Hochscherf J, Lindenblatt D, Steinkrüger M, Yoo E, Ulucan Ö, Herzig S, Issinger O-G, Helms V, Götz C, Neundorf I, Niefind K, Pietsch M: Development of a high-throughput screening-compatible assay to identify inhibitors of the CK2a/CK2b interaction.Anal Biochem468;2015:4–14.
Abstract: Increased activity of protein kinase CK2 is associated with various types of cancer, neurodegenerative diseases, and chronic inflammation. In the search for CK2 inhibitors, attention has expanded toward compounds disturbing the interaction between CK2α and CK2β in addition to established active site directed approaches. The current article describes the development of a fluorescence anisotropy-based assay that mimics the principle of CK2 subunit interaction by using CK2α1–335 and the fluorescent probe CF-Ahx-Pc as a CK2β analog. In addition, we identified new inhibitors able to displace the fluorescent probe from the subunit interface on CK2α1–335. Both CF-Ahx-Pc and the inhibitors I-Pc and Cl-Pc were derived from the cyclic peptide Pc, a mimetic of the C-terminal CK2α-binding motif of CK2β. The design of the two inhibitors was based on docking studies using the known crystal structure of the Pc/CK2α1–335 complex. The dissociation constants obtained in the fluorescence anisotropy assay for binding of all compounds to human CK2α1–335 were validated by isothermal titration calorimetry. I-Pc was identified as the tightest binding ligand with a KD value of 240 nM and was shown to inhibit the CK2 holoenzyme-dependent phosphorylation of PDX-1, a substrate requiring the presence of CK2β, with an IC50 value of 92 μM.
Commentary:The protein kinase CK2 is a heterotetrameric enzyme composed of two catalytic subunits (CK2α) and two regulatory subunits (CK2β). Regulation of CK2 is provided by a central CK2β dimer that modulates both the stability and substrate specificity of the catalytic domain. A cyclic peptide (Pc) has been identified that binds to the CK2α/CK2β interface, and the authors have previously solved the X-ray co-crystal structure of Pc bound to CK2 α1–335. This revealed that Pc mimics a β-hairpin loop found in the CK2β subunit and therefore Pc can be used as a probe of the CK2α/CK2β interface (see
figure
). Initially the Pc cyclic peptide was labeled directly with 6-carboxyfluorescein (CF), but this did not yield any appreciable binding to CK2α. A 6-aminohexanoic acid (Ahx) linker was added and the Cr-Ahx cyclic peptide showed a KD of 1.75 μM (confirmed by isothermal titration calorimetry [ITC]). A fluorescence anisotropy (FA) assay was established using 0.1 μM CF-Ahx-Pc and 3 μM CK2α which showed a Z′ of 0.72 in a 96-well plate format. The article demonstrates that the Pc probe binding is not influenced by either ATP or substrate competitive compounds. This assay was used to characterize several analogs of Pc with nonnatural amino acids at position Phe-190 that were designed based on docking studies. The article describes characterization of these Pc analogs using the FA assay, and the compounds were further confirmed by ITC and functional phorsphorylation assays. The functional assay revealed a large right shift in the potency of the Pc analog containing 3-iodo-L-Phe at position 190 (I-Pc). The potency of I-Pc in the binding assays is submicromolar, but I-Pc showed an IC50 of 92 μM in the phosphorylation assay suggesting that destabilization of an existing CK2 holoenzyme is more difficult to achieve then the preventing the association of the CK2α/CK2β subunits. The assay described here could be used to find improved inhibitors of this interaction. Contributed by Doug Auld.
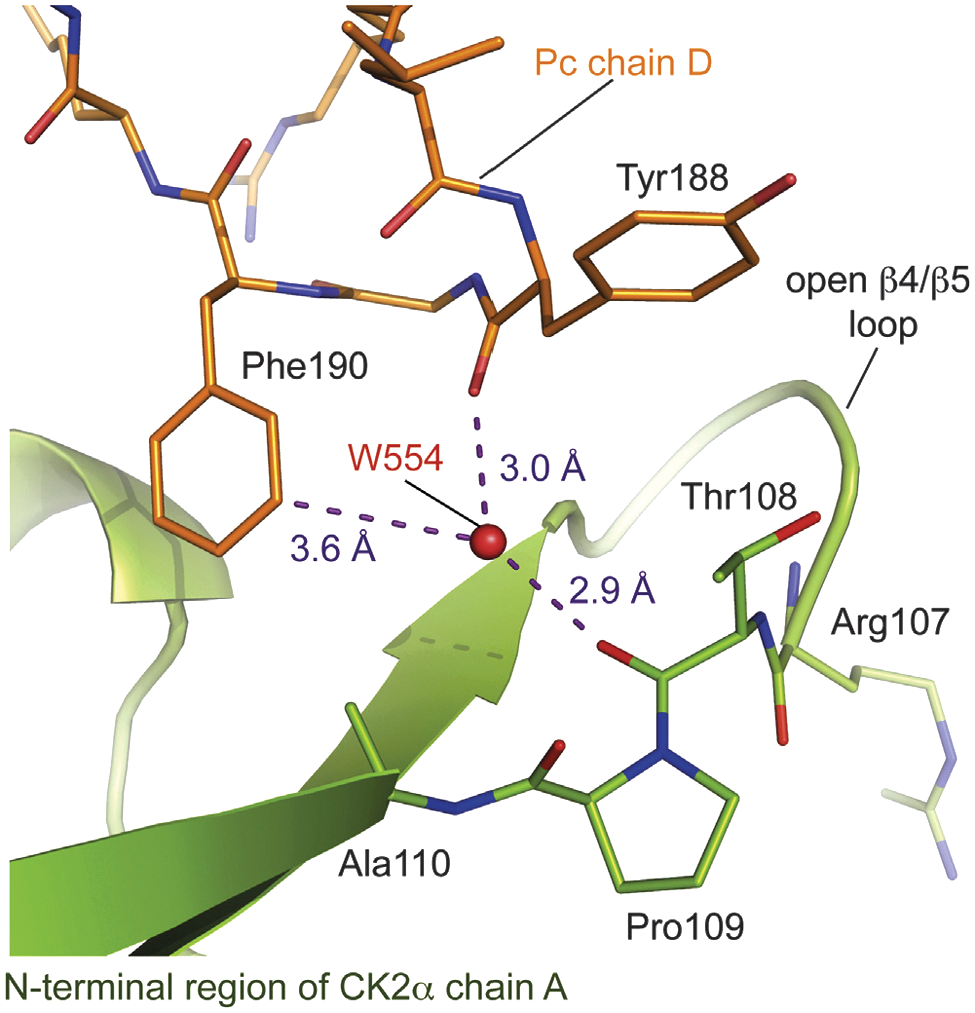
Binding site of Pc chain D at the CK2β interface of CK2α chain A in the CK2α1–335/Pc complex structure (PDB: 4IB5 [22]). The water molecule W554 (W119) is deeply buried in the cavity and is located in hydrogen bond distance to the carbonyl oxygen of CK2a's Thr108 and the carbonyl oxygen of Pc's residue Tyr188. Furthermore, W554 is located close (3.6 Å) to the meta position of Phe190 of the cyclic peptide. The figure was prepared with PyMOL (Schrödinger). Electron density pieces were contoured at 1.0 σ. Distances between atoms are indicated by purple dotted lines.
BUG DRUG PERMEABILITY
Davis TD, Gerry CJ, Tan DS: General platform for systematic quantitative evaluation of small-molecule permeability in bacteria.ACS Chem Biol2014;9:2535–2544.
Abstract: The chemical features that impact small molecule permeability across bacterial membranes are poorly understood, and the resulting lack of tools to predict permeability presents a major obstacle to the discovery and development of novel antibiotics. Antibacterials are known to have vastly different structural and physicochemical properties compared to nonantiinfective drugs, as illustrated herein by principal component analysis (PCA). To understand how these properties influence bacterial permeability, we have developed a systematic approach to evaluate the penetration of diverse compounds into bacteria with distinct cellular envelopes. Intracellular compound accumulation is quantitated using LC-MS/MS, then PCA and Pearson pairwise correlations are used to identify structural and physicochemical parameters that correlate with accumulation. An initial study using 10 sulfonyladenosines in Escherichia coli, Bacillus subtilis, and Mycobacterium smegmatis has identified nonobvious correlations between chemical structure and permeability that differ among the various bacteria. Effects of cotreatment with efflux pump inhibitors were also investigated. This sets the stage for use of this platform in larger prospective analyses of diverse chemotypes to identify global relationships between chemical structure and bacterial permeability that would enable the development of predictive tools to accelerate antibiotic drug discovery.
Commentary:Rules that help guide the optimization of compounds for oral bioavailability based on intestinal absorption data have been established, but little is known around the properties that govern intracellular compound concentration in standard cells lines or in bacterial cultures. In particular, bacteria have a cell envelope providing an additional barrier that aids in antibiotic resistance. Staining of bacteria with a crystal violet dye reveals types of bacteria that differ in their cell envelopes, and it is generally thought that gram-positive bacteria have a membrane that is permeable to certain compounds while gram-negative bacteria have a more complex cell envelope that is much less permeable. This article describes a study aimed at understanding the physicochemical properties that govern the bacterial cell penetration of compounds. Principle component analysis (PCA) employing 20 physicochemical parameters shows that antibacterial drugs have different properties than non–anti-infective drugs, which underscores why common rules for oral bioavailability cannot be applied to guide bacterial compound absorption. The authors then measure the intrabacterial compound concentration of several major anti-infective drug classes using liquid chromatography-tandem mass spectrometry (LC-MS/MS) and applied the PCA approach to define which physicochemical parameters govern bacteria compound absorption. Some bacteria have efflux pumps that pump compounds out of the bacteria, and the authors used known inhibitors of these pumps to determine how intracellular compound concentrations are influenced by efflux pumps. Importantly, for the validation of the LC-MS/MS method, the measured intracellular compound concentration could be increased by inhibiting certain efflux pumps, which supports that the LC-MS/MS procedure is measuring intracellular compound concentration and not compound adhered to the bacterial surface. Some interesting trends were observed, but these varied between different bacteria (see
figure
for analysis of sulfonyladenosine accumulation); however, the results generally support that hydrophobicity parameters alone are not sufficient to predict bacterial compound absorption. Within this sulfonyladenosine panel, ring content, size, and hydrophobicity parameters clustered with E. coli compound absorption; however, in B. subtilis, accumulation also clustered with molecular flexibility, and ring content was found to correlate with compound accumulation in M. smegmatis. The methods described in this article should provide a means for broader studies of bacterial compound absorption. Contributed by Doug Auld.
Multivariate analyses reveal correlations between sulfonyladenosine bacterial accumulation with structural and physicochemical parameters. PCA biplots showing relationships between 10 sulfonyladenosines, 20 structural and physicochemical descriptors, and accumulation in (A)E. coli, (B)B. subtilis, and (C)M. smegmatis; average sulfonyladenosine cellular concentration (μM) is noted in parentheses; ▲=sulfonyladenosines; ●=physicochemical parameters; ■=accumulation parameter; expanded PCA biplots for each bacterium are in Figures S8–S10 (Supporting Information).
ENABLING LIBRARY-AGAINST-LIBRARY SCREENING
Gu L, Li C, Aach J, Hill DE, Vidal M, Church GM: Multiplex single-molecule interaction profiling of DNA-barcoded proteins.Nature2014;515:554–557.
Abstract: In contrast with advances in massively parallel DNA sequencing, high-throughput protein analyses are often limited by ensemble measurements, individual analyte purification and hence compromised quality and cost-effectiveness. Single-molecule protein detection using optical methods is limited by the number of spectrally non-overlapping chromophores. Here we introduce a single-molecular interaction sequencing (SMI-seq) technology for parallel protein interaction profiling leveraging single-molecule advantages. DNA barcodes are attached to proteins collectively via ribosome display or individually via enzymatic conjugation. Barcoded proteins are assayed en masse in aqueous solution and subsequently immobilized in a polyacrylamide thin film to construct a random single-molecule array, where barcoding DNAs are amplified into in situ polymerase colonies (polonies) and analysed by DNA sequencing. This method allows precise quantification of various proteins with a theoretical maximum array density of over one million polonies per square millimetre. Furthermore, protein interactions can be measured on the basis of the statistics of colocalized polonies arising from barcoding DNAs of interacting proteins. Two demanding applications, G-protein coupled receptor and antibody-binding profiling, are demonstrated. SMI-seq enables “library versus library” screening in a one-pot assay, simultaneously interrogating molecular binding affinity and specificity.
Commentary:Being able to derive screening data from very large-scale experiments without the need to test every interacting pair separately (as is commonly done in small-molecule high-throughput screening) has remained largely an elusive goal. George Church and colleagues provide an elegant solution to the problem by preparing proteins that are barcoded with a DNA sequence attached to them through in vitro translation and ribosome display (
first figure
). In turn, the mixture of barcoded and displayed proteins could be analyzed through immobilization in a thin layer of polyacrylamide in which the individual protein nanospots were identified and quantified by amplification of the DNA barcode and subsequent sequencing, essentially visualizing and counting single-molecule occurrences (
second figure
). Using this technique, the authors performed large-scale “library-against-library” screens in which two distinct protein families were crossed with each other, and the outcome of selective binding and resultant co-localization of uniquely barcoded cognate partners was measured through the single-molecule barcode detection. The same experiment was also conducted in a hybrid format in which a small molecule library was screened against a family of related G-protein coupled receptors (GPCR), all barcoded and present together in each well of the microtiter plate, along with potential cognate β-arrestin partners that were also barcoded. The screen thus allowed the detection of specific small-molecule disrupters of the various GPCR/β-arrestin interactions without the need to test each protein–protein pair separately. Contributed by Anton Simeonov.
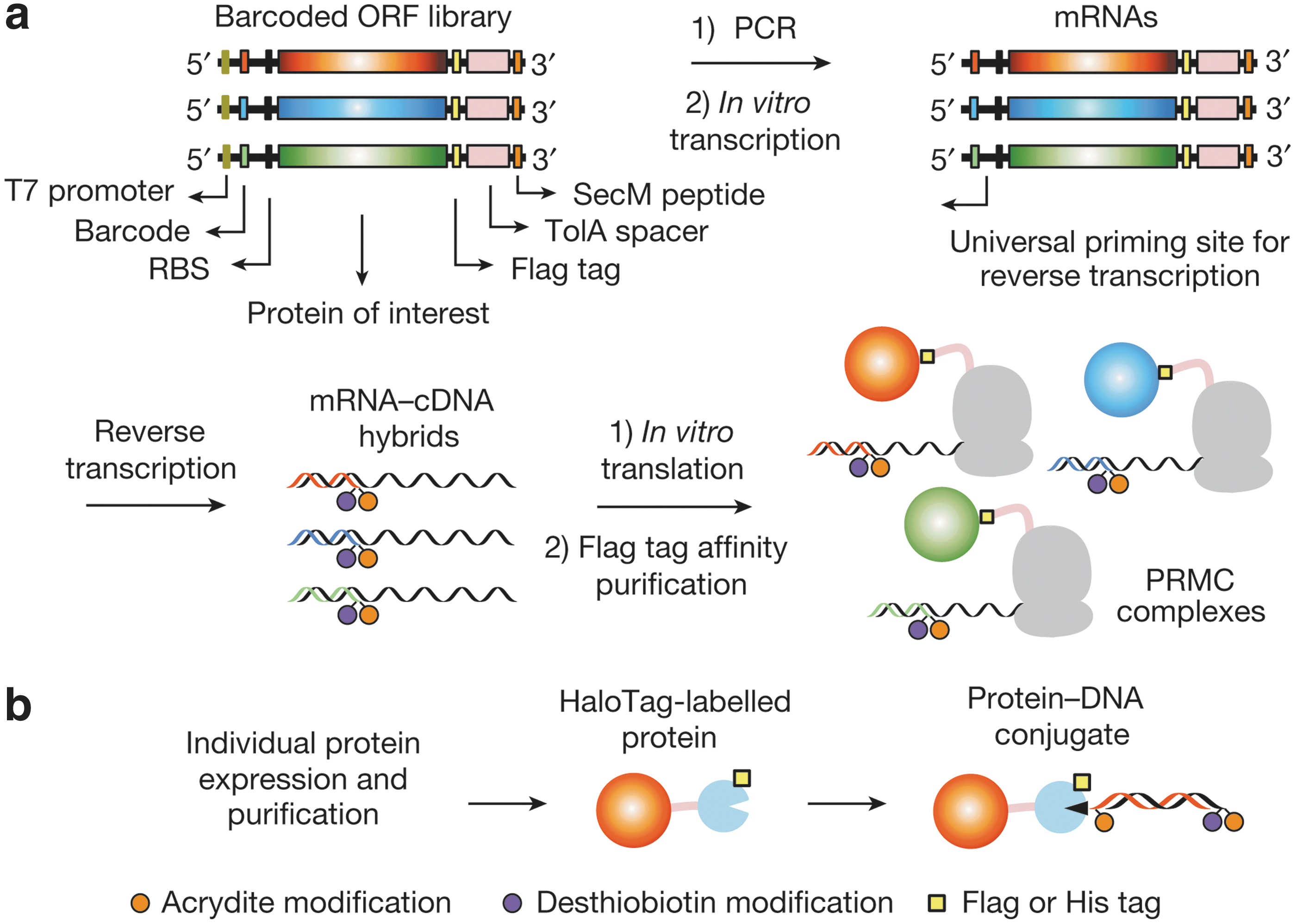
Schematics of protein barcoding methods. (a) Collective barcoding via ribosome display. A short synthetic barcoding sequence is joined to the 5′ end of DNA templates via PCR. PRMC complexes are formed via ribosome stalling triggered by a carboxy-terminal Escherichia coli SecM peptide. Displayed proteins bearing a C-terminal Flag tag are separated from the ribosomes by an E. coli TolA spacer domain. RBS, ribosomal binding site. (b) Individual barcoding via a HaloTag-mediated conjugation of proteins to a 220-base-pair (bp) double-stranded barcoding DNA with a HaloTag ligand modification (black triangle). Modifications are introduced to barcoding DNAs by PCR with modified primers.
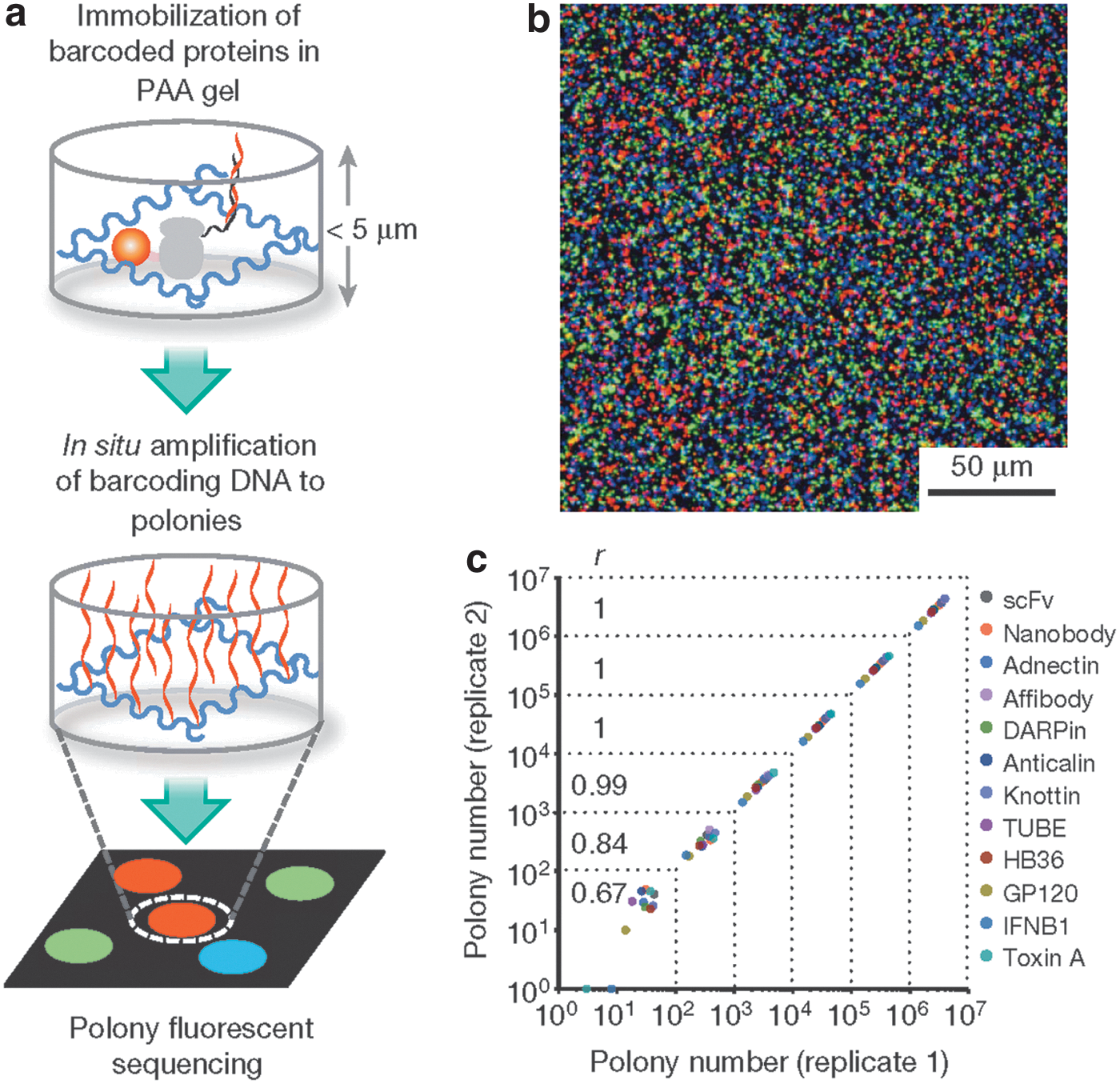
Amplification and quantification of barcoding DNAs. (a) Schematic of in situ polony amplification and sequencing. Barcoded proteins were immobilized in a polyacrylamide (PAA) gel matrix attached to a Bind-Silane-treated glass slide. The slide was assembled into a flow cell, where barcoding DNAs were amplified in situ into polonies for DNA sequencing. (b) Representative merged images of polonies hybridized with Cy5 (red), Cy3 (green) and fluorescein (blue)-labelled oligonucleotides (320 objective magnification). (c) Polony quantification of mixed protein binders and antigens. The Pearson correlation coefficient r was calculated for different coverages grouped by dotted lines.
A CHAPERONIN CAPTURE APPROACH TO TACKLE FRIEDREICH'S ATAXIA
Correia AR, Naik S, Fisher MT, Gomes CM: Probing the kinetic stabilities of Friedreich's ataxia clinical variants using a solid phase GroEL chaperonin capture platform.Biomolecules2014;4:956–979.
Abstract: Numerous human diseases are caused by protein folding defects where the protein may become more susceptible to degradation or aggregation. Aberrant protein folding can affect the kinetic stability of the proteins even if these proteins appear to be soluble in vivo. Experimental discrimination between functional properly folded and misfolded nonfunctional conformers is not always straightforward at near physiological conditions. The differences in the kinetic behavior of two initially folded frataxin clinical variants were examined using a high affinity chaperonin kinetic trap approach at 25°C. The kinetically stable wild type frataxin (FXN) shows no visible partitioning onto the chaperonin. In contrast, the clinical variants FXN-p.Asp122Tyr and FXN-p.Ile154Phe kinetically populate partial folded forms that tightly bind the GroEL chaperonin platform. The initially soluble FXN-p.Ile154Phe variant partitions onto GroEL more rapidly and is more kinetically liable. These differences in kinetic stability were confirmed using differential scanning fluorimetry. The kinetic and aggregation stability differences of these variants may lead to the distinct functional impairments described in Friedreich's ataxia, the neurodegenerative disease associated to frataxin functional deficiency. This chaperonin platform approach may be useful for identifying small molecule stabilizers since stabilizing ligands to frataxin variants should lead to a concomitant decrease in chaperonin binding.
Commentary:Friedreich's ataxia (FRDA) is a genetic mitochondrial disease, first described by physician Nikolaus Friedreich in 1863. Being a rare yet prevalent ataxia, FRDA affects 1 in 22,000–50,000 people in the general population, with the greatest incidence found in people of European descent. The disease affects multiple systems, with symptoms including gait disturbance and speech problems, and can also lead to heart disease (such as cardiomyopathy), and diabetes. The cause of the disease was identified in 1996 as a defect of the FXN gene located on chromosome 9Q13. FRDA is a triplet repeat disease in which expansion of GAA trinucleotide repeats within the first intron of the FXN gene interferes with mRNA transcription, leading to a reduction in the frataxin protein encoded by the FXN gene. One widely accepted function of frataxin in cells is to synthesize Fe-S cluster groups, while another property of the protein is to maintain iron level in mitochondria to alleviate the deleterious effect of hydroxyl radicals produced through Fenton reaction. Reminiscent of ferritin as an iron scavenger, researchers are still in search of the essential cellular role of frataxin in the mitochondria, which will aid in designing efficient therapeutic strategies for FRDA treatment. Currently no cure is available for FRDA. One promising approach is through drugs that either activate gene expression or inhibit histone deacetylation (HDAC), with the aim of increasing frataxin expression. Herein, Correia and co-workers studied the kinetic stabilities and differences between two missense FRDA mutants, with frataxin mutant Asp122Tyr linked to a mild phenotype, and another variant with an Ile154Phe mutation associated with a severe disease phenotype. The rationale was to establish a correlation between kinetic differences of the variants with FRDA prognosis severity. To probe the kinetic stability of FRDA clinical variants, the group utilized a high affinity (nucleotide free) form of the chaperonin GroEL (Hsp60 homolog in E. coli) to examine partition rate of the mutants to GroEL under different conditions. Both variants were shown to exhibit comparable thermal stability with similar Tm around 50°C, about 16°C lower than that of the wild type protein (see
figure
panel A). A GroEL bead-based capture platform was validated under 1 M urea and at elevated temperatures, both variants showed a time-dependent partition, while the wild-type frataxin exhibited little change in its soluble fraction over time (see
figure
panels B and C). Despite the similarities observed between the two variants in terms of their thermal stability, in a subsequent GroEL concentration-dependent partition study, the Ile154Phe variant exhibited a higher partition rate onto GroEL than the Asp122Tyr variant. Both partition rate profiles reasonably followed a pseudo-first-order decay, with the unfolding rate being the limiting factor at high GroEL concentrations. Additionally, protein solubility, stability and partition properties were also compared in presence of different osmolytes, including trimethylamine N-oxide (TMAO), trehalose, and glycerol. Addition of TMAO and glycerol led to a significant reduction in the magnitude of protein partition onto GroEL for the Asp122Tyr variant, whereas both osmolytes induced aggregation-dependent light scattering. This GroEL bead-based platform demonstrated its potential to evaluate stabilizers of kinetically metastable proteins, and it could be extended to a wide range of disease proteins. One caveat is the interference with GroEL partition due to solution protein aggregation, which can lead to false positives. One way to eliminate aggregation and circumvent limitations of the platform is to have a large excess of GroEL. This study also points to an alternative therapeutic approach for FRDA treatment through a search for global stabilizing agents for missense mutants of frataxin which would shift folding equilibria to correctly folded forms and increase functional protein levels. Contributed by Wendy Lea.
Comparison between the stability profile of wild-type (wt) frataxin (FXN) and two clinical mutants FXN-p.Asp122Tyr and FXN-p.Ile154Phe. (A) Thermal denaturation curves of (■) FXN (Tm = 66.3 ±0.1°C), (○) FXN-p.Asp122Tyr (Tm = 50.4 ± 0.1°C), and (Δ) FXN-p.Ile154Phe (Tm = 50.7 ± 0.1°C) (Curves redrawn from [11] to highlight variants used herein) demonstrate differences between wt FXN and the two mutants. (B–D) Effect of temperature on FXN partitioning profiles (starting concentration 2 μM frataxin) onto 2 μM immobilized GroEL oligomer beads in the presence of 1 M urea. The partitioning of (B) wt FXN, (C) FXN-p.Asp122Tyr (circles), and (D) FXN-p.Ile154Phe (triangles) was monitored by UV-visible spectroscopy at 25°C, 37°C, and 45°C to demonstrate that stability differences observed in (A) can be recapitulated with the GroEL chaperonin sink assay.
TARGETING KINASES ESSENTIAL TO MALARIA PARASITES
Derbyshire ER, Zuzarte-Luis V, Magalhães AD, Kato N, Sanschagrin PC, Wang J, Zhou W, Miduturu CV, Mazitschek R, Sliz P, Mota MM, Gray NS, Clardy J: Chemical interrogation of the malaria kinome.Chembiochem2014;15:1920–1930.
Abstract: Malaria, an infectious disease caused by eukaryotic parasites of the genus Plasmodium, afflicts hundreds of millions of people every year. Both the parasite and its host utilize protein kinases to regulate essential cellular processes. Bioinformatic analyses of parasite genomes predict at least 65 protein kinases, but their biological functions and therapeutic potential are largely unknown. We profiled 1358 small-molecule kinase inhibitors to evaluate the role of both the human and the malaria kinomes in Plasmodium infection of liver cells, the parasites' obligatory but transient developmental stage that precedes the symptomatic blood stage. The screen identified several small molecules that inhibit parasite load in liver cells, some with nanomolar efficacy, and each compound was subsequently assessed for activity against blood-stage malaria. Most of the screening hits inhibited both liver- and blood-stage malaria parasites, which have dissimilar gene expression profiles and infect different host cells. Evaluation of existing kinase activity profiling data for the library members suggests that several kinases are essential to malaria parasites, including cyclin-dependent kinases (CDKs), glycogen synthase kinases, and phosphoinositide-3-kinases. CDK inhibitors were found to bind to Plasmodium protein kinase 5, but it is likely that these compounds target multiple parasite kinases. The dual-stage inhibition of the identified kinase inhibitors makes them useful chemical probes and promising starting points for antimalarial development.
Commentary:A large unmet medical need exists for effective new treatments for malaria in the developing world. The malaria parasites initially travel from the site of the infected mosquito bite to the host's liver (called liver stage) where they reproduce and then infect red blood cells (called blood stage). Taking a cue from the effective treatments for a variety of human diseases that target one or more of the ∼530 human kinases, malaria researchers are taking aim at the <100 kinases present in malaria parasites, such as Plasmodium falciparum. In this article, the authors screened 1358 small-molecule kinase inhibitors that target diverse branches of the human kinome tree to assess their effect on Plasmodium parasite load in liver cells. After this screening identified candidate compounds, the compounds were tested in blood-stage malaria. Ideally, a drug that affected both blood-stage and liver-stage malaria while leaving normal human cells unaffected was desired. For the high-throughput screen, parasites from P. berghei (containing a transgenic luciferase reporter) mosquitos were used to infect the human liver cell line HepG2, and the effects of the compounds on parasite load were assessed. Another human liver cell line, Huh-7, along with an alternate detection method (immunofluorescence detection of anti-Plasmodium fluorescent antibodies) was used as a secondary assay.Compounds with IC50<10 μM (31 total) were considered to be hits and tested against blood stage malaria (P. falciparum) and for liver cell toxicity. All of the compounds were found to have a large window between effects on the parasite assay and the liver cell viability assay. Inhibitors of the human CDK family yielded the most frequent hits in the malaria assay. The authors note that this family of inhibitors has complications with the luciferase reporter system so the immunofluorescence assay was used predominantly. For each of the CDK inhibitors identified the IC50 in the malaria assay was elevated compared to the human enzyme so the authors undertook an earlier time point (12 hours); that is, before the expected duplication of the HEPG2 cells (doubling time is ∼48 hours) to see whether there was still an effect on parasite load, which doubles at a very fast rate. The effects were still there, and this indicated that the effect did not correlate with the effect of a CDK inhibitor on the human cell host cell cycle. PfPK5 kinase in Plasmodium parasites is the kinase most similar to CDK2, the most common human target of the CDK inhibitors identified as hits. The
figure
shows the docking of CDK1/2 to PfPK5. To confirm the docking prediction, all of the commercially available screening hits were tested in the commercially available competition binding assay for PfPK5 (DiscoveRx). CDK1/2, SNS-032 (currently in clinical trials for cancer indications), flavopiridol, and PIK-75 (a lipid kinase inhibitor) had Kd values for PfPK5 of <10 μM. CDK1/2, SNS-032, flavopiridol, and AT-5791 were tested and found to be safe and to inhibit liver-stage malaria in mice. SNS-032 was given prophylactically to mice prior to malaria infection and was found to reduce parasite load relative to vehicle control. The authors also tested CYC-116, an aurora inhibitor that was selective for liver-stage over blood-stage malaria, and found that it was also effective in reducing parasite load in the mouse model. It is fortunate that the predominantly human kinase KINOMEscan panel has this malaria kinase of interest here (PfPK5), and it would be helpful for the growing interest in malaria kinases if a comprehensive panel of malaria kinase assays was available as a companion to the human assay panel. In particular, it would be interesting to see how selective the above inhibitors are within the malaria kinome because many kinase inhibitors have extensive polypharmacology in humans. Contributed by Mindy I. Davis.
Binding to PfPK5. (A) Structure of CDK 1/2 docked onto the PfPK5 crystal structure (PDB ID: 1V0O). The protein kinase (pink) and CDK 1/2 carbon (gray), hydrogen (white), nitrogen (blue), sulfur (yellow), and oxygen (red) atoms are shown. Hydrogen bonds are predicted to form (black dotted lines) between the thioamide proton of CDK 1/2 and Gln84 and between the primary amine proton on aminotriazole and Asp85 (bond lengths <2.5 Å). (B) Percentage binding of commercially available kinase inhibitors to PfPK5 at 10 μm. Significant binding (>50%) is only observed with the known CDK inhibitors, flavopiridol, SNS-032, and CDK 1/2, and with the PIK inhibitor PIK-75. (C) Competitive binding plots of CDK 1/2 (red curve), flavopiridol (black curve), and SNS-032 (blue curve) to PfPK5, where decrease in relative signal correlates with compound binding. Data in (C) were fit to a nonlinear regression equation (curves shown) to obtain Kd values.
KINETIC BINDING PARAMETERS DERIVED FROM SIZE-EXCLUSION CHROMATOGRAPHY
Bao J, Krylova SM, Cherney LT, LeBlanc JC, Pribil P, Johnson PE, Wilson DJ, Krylov SN: Kinetic size-exclusion chromatography with mass spectrometry detection: an approach for solution-based label-free kinetic analysis of protein–small molecule interactions.Anal Chem2014;86:10016–10020.
Abstract: Studying the kinetics of reversible protein–small molecule binding is a major challenge. The available approaches require that either the small molecule or the protein be modified by labeling or immobilization on a surface. Not only can such modifications be difficult to do but also they can drastically affect the kinetic parameters of the interaction. To solve this problem, we present kinetic size-exclusion chromatography with mass spectrometry detection (KSECMS), a solution-based label-free approach. KSEC-MS utilizes the ability of size-exclusion chromatography (SEC) to separate any small molecule from any protein–small molecule complex without immobilization and the ability of mass spectrometry (MS) to detect a small molecule without a label. The rate constants of complex formation and dissociation are deconvoluted from the temporal pattern of small molecule elution measured with MS at the exit from the SEC column. This work describes the concept of KSEC-MS and proves it in principle by measuring the rate constants of interaction between carbonic anhydrase and acetazolamide.
Commentary:The quest to develop even-simpler platforms for interrogation of protein–small molecule interactions continues. Despite dramatic advances in surface plasmon resonance and related techniques, the need still remains for approaches that do not require surface immobilization of proteins because this step has remained the most difficult aspect of such experiments. Here, the authors provide a simple chromatography-based technique, coupled with novel data analysis, which allows the derivation of binding kinetics of small molecules interacting with proteins. In size-exclusion chromatography, an unbound small molecule travels slowly, eluting from the column last (together with salts, dyes, and/or other small molecule buffer components of the injected sample), while a fully bound ligand is expected to elute relatively fast as a part of its complex with the protein of interest (exact elution time of the latter depending on the molecular weight of the protein). The authors recognized that between these extreme cases lies the typical situation in which given certain konand koffof the complex, during migration through the size-exclusion column, the small molecule is likely to dissociate from the complex gradually and to begin to lag behind the protein, thus creating a specific profile of its concentration as a function of elution volume. The team used mass spectrometry for detection and quantitation of the small molecule in order to construct the profile (see
figure
) and then applied a series of equations to derive the kinetics of interaction. Data obtained from this method, which entails only injecting the complex and following the small molecule through mass spectrometry detector, compared favorably with results obtained through traditional means. The extreme simplicity of this new approach should make it an attractive alternative, especially in cases where the protein to be immobilized displays extreme sensitivity to such treatment. Contributed by Anton Simeonov.
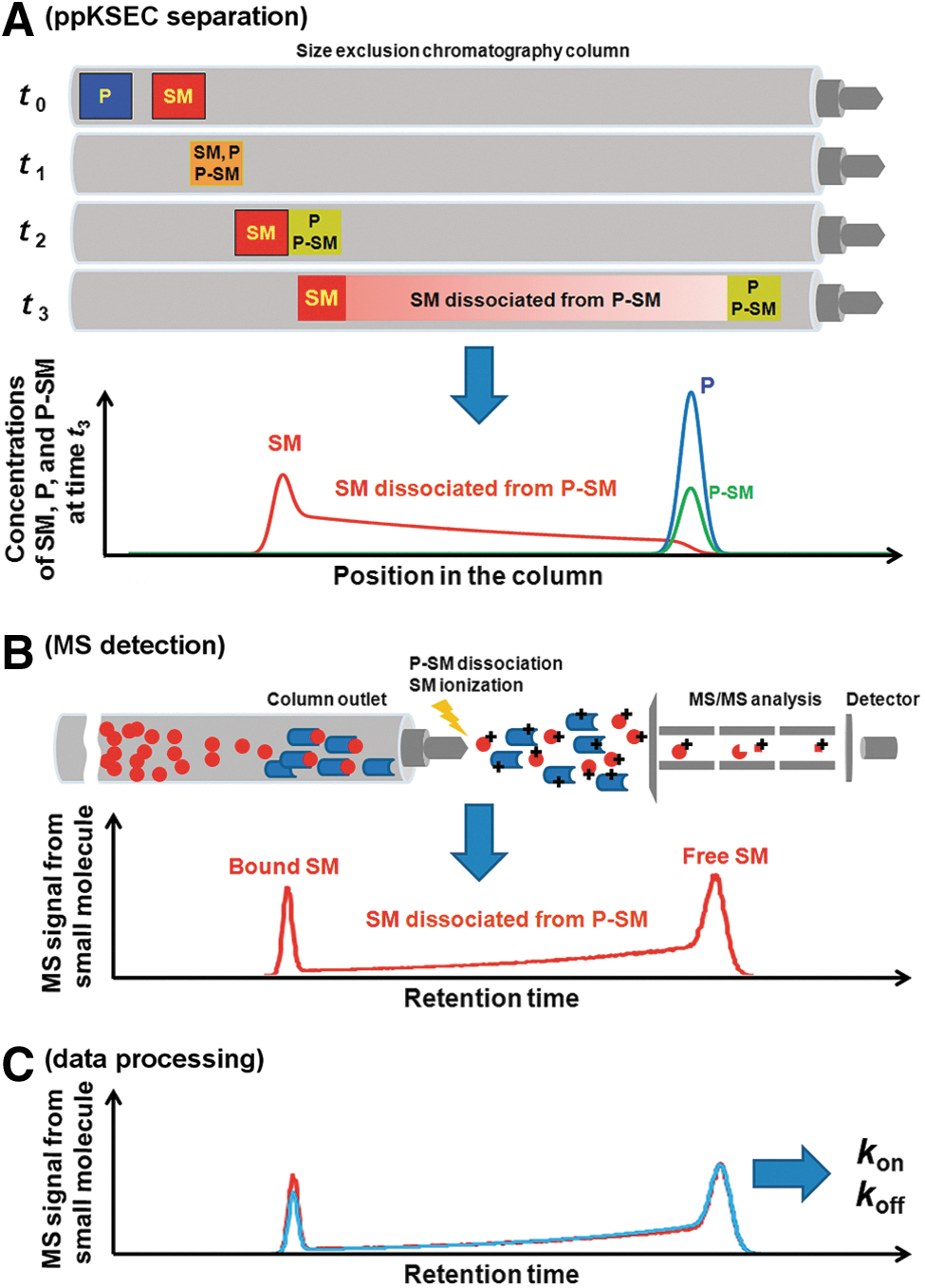
Conceptual depiction of ppKSEC-MS. (A) The small molecule, SM, and protein, P, are sequentially injected into a SEC column separated by a spacer of the buffer (t0). P moves faster than SM, and then, P meets SM; they can bind each other forming complex P–SM (t1). After P and P–SM leave the zone of SM (t2), P–SM starts dissociating. The released SM is continuously separated from P–SM and creates a bridge between the zones of P/P–SM and SM (t3). The graph illustrates the corresponding concentrations of P, SM, and P–SM at time t3. (B) The eluate from the column is continuously sampled into the ion source of a tandem MS/MS mass spectrometer, which is tuned to detect SM. The blue rods represent P while the red dots represent SM. SM dissociates to form P–SM, which facilitates indirect detection of intact P–SM exiting the column. The resulting ppKSEC-MS chromatogram contains two peaks and a bridge between them. (C) The experimental ppKSEC-MS chromatogram is numerically fitted with a computer-simulated one. The values of kon and koff are used as fitting parameters, and the best fit corresponds to the sought correct values of kon and koff.