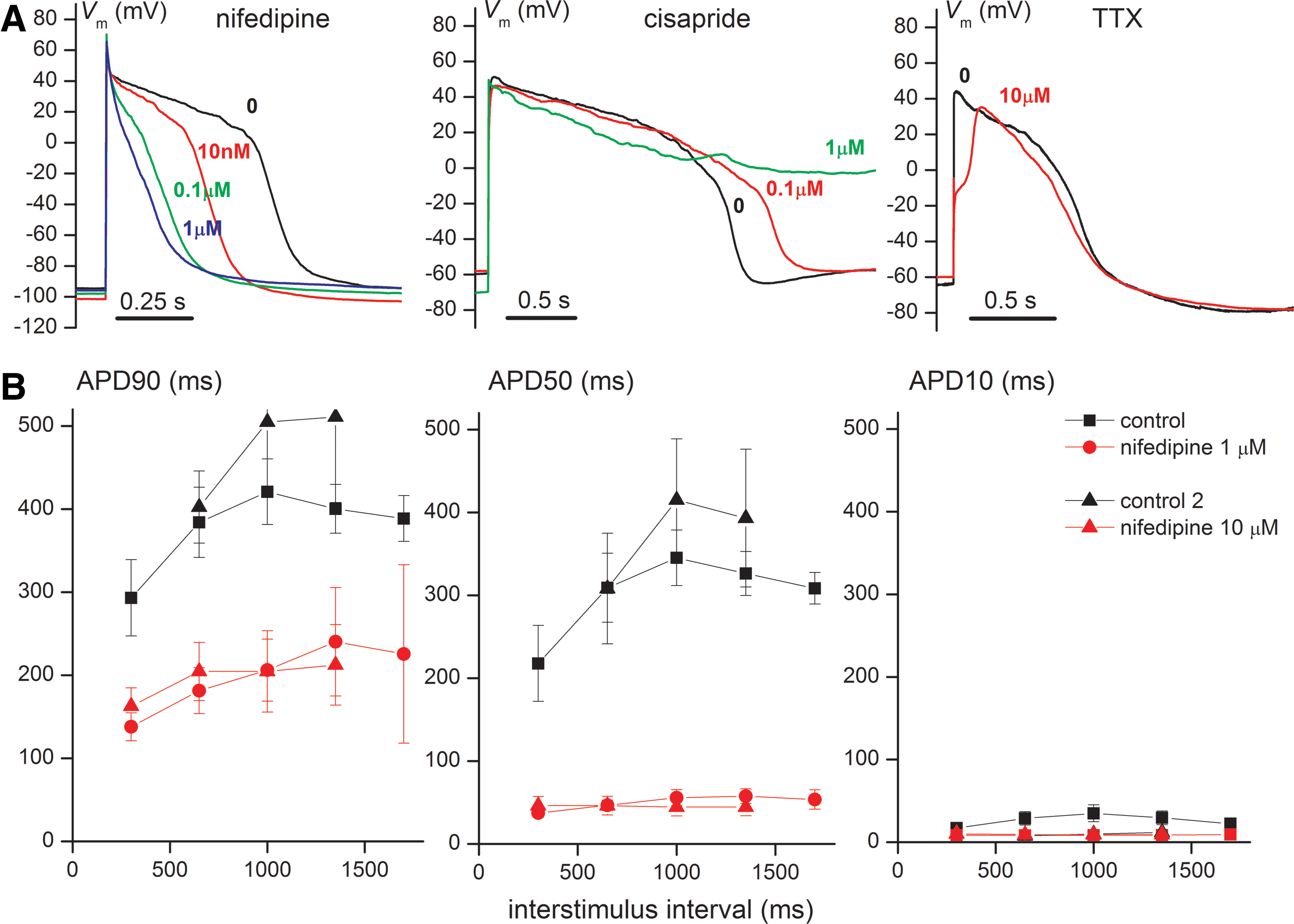
Pharmacological effects of nifedipine, cisapride, and tetrodotoxin (TTX).

Effects of nifedipine, cisapride, and TTX on different AP characteristics: maximal slope of fast depolarization (phase 0) (dV
m/dt
max) (relative to control), APD50 and APD90 (relative to control), and the ratio APD50/APD90. For the first three parameters control values were considered as 100%. Control data
Pharmacological Effects on AP Characteristics
aAssays performed on Cor.4U® cells; data are expressed as mean±SD; number of cells included: n=6 for nifedipine 1 μM, n=4 for nifedipine 0.1 μM, n=2 for cisapride 0.1 μM, n=6 for cisapride 1 μM, and n=6 for TTX 10 μM; figures in bold indicate main pharmacological effects.
bWe applied the variant for independent and not-paired samples because parameter values for each cell and in each tested condition (initial control values and during drug application) represent averages of average values at different pacing frequencies, and in certain experimental conditions we could not measure parameters at all pacing rates due to incomplete repolarization; however, the statistical significance would have been even stronger and the P values lower if we would have applied the paired tests to complete datasets.
RP, resting potential; dV m/dt max, maximum slope of AP phase 0 (upstroke); V peak, peak depolarization; APD10, 50, 90, duration from moment of maximum depolarization slope to recovery of 10%, 50%, or 90% of the difference between V peak and RP.
Discussion
In this study we have proven that automated whole-cell patch clamp recordings of iPSC-derived CMs can be performed to characterize cardiac APs and to assess drug effects on the APs. In particular we have shown that whole-cell recordings with high GΩ seal resistances can be obtained reproducibly with the CytoPatch 2 equipment, and that cardiac AP can be induced and recorded, offering comparable results with those obtained in manual patch-clamp studies. 22,28 –30
In particular it is possible to characterize and quantify cell fractions in terms of the quantitative features of the AP, that is, to distinguish between atrial, nodal, and ventricular types of AP, and to determine the yield of successful recordings. In this respect, it was encouraging for us to find proportions of ventricular, atrial, and nodal CMs, according to AP morphology, 30 –33 as well as quantitative AP parameters, 28,30 –36 similar to those reported in previous electrophysiology studies on stem-cell-derived CM preparations. Therefore, this method is adequate for use in the batch control of iPSC-CMs, for example, during the in-production control or the validation before using a batch of these cells in high-throughput assays. As an integral part of the optimization process for iPSC-CM differentiation, it can be applied to help drive derivation procedures to a particular cell type. It is possible that the peculiar S-type encountered in our experiments (Fig. 1 and Table 2)—that is, CMs with fast ventricular-like phase 0, but lacking a plateau—is a consequence of increased production or impaired scavenging of reactive oxygen species, 37 and that further precautions aimed to diminish these phenomena may increase the percentages of mature ventricular-like iPSC-CMs. However, our assay is not limited to the development of iPSC-derived CMs, but is expected to be applicable as well for patient-specific stem-cell-derived CMs, 38 –40 as preliminary experiments suggest, 41 or ESC-derived CMs.
We have shown that spontaneous firing activity, that is, the occurrence of APs, can be efficiently suppressed by constant injection of a small negative current during the current-clamp recordings. There is considerable experimental support and a solid theoretical background justifying this method of external current injection to suppress spontaneous rhythmic firing. Thus, previous studies based on dynamic system analysis and bifurcation theory methods applied to various models of sinoatrial or ventricular CMs, 42 –47 taking a constant externally applied current as parameter and assessing its influence over a wide range of values, have shown distinct intervals of stable dynamic equilibrium at a steady V m or stable limit cycles representing sustained oscillations, that is, spontaneous pace-making. The transitions between these regimes occur at Hopf bifurcation points in the parameter space of the model. In pacemaker cells slow diastolic depolarizing currents, such as I f, exert the function of driving the system away from a stable equilibrium point to a sustained oscillatory regime; therefore, opposing this action via a steady injection of a faint hyperpolarizing current seems perfectly reasonable. In addition to suppressing spontaneous pacing, this method has the advantage of rendering V m more negative, close to a typical ventricular RP of −80 to −90 mV, rendering more physiological occupancies of various kinetic states of voltage-dependent channels during RP, and removing more channels from voltage-dependent inactivation, thus increasing their availability for the next AP. By this it is possible to apply a well-defined activation pattern as needed for a systematic assessment of drug effects on ventricular APs. This is of particular interest as the FDA is working together with other drug authorities and pharmaceutical industry and academia representatives on a new paradigm for the cardiac safety screening 18 (the CiPA initiative), and the assessment of drug effects on cardiac APs measured in whole-cell current-clamp recordings in combination with multiple voltage-clamp protocols, as proposed by the MICE approach, 17 may become an integral part of it.
A problem in current-clamp AP recordings with patch-clamp amplifiers, especially in the case of fast neuronal APs, is the distortion of AP shape by the resistive feedback amplifier headstage during rapid V m changes due to limited input impedance. The problem is alleviated to a great extent in modern patch-clamp amplifiers by injection in current-clamp mode of the C fast cancellation signal to the I–V converter input via capacitive coupling. In this setting, however, complete C fast compensation may result in overshoot and oscillations during rapid variations in V m, as we encountered in most recordings during phase 0 of the AP (Fig. 3). Recommendations of other amplifier manufacturers to avoid this problem include reduction of C fast compensation by 0.5 to 1 pF compared to the value found in voltage-clamp mode, or even inclusion of C slow compensation, and a low-pass filtering of rectangular stimulus signals to decrease in steepness the ascending wavefront. We found as a convenient solution for removal of oscillations during fast V m rises a numeric low-pass filtering of the acquired signal during analysis of recordings, instead of changing the C fast settings automatically established within voltage-clamp protocols, since the effects on signal frequency bandwidth should be similar. To avoid the resulting reduction in maximal V m slope during phase 0, we measured this parameter on unfiltered recordings, and the average values obtained in control conditions, around 100 V/s, were similar to expected values at room temperature for typical adult ventricular CMs, given the fast upstroke velocity of 100–200 V/s found in AP simulations based on I Na kinetics measured in early patch-clamp experiments on isolated CMs. 48 –50 Another good agreement with simulation studies using advanced ventricular CM electrophysiology models, for example, the Luo-Rudy I model, 51 was the variable pattern of response to external pacing stimuli with fast repetition rate; we retrieved stimulus:response (S:R) ratios of 5:1 to 2:1 for basic cycle length values of 300 ms, and the well-known beat-to-beat APD alternans (Fig. 3B). Likewise, pharmacological effects on AP characteristics of the three tested compounds were in good agreement with those reported in previous studies on hiPSC-CMs 22,30 ; for nifedipine, marked reductions in plateau durations (with APD90 and especially APD50 shortening) and a triangularization of AP shape reflected in decrease of APD50/APD90 ratios; for cisapride, APD lengthening and proarrhythmogenic phenomena such as EADs; and, for TTX at 10 μM, large reductions in phase 0 slope and peak depolarization potential, and a moderate decrease in APD90, due to its well-known blocking effects, in the micromolar range, on voltage-dependent Na+ channels, including TTX-sensitive Ca2+ currents via a subpopulation of cardiac Na+ channels. 52
Another important issue in iPSC-CM electrophysiology experiments is the stability in time of approached cells, including seal parameters and access resistance that were carefully monitored in each voltage-clamp recording; lack of mechanical contractility, and lack of significant rundown of various transmembrane ion current components due to changes in intracellular signaling pathway state of activation; membrane trafficking of channel proteins; and a variety of other modulatory factors. A challenge in many electrophysiology studies on these stem-cell-derived CM preparations is the progressive rundown of I CaL, in connection with variable phosphatidyl-inositol (4,5) bis phosphate (PIP2) levels, variable intracellular Ca2+ levels leading to calcium-dependent inactivation of L-type Ca2+ channels, 53 –55 and variable phosphatidyl inositol (3) kinase (PI3K) activity levels, leading to changes in phosphorylation and activity of protein kinase B (or Akt1) 56 and calmodulin kinase II (CaMKII), 57,58 a known modulator of Cav1.2 and Nav1.5 cardiac-voltage-gated channels. In our experimental conditions, using a physiological intracellular solution without fluoride but with high calcium buffering capacity (11 mM EGTA) and sufficient energy and substrate supplies for phosphorylation, L-type Ca2+ current rundown could be kept under fairly good control, and APD attained stable values in control conditions over the entire duration of the current-clamp assay due to the stable seals obtained with the Cytocentering technology.
In conclusion, using a special cytocentering technology implemented in the CytoPatch 2 automated patch-clamp equipment, 26,59,60 we could systematically approach human iPSC-derived CM preparations from commercial suppliers, obtaining very high seal resistances and whole-cell membrane resistances with physiological external and internal solutions. Voltage-clamp and current-clamp protocols were combined in a complex assay devised for quantitative characterization of drug effects on ventricular AP and in-depth screening of their proarrhythmogenic risk. Although further improvements in cellular preparation and assay are necessary, the preliminary results are promising. Further, our assay can be applied to human patient iPSC-derived CMs, offering new perspectives for diagnosis and treatment of cardiac channelopathy patients.
Footnotes
Acknowledgments
The authors gratefully acknowledge Christian Scherpeltz, Ingrid Rosenkranz, and Juliane Böttcher for expert technical assistance; Jens Wolgast, Stefan Brenner, and Stephan Wierschke for help with computing infrastructure; and the entire Cytocentrics team for support. We are indebted to Axiogenesis for provision of human iPSC-CM preparations.
Disclosure Statement
None of the authors have conflicting interests concerning the present study, except for representing the Cytocentrics company.