
Abstract
Conformational remodeling of chromatin in cells is known to alter gene expression. The histone code hypothesis postulates that multiple modifications present on histone tails can regulate gene expression both through direct effects on chromatin compaction as well as through recruitment of unique complexes that signal specific downstream functions. Histone methylation is an important component of the histone code, and the dysregulation of histone methylation in disease makes methyltransferases and demethylases viable targets for drug discovery. We developed a biochemical assay platform, which takes advantage of the fact that protein methyltransferases (PMTs) all utilize the cofactor S-Adenosyl-L-methionine (SAM) as the methyl donor. The platform utilizes the High-throughput Mass Spectrometry (MS) technology to measure SAM and the S-Adenosyl-L-homocysteine product in a label-free manner. The platform has all the advantages of a label-free system coupled with the benefit of substrate agnostic measurements making it an ideal setup for PMT biochemical studies and drug discovery. In addition, MS is ideally suited for detecting multiple modification events within the same substrate. The ability to adjust the detection to monitor the methyl acceptor product allows for real-time measurements of multiple product species simultaneously, a distinct advantage over other commonly used assay formats.
Introduction
Epigenetics is defined as heritable changes in the phenotype that cannot be explained by changes in the DNA sequence. Chromatin, the physiological substrate for all DNA-templated processes, is highly dynamic and exists in transcriptionally active and inactive forms. Modifications to DNA and histone proteins, including methylation and acetylation, cause changes in the chromatin structure and recruit additional proteins to specific chromatin regions where they act as effectors of chromatin modification. 1,2 These epigenetic events are critical in the regulation of all DNA-templated events, including gene transcription. Deregulation of gene expression through alteration of DNA methylation and histone modification contributes to oncogenesis through suppression of tumor suppressor genes and genes involved in differentiation, as well as activation of transcription at normally silent genes. 2 –5 In excess of 90, protein methyltransferases (PMTs) have been identified in the human genome, 6 many of which are known to be mutated and/or overexpressed in cancer, making this an attractive target class for pharmacological intervention. 7,8
While PMTs have proven to be druggable, 7,9 –12 identification of bone-fide leads with good ligand efficiency and physicochemical properties has proven challenging for many enzymes in this class. As a result, we decided to include an experimental druggability assessment as part of our target evaluation. Fragment screening has been described as a method to experimentally evaluate druggability, 13 and therefore, we chose to include fragment screening as part of our druggability assessment. To take advantage in the similarities across this large class of related enzymes and evaluate multiple different methyltransferases in parallel, we needed an assay platform that was substrate agnostic and capable of screening medium-sized chemical libraries and our fragment library.
Multiple assay formats for PMTs have been described each with its own advantages and disadvantages. 14 We prefer the radioactive filter-binding assay (RFA) format because it is free from issues related to compound interference and inhibition of coupling enzymes and is very sensitive and largely substrate agnostic. Like all assay formats, however, there are downsides to the RFA. The use of radioactivity necessitates permits and procedures for use, storage, and disposal of radioactive materials, which contributes significantly to the overall cost and ease of implementation, particularly for small organizations. The filtration step necessitates capture of the methyl acceptor product on the filter. This works quite well overall and is easy to optimize; however, care must be taken when comparing the activity of a specific enzyme across a series of diverse substrates to ensure that differences in substrate binding to the filter are not contributing to apparent activity differences. This is especially true when evaluating multiple different peptide substrates where the overall charge of the peptide can impact the capture efficiency. 15 Finally, the biggest drawback to the RFA is the fact that the location of the transferred methyl group cannot be determined. For example, following PRMT5 methylation of H4 (1–21) peptide, it is not possible to determine the relative amounts of H4R3me1 versus H4R3me2 formed, only to quantitate the total number of methyl groups transferred.
Mass spectrometry (MS) is a technology that has been used to quantitate multiple different peptide species from the same sample 16 –19 and has been used successfully to quantitate changes in the methylation status on peptides and nucleosomes. 20 However, it requires specialized equipment and expertise. Furthermore, until recently, the throughput of MS methods was insufficient to be useful for primary screening or iterative drug discovery. The development of high-throughput MS technologies, including the RapidFire SPE-MS/MS system from Agilent, 21,22 the ADDA LC-MS/MS system from Apricot Designs, 23 and SAMDI, 24 enables the use of MS as a platform for drug discovery and medium-throughput screening.
We chose to implement the Agilent RapidFire SPE-MS/MS system because it was compatible with existing equipment in our laboratory and also because at that time Agilent provided assay development services as well as a fee for service model for testing assay plates. This allowed us to evaluate the RapidFire technology before purchasing the system. For methyltransferase assays, detection of the S-Adenosyl-L-methionine (SAM) substrate and S-Adenosyl-L-homocysteine (SAH) product affords a highly sensitive, methyl acceptor substrate agnostic, nonradioactive, label-free, and coupling enzyme-free system for biochemical characterization and screening of PMTs. A simple adjustment of the mass spectrometer to monitor the methyl acceptor substrate and product rather than SAM/SAH, allows for quantitation of the various different methylated product species simultaneously.
In this study, we compare the SPE-MS/MS assay format to the gold standard RFA format using protein arginine methyltransferase 5 (PRMT5) as a test case. PRMT5 is a type II arginine methyltransferase, which catalyzes the formation of ω-NG-monomethyl and ω-NG, N′G-symmetric dimethylarginine residues. Along with its binding partner MEP50, PRMT5 is known to form multiple different complexes, which facilitate methylation of a large number of different cytoplasmic and nuclear proteins, in a context-specific manner. 17,25 Some of the known substrates for PRMT5 include epidermal growth factor receptor, p53, Sm proteins, Histone H2A, Histone H3, and Histone H4. This work utilizes a 21-mer peptide based on the Histone H4 sequence as the substrate, but the results are indicative of results obtained with rH2A, rH4, rH3/rH4 tetramer, and SmD3/pICLn protein substrates, as well as alternative peptide substrates.
Materials and Methods
Materials
SAM and recombinant histone H2A and H3 were purchased from New England Biochemical. SAH and Sinefungin were purchased from Sigma. Recombinant H3/H4 tetramer was from BPS Biosciences. MS solvents were purchased from Sigma and Burdick & Jackson. Custom peptides were synthesized by CPC Scientific. Ultrapure SAM was purchased from Cisbio.
Expression and Purification of PRMT5/MEP50 Protein Complex
Flag tagged full-length PRMT5 and full-length untagged MEP50 were cloned into the insect cell expression vector pFASTBac Dual (Life Technologies). In addition, MEP50 was subcloned into the pFASTBac1 vector. Viruses were generated using standard Bac-to-Bac viral generation protocols (Life Technologies). Protein overexpression was conducted in exponentially growing Sf21 cells infected at 2×106 with P2 viral stock at MOI=1 of PRMT5-Mep50 dual construct virus and Mep50 construct virus at 1:1 ratio. The coexpression protocol was used to supplement additional Mep50 for FlagPRMT5-Mep50 heterodimer formation. The FlagPRMT5-Mep50 complex was purified from the cell lysate using Flag affinity chromatography. FlagPRMT5-Mep50 was further purified using the S300 26/600 column (GE Healthcare).
Radioactive Filter-Binding Assay
PRMT5/MEP50 (10–25 nM) was combined with the H4 (1–21) peptide (SGRGKGGKGLGKGGAKRHRKV) in assay buffer (50 mM Tris pH 8.5, 50 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT), and 44 μL was added to a microtiter plate containing 1 μL of 100% DMSO or test compound. SAM was prepared by combining 3H-labeled SAM with unlabeled SAM in the assay buffer such that the final SAM concentration was 1 μM and the specific activity was 0.2 μCi/μL. The reaction was initiated by adding 5 μL of SAM stock to the microtiter plate. Reactions were run at room temperature either as time course experiments where velocity was determined by linear regression or at a single 15-min time point. The reaction was stopped with the addition of 100 μL of 20% trichloroacetic acid. The 3H-peptide product was captured using a 96-well filter plate (MSIPN4B50; Millipore) and washed five times with a phosphate-buffered saline buffer. Scintillation fluid (50 μL) was added to the dried filter plate and counted in a liquid scintillation counter. IC50 values were determined by fitting the data to the standard four-parameter dose–response equation using Prism® software (GraphPad). For conversion of CPM to pmoles of product, 5 μL of a 1/10 dilution of the 3HSAM stock was added to five individual empty wells in the plate after filtration and after scintillation fluid was added. Counts were averaged and divided by the pmoles of SAM in the well to get specific activity in CPM/pmol.
MS Assay
Experiments were run as described for the radioactive filtration assay. Following incubation for the appropriate time, reactions were stopped by the addition of formic acid. Plates were then sealed and analyzed by RapidFire-MS/MS.
RapidFire Solid-Phase Extraction
Enzymatic reactions were analyzed utilizing a RapidFire 350 high-throughput SPE chromatography system coupled to a 6495 triple quadrupole mass spectrometer (Agilent Technologies). Detection of SAM and SAH was accomplished following injection of 42 μL of reaction mix (injection loop volume is 10 μL) onto an Agilent Graphite Type D cartridge in 0.1% trifluroacetic acid (TFA) and eluted using 80% acetonitrile and 0.1% TFA. For detection of peptides, the reaction mix was injected onto a C18 cartridge in 0.09% formic acid and 0.01% trichloroacetic acid and eluted in the same buffer plus 80% acetonitrile. The finalized RapidFire settings were as follows: aspiration time: 600 ms or until the loop is full per the sip sensor, load time: 3,000 ms, elution time: 5,000–8,000 ms, and re-equilibration time: 500 ms at a flow rate of 1.5 mL/min.
Mass Spectrometry
Following RapidFire SPE, samples were eluted into an Agilent 6495 triple quadrupole mass spectrometer with an Agilent Jet Stream source with ion funnel technology, set in the positive ion mode. A multiple reaction monitoring (MRM) protocol was optimized employing Q1 m/z ratios of 399.0 and 385.1 for SAM and SAH, respectively. The second quadrupole (Q2) was used as a collision chamber employing house nitrogen as the collision gas. The third quadrupole (Q3) was set to select the product ions of SAM (m/z=250.1 and 136.1) and SAH (m/z=136.1). The MRM method for detecting H4(1–21) peptides used m/z ratios of 523.7, 527.0642, and 530.5681 for unmethylated, monomethylated, and dimethylated peptides, respectively. The product ion for all peptides was 129.1 m/z. Fragmentor voltage was 380 V, collision energy settings were 10 V for SAM, 20 V for SAH, and 43 V for peptides. Area under the curve (AUC) for SAM, SAH, and peptide was quantitated using RapidFire Integrator software (Agilent). For conversion of AUC to pmoles of analyte, standard curves were generated over a range of concentrations from 1 nM up to 1 μM. For inhibition studies, the peptide product was observed as monitoring the SAH product is not feasible when SAH is the inhibitor. It is possible to quantitate SAH inhibition by monitoring loss of SAM (data not shown).
Results
MS Method Development
LC-MS/MS methods to detect SAM, SAH, and various methylated forms of histone peptides have been previously described. 21,26,27 Direct infusion of SAM, SAH, and the peptide substrate into the triple quadrupole mass spectrometer resulted in detection of the expected parent and product ions.
To meet the throughput requirements of drug discovery, an MS-based enzymatic assay has to be capable of analyzing a 96-well plate in well under an hour. Traditional LC-MS/MS methods typically take at least 1–2 min per sample depending on the LC method used and the time required to regenerate the column before injecting the next sample. This low throughput is insufficient to support the testing of many compounds typical of a drug discovery project. This is especially true if one wishes to evaluate multiple enzyme targets or substrates in parallel or run a screening campaign. We chose to utilize the RapidFire system and have consistently seen cycle times of 10–12 s. This results in analysis times of 15–20 min for a 96-well plate. Following optimization of RapidFire trap and elute parameters (see Materials and Methods section), both SAM and SAH in the assay buffer were analyzed. A linear correlation between the analyte peak area and analyte concentration with a limit of quantitation of 2 nM for both SAM and SAH (Fig. 1) was observed.

Evaluation of S-Adenosyl-L-methionine (SAM) and S-Adenosyl-L-homocysteine (SAH) by mass spectrometry (MS). SAM and SAH were infused directly into the mass spectrometer. Peaks at 399.0 and 385.1 m/z are observed for SAM and SAH, respectively. Fragmentation in quadrupole 2 resulted in characteristic peaks for SAM (250.1 and 136.1) and SAH (136.1). SAM and SAH standard curves in the assay buffer following trap and elute on the RapidFire were generated by plotting the AUC of the fragment ions. The limit of quantitation was determined to be 12,000 AUC and is calculated from 3×the average value of six data points collected from blank injection samples taken after a nonblank sample. SAM data are represented by squares and SAH by circles. AUC was determined for each injection at the indicated concentration and is represented with closed symbols. The carryover between injections was quantitated and is represented with open symbols. The figure inset highlights the data at a low analyte concentration. AUC, area under the curve.
These methods were subsequently used to quantitate the enzymatically generated production of SAH as well as the amount of SAM remaining in the sample. Similar results were obtained monitoring the peptide substrate with a limit of quantitation of 5 nM (data not shown).
Validation of HT-MS/MS Assay
The PRMT5 activity was monitored using the high-throughput MS/MS (HT-MS/MS) assay, and results were compared to results generated using RFA. The RFA has many of the same advantages of the MS assay, including high sensitivity and direct measurement of the enzymatic product. 14 Therefore, we used the radioactive assay format as a benchmark to evaluate the high-throughput mass spectrometry (HT-MS/MS) assay. Inspection of the progress curve (Fig. 2A) indicates that both assay formats are able to detect the enzymatically generated product, SAH in the case of the HT-MS/MS assay and methylated H4(1–21) peptide in the case of the RFA, with high sensitivity.
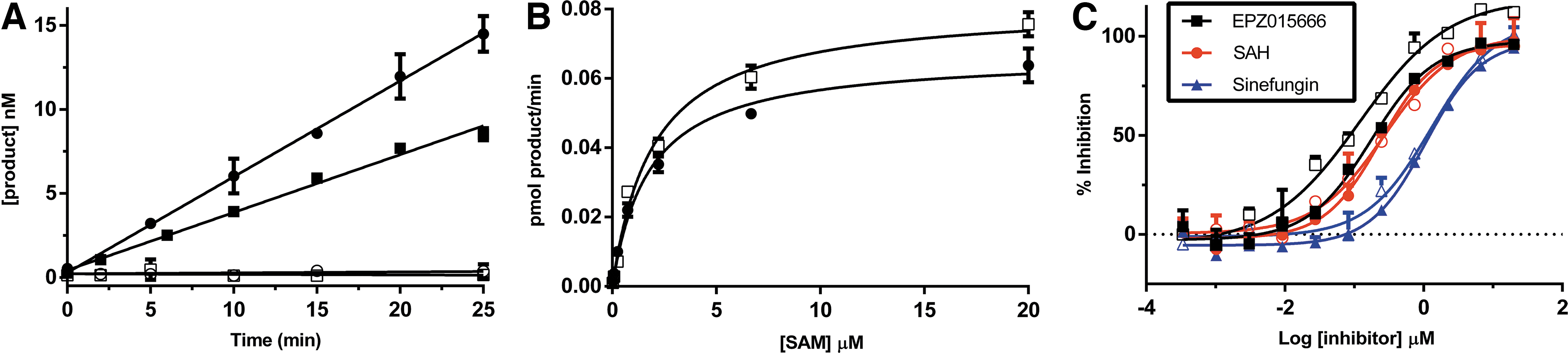
PRMT5 reaction kinetics. PRMT5 enzyme activity was quantitated using both radioactive filtration and HT-MS/MS assay formats. Data are representative of multiple independent experiments. Each data point is the average of three replicates; error bars represent the standard deviation.
A broad characterization of PMTs shows that this class of enzymes tends to exhibit a relatively poor catalytic activity coupled with high-affinity binding of the methyl acceptor substrate. 14 The observed low Km values result in a low substrate concentration in assays designed to identify all possible inhibitors, including those competitive with the substrate. As a result, the amount of product produced under initial velocity conditions necessitates a very sensitive assay. The limit of detection observed with the HT-MS/MS assays is compatible with the sensitivity requirements for even the most difficult PMTs. For PRMT5, the observed Km for the H4 (1–21) peptide in the RFA is 1 μM. To stay below 10% turnover to satisfy initial velocity conditions, an assay needs to be able to reproducibly quantitate a product below 100 nM. Figure 2A shows that both the HT-MS/MS and RFA assay formats are able to quantitate SAM and SAH at concentrations below 100 nM.
Comparison of PRMT5 kinetics (Fig. 2B, C; Tables 1 and 2) demonstrates that the HT-MS/MS assay is able to produce similar results both with respect to kinetic constants as well as inhibition by the product inhibitor SAH, the SAM analog sinefungin, and by EPZ015666, a potent and selective PRMT5 inhibitor. 28 The IC50 values generated here for EPZ015666 are higher than those reported in the literature. This is likely due to the fact that we used a higher peptide concentration in our assay, and this compound is reported to be a peptide competitive inhibitor.
Comparison of Kinetic Constants for PRMT5
PRMT5 catalyzed methylation of H4 (1–21) peptide was monitored using the RFA or HT-MS/MS. Assay conditions, including enzyme concentration, were identical for both assay formats.
HT-MS/MS, high-throughput mass spectrometry assay; RFA, radioactive filter-binding assay; SAM, S-Adenosyl-L-methionine.
Comparison of Inhibition Constants
IC50 values determined in both assay formats displayed in μM. The values are averages of three independent measurements±SD.
SAH, S-Adenosyl-L-homocysteine
Quantitation of Methyl Peptide Product
Quantifying methyltransferase activity by monitoring the conversion of SAM to SAH ignores one powerful aspect of product detection by MS, the ability to easily resolve multiple methylation states of the substrate. Since many PMTs are known to transfer more than one methyl group to a target lysine or arginine, the ability to measure the levels of each methylated species can provide valuable information. Unlike the RFA, HT-MS/MS can individually quantitate product peptides with different methylation states from the same sample simultaneously and has the capability to watch the conversion of methylated species in real time. To illustrate this, we show the methylation of the histone H4 (1–21) peptide over time (Fig. 3). In this study, you can see the amount of unmethylated, monomethylated, and dimethylated peptides present at each time point. This experiment can be done in a continuous format with repetitive sampling from the same tube or in the discontinuous format where the reaction is stopped with formic acid at the desired time points followed by sample analysis. Quantitation of each peptide over time shows the expected build-up of monomethylated peptide before formation of dimethylated peptide. Studies of this type have been used to suggest a distributive mechanism for PRMT5 methylation. 17
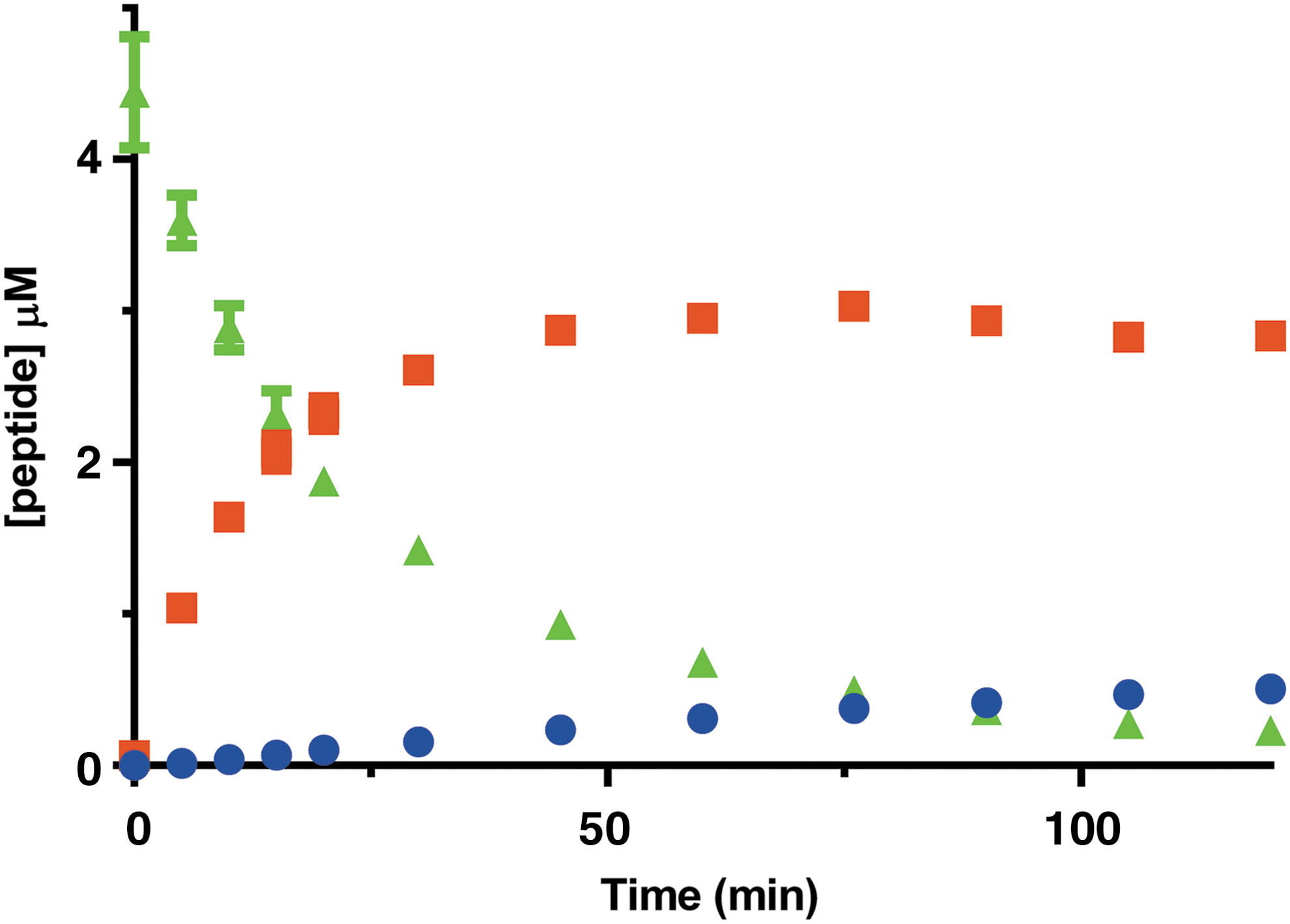
Simultaneous detection of peptide substrate and products. Quantitation of all three H4(1–21) methylated species as a function of time. H4(1–21)me0 is in green, H4(1–21)me1 is in orange, and unmethylated H4(1–21) peptide is in blue. Data points are averages of three replicates and error bars are the SD.
Conclusion
We have implemented a HT-MS/MS assay platform for evaluation of epigenetic enzyme targets. This technology is well suited for the study of PMTs where high-sensitivity and label-free methods are required. We find that this assay format in combination with an RFA provides us with sufficient tools to perform most of the enzymatic analyses required for drug discovery.
We utilize the radioactive format for routine structure–activity relationship needs because it is simple to run, has sufficient throughput, and meets our reproducibility needs. It is also easier to outsource, as many vendors have the capability to run radioactive assays, but fewer vendors are able to support HT-MS/MS protocols. We find the HT-MS/MS assay format to be preferable for screening of medium-sized libraries (<40,000). The HT-MS/MS throughput is similar to that of the RFA, but does not require a filtration step. While it is possible to automate the filtration step, there can be issues with some wells taking significantly longer to filter than others. We find working in a 384-well format to be easier using the HT-MS/MS system, and the ability to detect both the product and substrate in the same sample allows for ratiometric quantitation, which can significantly improve assay quality. Both assay formats allow for substrate agnostic evaluation of enzyme activity. We routinely run substrate comparison studies in the HT-MS/MS assay to avoid complications associated with differential binding to the filtration membrane.
Finally, the ability to look at differentially methylated peptide products simultaneously in the HT-MS/MS format provides a distinct advantage over the RFA. This approach allows a more detailed evaluation of the kinetics of methyl transfer by allowing visualization of the individual products. In addition, in cases where very high SAM concentrations are necessary, such as when working with tight-binding inhibitors or with site-directed mutants with poor SAM Km values, detection of the peptide product has distinct advantages. In these cases, dilution of the radiolabel can limit the sensitivity of the RFA, but has no consequence in the HT-MS/MS assay.
Footnotes
Acknowledgments
The authors would like to thank Drs. Lauren E. Frick, Peter T. Rye, and William A. LaMarr for their help in establishing the HT-MS/MS platform at Pfizer La Jolla. Their help with RapidFire assay development as well as running screening plates at Agilent was instrumental to success building our MS laboratory.
Disclosure Statement
No competing financial interests exist.