NO SUGAR FOR YOU: BLOCKING HEXOSE UPTAKE IN MALARIA
Ortiz D, Guiguemde WA, Johnson A, Elya C, Anderson J, Clark J, Connelly M, Yang L, Min J, Sato Y, Guy RK, Landfear SM: Identification of selective inhibitors of thePlasmodium falciparumhexose transporter PfHT by screening focused libraries of anti-malarial compounds.Plos One2015; DOI: 10.1371/journal.pone.0123598.
Abstract: Development of resistance against current antimalarial drugs necessitates the search for novel drugs that interact with different targets and have distinct mechanisms of action. Malaria parasites depend upon high levels of glucose uptake followed by inefficient metabolic utilization via the glycolytic pathway, and the Plasmodium falciparum hexose transporter PfHT, which mediates uptake of glucose, has thus been recognized as a promising drug target. This transporter is highly divergent from mammalian hexose transporters, and it appears to be a permease that is essential for parasite viability in intra-erythrocytic, mosquito, and liver stages of the parasite life cycle. An assay was developed that is appropriate for high throughput screening against PfHT based upon heterologous expression of PfHT in Leishmania mexicana parasites that are null mutants for their endogenous hexose transporters. Screening of two focused libraries of antimalarial compounds identified two such compounds that are high potency selective inhibitors of PfHT compared to human GLUT1. Additionally, 7 other compounds were identified that are lower potency and lower specificity PfHT inhibitors but might nonetheless serve as starting points for identification of analogs with more selective properties. These results further support the potential of PfHT as a novel drug target.
Commentary:Malaria affects millions of individuals worldwide, and several hundred thousand people, predominantly children, die each year from malaria infections. There are drugs available to treat malaria, but malaria parasites are becoming resistant to some of the therapies, and some have unpleasant side effects. There is a need for new drugs to treat malaria at the various parasitic stages, as well as to identify novel therapeutic targets for malaria to help counter resistance. Malaria parasites must gather significant quantities of glucose from the host as the glycolytic pathway is the predominant source of energy production in parasites, rather than the TCA cycle. Malaria obtain glucose from the host through transporters such as PfHT (Plasmodium falciparum hexose transporter), which shares only 28% sequence identity to the closest human glucose transporter (GLUT1). PfHT was shown previously to be essential to the parasite and therefore an intriguing drug target. The authors developed a screening strategy (see
figure
) to identify inhibitors of parasite growth and glucose uptake. They engineered in PfHT or GLUT1 into Leishmania mexicana parasites that had their glucose transporter deleted. Their initial 384-well high-throughput SYBR green assay identified small molecule compounds that inhibited the growth of the parasites. In a lower throughput 96-well secondary screening assay, hits were tested for their ability to decrease the uptake of tritiated glucose into the parasite. As a counterscreen, they identified compounds that also affected the uptake of tritiated proline, an alternate carbon source for the parasites, to eliminate compounds that were not specific for PfHT. Through analysis of kinetics, Compound 1 was identified as a likely non- or mixed-competitive inhibitor, meaning that it does not need to compete with the high glucose levels found in the host. Compound 1 was quite active in the cell-based assays with inhibition of proliferation of 0.97–1.24 μM in a multiresistant and nonresistant strain of malaria. The inhibition of glucose uptake by Compound 1 via PfHT was 0.039 μM. This work has shown that this transporter can be targeted by small molecules with structures unique from sugar analogs, which were previously known to be competitive inhibitors. The assays have good Z′ and CV values and are amenable to additional screening of new libraries or SAR exploration of the current hits. Contributed by Mindy I. Davis.
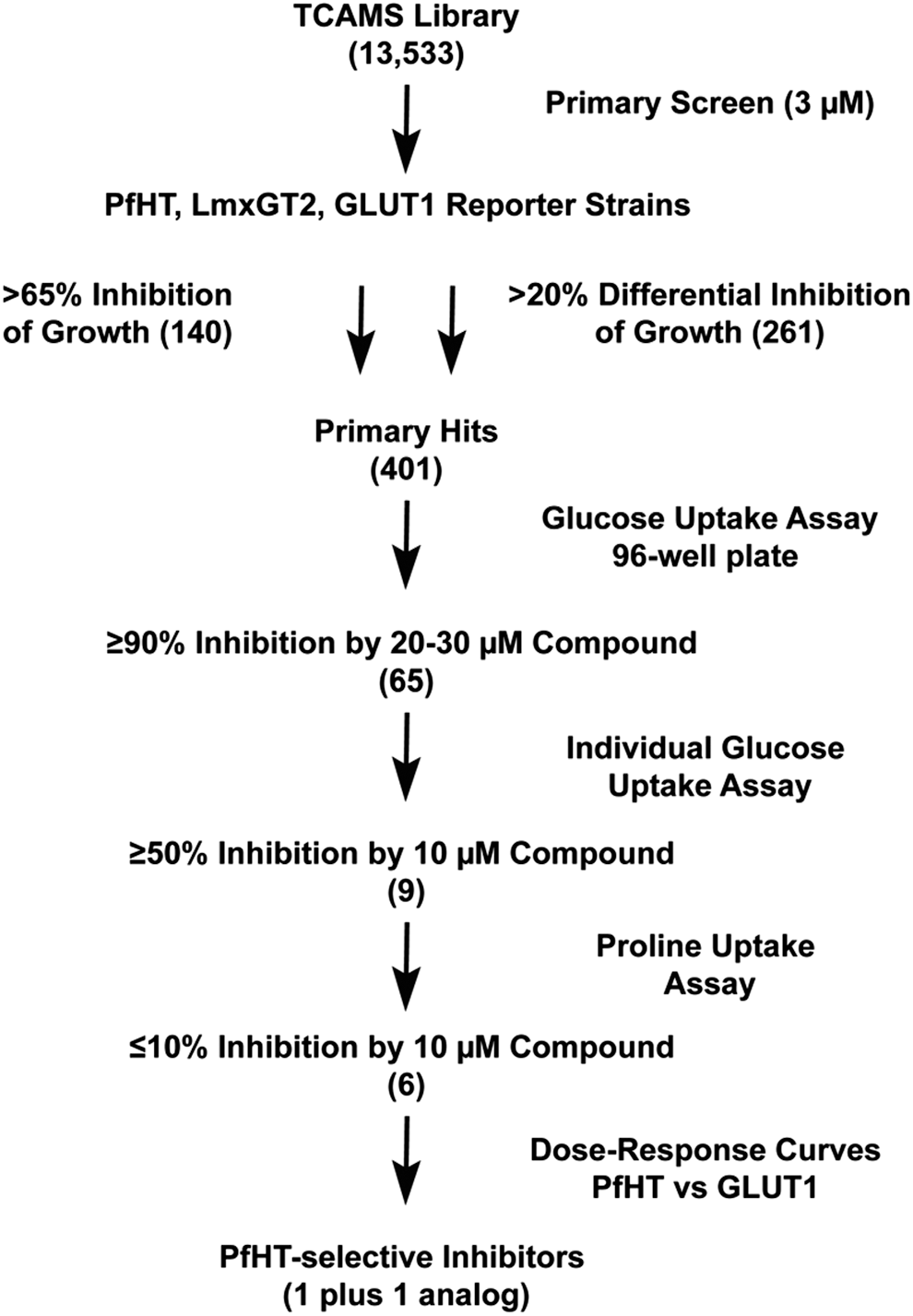
Flow chart for screen of TCAMS library. The TCAMS library of 13,533 compounds with demonstrated growth inhibitory activity against intraerythrocytic Plasmodium falciparum parasites was screened by sequential criteria. The steps included: (1) proliferation inhibitory screen (>65% inhibition at 3 μM concentration or >20% differential inhibition among the three strains) of PfHT, LmxGT2, and GLUT1 reporter strains to produce 401 primary hits; (2) 96-well plate assays for compounds (20–30 μM) that inhibited uptake of 200 μM [3H] D-glucose by ≥90%; (3) individual uptake assays for compounds (10 μM) that inhibited glucose uptake by ≥50%; (4) individual uptake assays for compounds (10 μM) that inhibited uptake of 100 μM [3H] L-proline by ≤10%; (5) dose–response curves for compounds that selectively inhibited uptake of glucose through PfHT versus GLUT1 (1 compound plus 1 additional hit that emerged from analysis of analogs). Numbers in parentheses represent the number of positive hits obtained after each sequential step.
FAPS FOR LIVE CELL IMAGING
Telmer CA, Verma R, Teng H, Andreko S, Law L, Bruchez MP: Rapid, specific, no-wash, far-red fluorogen activation in subcellular compartments by targeted fluorogen activating proteins.ACS Chem Biol2015;10:1239–1246.
Abstract: Live cell imaging requires bright photostable dyes that can target intracellular organelles and proteins with high specificity in a no-wash protocol. Organic dyes possess the desired photochemical properties and can be covalently linked to various protein tags. The currently available fluorogenic dyes are in the green/yellow range where there is high cellular autofluorescence and the near-infrared (NIR) dyes need to be washed out. Protein-mediated activation of far-red fluorogenic dyes has the potential to address these challenges because the cell-permeant dye is small and nonfluorescent until bound to its activating protein, and this binding is rapid. In this study, three single chain variable fragment (scFv)-derived fluorogen activating proteins (FAPs), which activate far-red emitting fluorogens, were evaluated for targeting, brightness, and photostability in the cytosol, nucleus, mitochondria, peroxisomes, and endoplasmic reticulum with a cell-permeant malachite green analog in cultured mammalian cells. Efficient labeling was achieved within 20–30 min for each protein upon the addition of nM concentrations of dye, producing a signal that colocalized significantly with a linked mCerulean3 (mCer3) fluorescent protein and organelle specific dyes but showed divergent photostability and brightness properties dependent on the FAP. These FAPs and the ester of malachite green dye (MGe) can be used as specific, rapid, and wash-free labels for intracellular sites in live cells with far-red excitation and emission properties, useful in a variety of multicolor experiments.
Commentary:Reagents that can be used to label cell compartments using no-wash protocols can greatly facilitate live cell high-content imaging assay implementation. This paper describes a series of fluorescent proteins that can be used to label five cellular compartments. The labels are based on fluorescent activating proteins (FAPs) and a fluorogen, in this case a compound that becomes fluorescent upon binding to the FAP. The construction of FAPs has been described and involves directed evolution of single-chain antibodies to find variants that bind fluorogens with high affinity, resulting in the generation of strong fluorescent signals. One such fluorogen is malachite green (MGe) and the ester of malachite green, which can penetrate cells. This paper constructed fusion proteins of organelle targeting sequences to a FAP that was also fused to the fluorescent protein mCerulean3 (mCer3). Labeling of the correct cellular compartment was then determined by confocal microscopy by examining co-localization of the mCer3 label with the FAP-bound fluorogen label (see
figure
). Due to the high affinity of the FAP for the MGe fluorogen, low concentrations of MGe could be used in the assay (∼25 nM). No phototoxicity was observed, but photobleaching in the presence of excess MGe differed between the cell compartments. The most photostable FAPs were found to be p13-CW and DL5**, with the latter having the most generally desirable properties for labeling. The half time for labeling of the FAPs with MGe using a no-wash protocol was found to be 15 min or less. Previous applications of FAPs in imaging applications have focused on cell surface staining, but this paper demonstrates a series of reagents for labeling intracellular compartments. Furthermore, the fluorescent emission is in the far-red, which will facilitate multiplexing with other labels. Contributed by Doug Auld.
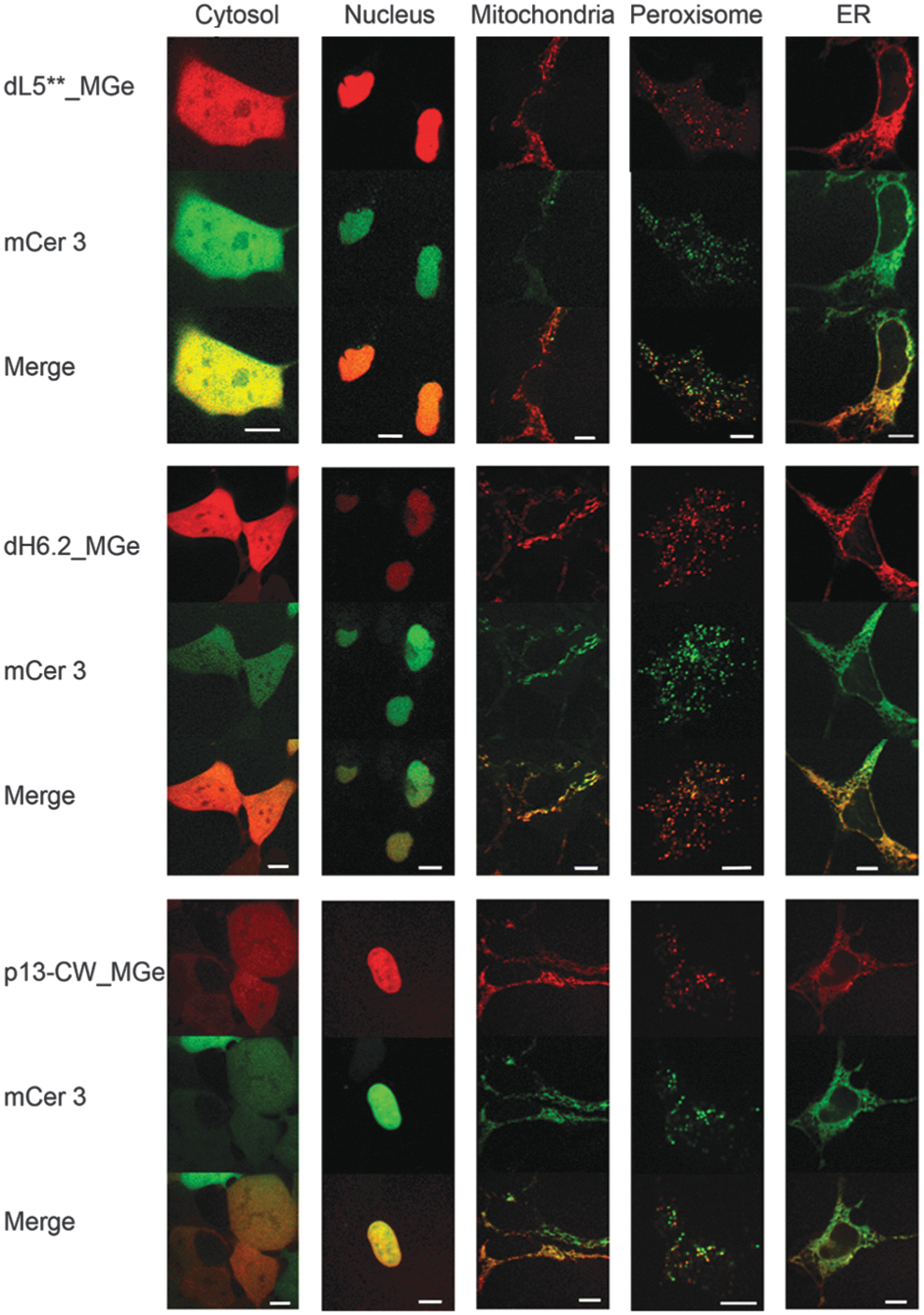
Confocal images showing the fluorescence pattern following expression of three different FAP-mCer3 tandems (side labels) targeted to five different cellular locations (top label). Signal from FAP_MGe (red) colocalizes with that of mCer3 (green), resulting in a yellow color in the merged images. Scale bars are 10 μm.
DISENTANGLING VEGF EFFECTS
Yang X, Zhang Y, Hosaka K, Andersson P, Wang J, Tholander F, Cao Z, Morikawa H, Tegner J, Yang Y, Iwamoto H, Lim S, Cao Y: VEGF-B promotes cancer metastasis through a VEGF-A-independent mechanism and serves as a marker of poor prognosis for cancer patients. Proc Natl Acad Sci U S A 2015;112:E2900–E2909.
Abstract: The biological functions of VEGF-B in cancer progression remain poorly understood. Here, we report that VEGF-B promotes cancer metastasis through the remodeling of tumor microvasculature. Knockdown of VEGF-B in tumors resulted in increased perivascular cell coverage and impaired pulmonary metastasis of human melanomas. In contrast, the gain of VEGF-B function in tumors led to pseudonormalized tumor vasculatures that were highly leaky and poorly perfused. Tumors expressing high levels of VEGF-B were more metastatic, although primary tumor growth was largely impaired. Similarly, VEGF-B in a VEGF-A-null tumor resulted in attenuated primary tumor growth but substantial pulmonary metastases. VEGF-B also led to highly metastatic phenotypes in Vegfr1 tk(–/–) mice and mice treated with anti-VEGF-A. These data indicate that VEGF-B promotes cancer metastasis through a VEGF-A-independent mechanism. High expression levels of VEGF-B in two large-cohort studies of human patients with lung squamous cell carcinoma and melanoma correlated with poor survival. Taken together, our findings demonstrate that VEGF-B is a vascular remodeling factor promoting cancer metastasis and that targeting VEGF-B may be an important therapeutic approach for cancer metastasis.
Commentary:Vascular endothelial growth factor (VEGF) has been studied extensively during the past decades, and its role in promoting tumor angiogenesis has been well documented. Agents targeting the VEGF signaling system to reduce tumor vascularization and growth already exist. In recent years, however, a more complex picture has emerged where the angiogenesis properties of VEGF have been specifically mapped to its type A isoform (VEGF-A), while the functions of the VEGF-B counterpart have remained poorly characterized. VEGF-A binds to two cognate receptors—VEGF receptor 1 and 2 (VEGFR1 and VEGFR2)—with VEGFR2 mediating most of the VEGF-A's effects on vasculature, while VEGF-B binds only to VEGFR1. Here, the team used a combination of cell biology, clinical data analysis, as well as mouse and zebrafish tumor models to dissect the role of VEGF-B. Surprisingly, VEGF-B was shown to have a distinct effect on tumor vasculature and to promote metastasis in human and mouse tumor models. Instead of promoting angiogenesis, a role clearly being played by VEGF-A, VEGF-B negatively regulated angiogenesis and caused vasculature remodeling, leading to leaky vessels and tumor cell intravasation, which in turn was associated with significant metastasis (see
figure
). Thus, in addition to outlining a unique role for VEGF-B, this work demonstrates the uncoupling of metastasis from tumor growth, and potentially paves the way to targeting metastasis by blockade of VEGF-B specifically. Contributed by Anton Simeonov.
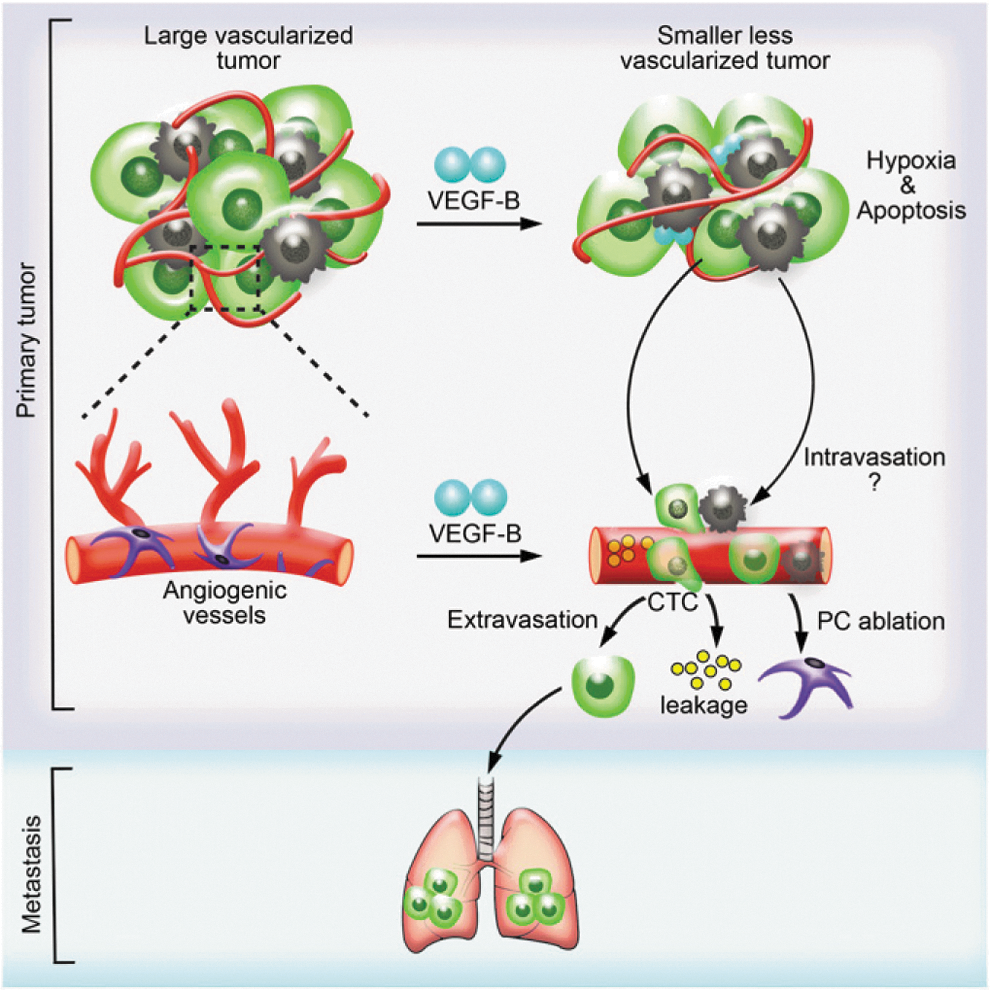
Schematic illustration of VEGF-B-induced tumor vessel remodeling and metastasis. The tumor-derived high VEGF-B level leads to the suppression of tumor angiogenesis and increased cell death. As a consequence, primary tumor growth is impaired, and the tumor microenvironment becomes more hypoxic. However, VEGF-B facilitates tumor cell intravasation by generating highly leaky vasculature in tumor and recruiting M2-like macrophages, thus giving rise to more severe pulmonary metastasis.
USING PHAGE TO REPROGRAM A BACTERIAL POPULATION
Yosef I, Manor M, Kiro R, Qimron U: Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria.Proc Natl Acad Sci U S A 2015;112:7267–7272.
Abstract: The increasing threat of pathogen resistance to antibiotics requires the development of novel antimicrobial strategies. Here we present a proof of concept for a genetic strategy that aims to sensitize bacteria to antibiotics and selectively kill antibiotic-resistant bacteria. We use temperate phages to deliver a functional clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR associated (Cas) system into the genome of antibiotic-resistant bacteria. The delivered CRISPR-Cas system destroys both antibiotic resistance-conferring plasmids and genetically modified lytic phages. This linkage between antibiotic sensitization and protection from lytic phages is a key feature of the strategy. It allows programming of lytic phages to kill only antibiotic-resistant bacteria while protecting antibiotic-sensitized bacteria. Phages designed according to this strategy may be used on hospital surfaces and hand sanitizers to facilitate replacement of antibiotic resistant pathogens with sensitive ones.
Commentary:Bacteria eradication methods using antibiotics or the much less popular lytic phage have a major weakness in causing the emergence and establishment of treatment-resistant bacterial populations, particularly in hospital settings. A new approach presented here attempts to change the course of nosocomial infections by making the bacteria more susceptible rather than more resistant to antibiotics. The team accomplishes this by using CRISPR-Cas system to modify traditionally lytic lambda phage rendering them lysogenic and concomitantly able to destroy specific bacterial plasmids that confer antibiotic resistance while simultaneously providing advantage to the antibiotic-sensitized bacteria by making them resistant to the lytic phage. Thus, by application of the new phage to a bacterial population, one aims to transform the population from lytic phage-sensitive and antibiotic-resistant to predominantly antibiotic-sensitive without concomitant loss of fitness. Enrichment of antibiotic-sensitized bacteria was achieved by mixing a bacterial culture with the new lysogenizing phage to produce a mixture of lysogens and nonlysogens in the culture. Subsequently, application of lytic phage leads to a selective elimination of the nonlysogens leaving behind lysogens that are both antibiotic-sensitized and phage resistant, as the CRISPR-Cas system degrades the antibiotic resistance-conferring plasmid and the lytic-phage chromosome (see
figure
). The authors propose the intended practical application of the method as consisting of treating the skin of patients and hospital staff, as well as common surfaces, with a topical preparation of the phage to shift the predominant populations gradually from antibiotic resistance to sensitive. Contributed by Anton Simeonov.
Enrichment of antibiotic-sensitized bacteria by lytic phages; schematics of the procedure to enrich for antibiotic-sensitized bacteria. A bacterial culture is mixed with lysogenizing phages, resulting in both lysogens and nonlysogens in the culture. Lysogens are both antibiotic sensitized and phage resistant as the CRISPR-Cas system degrades the antibiotic resistance-conferring plasmid and the lytic-phage chromosome. The treated culture is inoculated on agar-containing lytic phages that selectively kill the nonlysogens and thus enrich for antibiotic-sensitized bacteria.
EDITING RGAS FOR IMPROVED ANALYSIS
Hasson SA, Fogel AI, Wang C, MacArthur R, Guha R, Heman-Ackah S, Martin S, Youle RJ, Inglese J: Chemogenomic profiling of endogenous PARK2 expression using a genome-edited coincidence reporter.ACS Chem Biol2015;10:1188–1197.
Abstract: Parkin, an E3 ubiquitin ligase, is a central mediator of mitochondrial quality control and is linked to familial forms of Parkinson's disease (PD). Removal of dysfunctional mitochondria from the cell by Parkin is thought to be neuroprotective, and pharmacologically increasing Parkin levels may be a novel therapeutic approach. We used genome editing to integrate a coincidence reporter into the PARK2 gene locus of a neuroblastoma-derived cell line and developed a quantitative high-throughput screening (qHTS) assay capable of accurately detecting subtle compound-mediated increases in endogenous PARK2 expression. Interrogation of a chemogenomic library revealed diverse chemical classes that up-regulate the PARK2 transcript, including epigenetic agents, drugs controlling cholesterol biosynthesis, and JNK inhibitors. Use of the coincidence reporter eliminated wasted time pursuing reporter-biased false positives accounting for ∼2/3 of the actives and, coupled with titration-based screening, greatly improves the efficiency of compound selection. This approach represents a strategy to revitalize reporter-gene assays for drug discovery.
Commentary:Reporter gene assays (RGAs) are a commonly used assay format to construct cell-based assays aimed at measuring pathway activity through measuring a transcriptional response. However, many compounds in typical screening libraries can interfere with luciferases such as firefly luciferase (FLuc) or Renilla luciferase (RLuc), leading to a large enrichment of this unwanted activity in hit lists derived from RGAs. Counterscreens against the reporter enzyme itself or other cell-based assays using the same reporters only serve to deselect compounds that interfere with the reporter systems and do not answer if such compounds may have activity of interest. The best strategy to address this interference is to use an orthogonal reporter enzyme known to have a different inhibitor profile, but this requires constructing a separate cell line in an isogenic fashion. This paper provides a solution to improve the interpretation of RGA data by incorporating two orthogonal luciferases in the same cell. The so-called “coincident reporter” system involves expressing both reporters from one mRNA where the two reporters are separated by a ribosomal skipping peptide, resulting in near-equal expression of both reporters as separate enzymes in cells (see
figure
). In this way, a built-in orthogonal assay is achieved by detecting both reporters. Desirable compound activity should show near equal responses from both reporter enzymes, while compounds interfering with one of the reporter enzymes will show a biased effect. The authors further improved this system through introducing the reporters with gene editing. In the paper, the neuroblastoma cell line (BE(2)-M17) was engineered using TALEN-mediated genome editing to express both FLuc and a PEST-destabilized form of NanoLuc (NLucP) at the PARK2 locus. A FLuc/NLuc dual detection reagent was developed with Promega Corp. so that FLuc activity can be read first, followed by quenching of the FLuc signal and detection of NLucP. Titration-based screening (qHTS) was then used to panel a chemogenomic library for regulators of PARK2 expression, a gene that is involved with maintenance of mitochondria. Retrospective analysis of the activity at a single screening concentration showed that the coincident RGA reduced the number of actives by 77% if FLuc alone was used, and by 72% if NLuc alone was used. Therefore, the coincident reporter assay saved time and greatly improved the interpretation of the screening results. A series of dihydropyridines inhibited and stabilized NLucP in cells leading to increases in NLuc activity but not FLuc activity in the coincident RGA. Such noncoincident activity was readily identified in the assay. Among compounds showing a coincident activity profile were inhibitors of epigenetic targets including Brd4 and inhibitors of JNK. Interestingly, another PARK2 pathway identified included the cholesterol biosynthetic pathway perhaps providing a link between prolonged statin dosing and the observed lower rates of Parkinson's disease through a neuroprotective effect mediated by PARK2 expression. Contributed by Doug Auld.
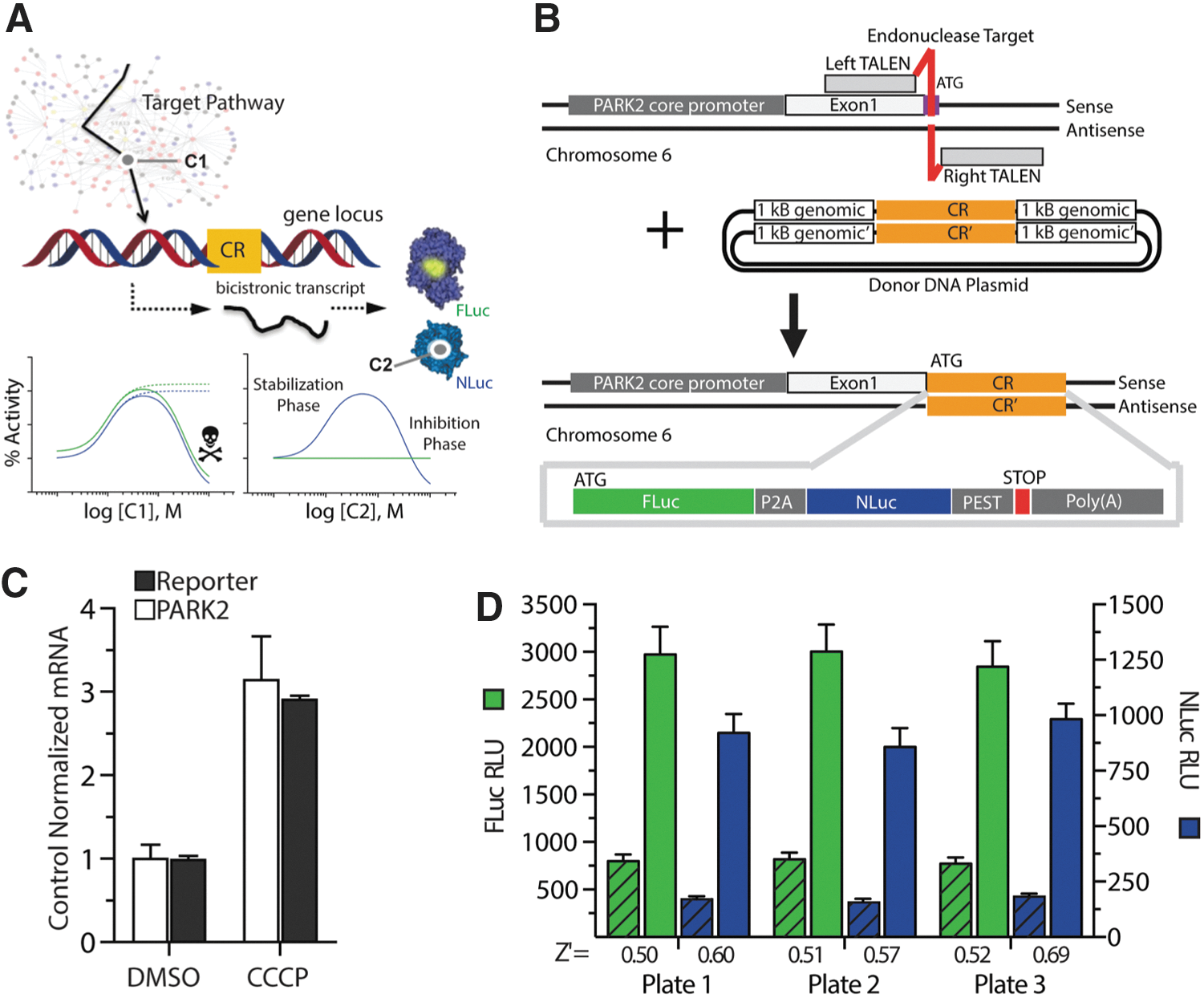
Development of a coincidence reporter qHTS assay for endogenous PARK2 expression. (A) Overview of coincidence reporter (CR) concept for chemical library HTS. Combined outputs from individual reporters encode the mechanism of compound activity as shown conceptually in model concentration response plots. For example, compound C1 acts on a target within a cellular pathway, as reporters respond similarly in an unbiased manner, while the noncoincident response of C2 indicates stabilization of the NLuc reporter. (B) Graphical representation of the TALEN mediated genome editing strategy for coincidence reporter insertion into the PARK2 locus. In a BE(2)-M17 neuroblastoma cell line, a TALEN pair was used to generate double-strand DNA breaks (site in red near the ATG start codon) and stimulate homologous recombination of the sequence containing the FLuc-P2A-NLuc-PEST coincidence reporter from an HDR vector. All primers used to create the HDR vector are listed in Table S1. (C) qRT-PCR analysis of mRNA from PTRC6 cells after treatment with 10 μM CCCP or DMSO for 24 h. Black bars represent levels of the coincidence reporter mRNA, and white bars represent levels of the wild-type PARK2 mRNA. (D) Bioluminescent output from assay validation plates (sample image in Figure S1E) demonstrate assay performance in the FLuc (green; left y-axis) and NLuc (blue; right y-axis) channel. Data shown for three replicate plates, left bar graph in FLuc or NLuc represents DMSO control (hatched) and treatment with 500 nM panobinostat (solid). Bars in C and D represent mean ± SD of three independent experiments.