
Abstract
Kynurenine 3-monooxygenase (KMO), a pivotal enzyme in the kynurenine pathway, was identified as a potential therapeutic target for treating neurodegenerative and psychiatric disorders. In this article, we describe a surface plasmon resonance (SPR) assay that delivers both kinetics and the mechanism of binding (MoB) data, enabling a detailed characterization of KMO inhibitors for the enzyme in real time. SPR assay development included optimization of the protein construct and the buffer conditions. The stability and inhibitor binding activity of the immobilized KMO were significantly improved when the experiments were performed at 10°C using a buffer containing 0.05% n-dodecyl-β-d-maltoside (DDM) as the detergent. The K D values of the known KMO inhibitors (UPF648 and RO61-8048) from the SPR assay were in good accordance with the biochemical LC/MS/MS assay. Also, the SPR assay was able to differentiate the binding kinetics (k a and k d ) of the selected unknown KMO inhibitors. For example, the inhibitors that showed comparable IC50 values in the LC/MS/MS assay displayed differences in their residence time (τ = 1/k d ) in the SPR assay. To better define the MoB of the inhibitors to KMO, an SPR-based competition assay was developed, which demonstrated that both UPF648 and RO61-8048 bound to the substrate-binding site. These results demonstrate the potential of the SPR assay for characterizing the affinity, the kinetics, and the MoB profiles of the KMO inhibitors.
Introduction
The ability of drug binding to evoke a pharmacological response is dependent on how efficiently drug binding is coupled to physiology. Mechanism of binding (MoB) and binding kinetics, each plays a significant role in shaping clinical outcomes. The role of the target and importance of drug concentration (pharmacokinetics) at the site of action are well established; however, the roles of MoB and kinetics in shaping the pharmacological response and clinical outcomes are rarely understood. The concept of binding kinetics is often overlooked in the early phase of drug discovery; nevertheless, the incorporation of this parameter could help decrease the attrition rate in later stages of drug development. 1
Most traditional medicinal chemistry efforts use simple K i or IC50 measurements to drive structure activity relationships during lead optimization campaigns. Although this paradigm has been successful, a number of recent reviews have suggested the importance of determining the rate constants of association (k a), dissociation (k d), and residence time (RT) for compounds as relevant parameters for optimization. 2 MoB and kinetics clearly contribute to the clinical effectiveness and differentiation of many drugs. 3 Memantine provides a tolerable safety profile that could not be achieved with slower dissociation compounds. The relatively fast dissociation rate prevents the drug from accumulating in ion channels and interfering with subsequent normal synaptic transmission. 4 The rapid dissociation kinetics of memantine provides differentiation from a slow dissociation antagonist such as MK-801 and contributes to the improved safety and corresponding therapeutic usefulness of memantine. These data highlight the need for improved methods and technologies to enable cost-effective, convenient, and high-throughput access to kinetic data to characterize drug–target interactions.
MoB can affect how efficiently a binding interaction is coupled to the functional response. For instance, resistance to the ATP-competitive kinase inhibitors gefitinib and erlotinib, which target the epidermal growth factor receptor (EGFR) kinase, is caused by mutations that alter the ATP-binding site in such a way that the affinity of the EGFR kinase domain for ATP is increased. The functional consequence of these resistance mutations is, therefore, to enable ATP to compete more efficiently with gefitinib and erlotinib. 5 This fact provides an explanation for the mechanism of resistance to these rapidly acting ATP-competitive inhibitors as well as the way in which irreversible covalent binding inhibitors overcome this resistance. 5 MoB can also differentiate similar drugs with respect to their therapeutic indications. At the structural level, aspirin is an irreversible inhibitor of cyclooxygenases, whereas ibuprofen and naproxen are reversible inhibitors. All three molecules bind to cyclooxygenase enzymes at the same substrate-binding site. However, the irreversible MoB of aspirin differentiates its functional use as an antiplatelet drug from reversible inhibitors, because this MoB translates into a long-lasting action of aspirin in platelets, as platelets do not have the capacity to resynthesize new enzymes. 6
The tryptophan/kynurenine metabolic pathway produces several biologically active intermediates with diverse physiological actions. These metabolites are predicted to participate in the pathogenesis of various psychiatric and neurodegenerative disorders, 7 as well as have a well-defined role in immune regulation. 7 One important enzyme in this pathway is the NADPH-dependent flavoprotein hydroxylase, kynurenine 3-monooxygenase (KMO) (EC 1.14.13.9). This enzyme is believed to be localized in the outer mitochondrial membrane because of the presence of a proposed transmembrane domain near its C-terminus. KMO is predominantly expressed in the liver, kidney, macrophages/monocytes, and microglia cells in the brain. This enzyme catalyzes the conversion of kynurenine to 3-hydroxykynurenine (3-HK). The product 3-HK exhibits toxicity to cells through reactive oxygen species generation, the cross-linking of proteins, and mitochondrial respiratory chain inhibition. 8 Indeed, KMO has recently been implicated as a therapeutic target for several neurological disorders, where oxidative stress and mitochondrial dysfunction are associated with the disease pathology. 9,10
Previously described assays for KMO, such as the measurement of absorbance of NADPH, 11 a radiometric assay based on the release of tritiated [ 3 H] water, 12 and recently published hit discovery for KMO inhibitors by LC/MS/MS, 13 are not suitable for the kinetic characterization of compounds. In the past 10 years, several label-free biophysical binding assays that aim to identify small molecule interactions with target proteins have been developed. Among these binding assays, surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), analytical mass spectroscopy, and isothermal titration calorimetry appear to be of great interest for drug discovery. Enzymatic assays are used to calculate the enzyme's K m for a specific substrate, V max, and K i for inhibitors. However, the binding assays provide complementary data on the fundamental properties of the target protein–ligand interaction, which are important for confirming binding and kinetic data for novel chemical entities interacting with drug targets.
SPR-based biosensing is among the most informative technologies for this purpose, giving access to binding affinity, kinetics, and stoichiometry. Furthermore, SPR is particularly suited for the detection of low- to high-affinity interactions expected in compound screening because the formation of a compound–target complex is monitored in real time. It can be run at high throughput with low-target consumption. Moreover, it can detect promiscuous binders that inhibit enzymes by nonspecific aggregation-driven binding. 14
In this study, we used a Biacore T200 instrument (GE Healthcare, Biacore) to characterize KMO inhibitors. In all cases, the sensor surface was prepared by capturing chemically biotinylated KMO on a NeutrAvidin-coated sensorchip. The objectives were to investigate the suitability of SPR for the evaluation of the physical interactions of inhibitors with the KMO enzyme, establish optimized procedures that provide reliable quantitative data, and determine the binding mechanism and kinetics of these KMO inhibitors.
Materials and Methods
Materials
N-terminal GST-KMO was expressed in an insect cell expression system. A modified version of the pFastBac series was used for the production of the recombinant virus (Proteros). The enzyme was purified by a three-step chromatographic procedure comprising affinity chromatography and size exclusion chromatography steps. A full-length KMO without GST expressed in HEK293s cells was purchased from Origene. The purity of the enzymes (>90%) was assessed by SDS-PAGE gel electrophoresis and the protein concentration was quantified by Bradford's method. The catalytic activity of the enzymes was confirmed by a biochemical assay and the Kynurenine K m was 15 ± 3.2 μM, which is in agreement with the literature data. 15
The following reagents were purchased from commercial vendors: Tris-HCl (Sigma T5941), KCl (Sigma P9541), EDTA-Invitrogen (Gibco-15575-038), NADPH (Enzo ALX-480-004-M050), glucose-6-phosphate (Sigma G7250), glucose-6-phosphate dehydrogenase (Sigma G4134), L-kynurenine (Sigma K8625), trichloroacetic acid (Sigma T0699), formic acid, acetic acid, 3-HK, and KYN were all purchased from Sigma Aldrich. 13 C6-3-HK and 13 C6-KYN were purchased from AlsaChim France. High-purity acetonitrile (B&J) was purchased from Fisher Scientific and water was purified in house using a MilliQ system (Millipore) equipped with a biofilter. NeutraAvidin, Sulfo NHS-LC-LC-Biotin, and 3 × CM5 sensorchips were purchased from Thermo Scientific and GE. The following buffer composition was used in the study. Biotinylation buffer: 10 mM HEPES pH 7.5, 150 mM NaCl, and 2 mM DTT, 0.05% n-dodecyl-β-D-maltoside (DDM). Immobilization buffer: 20 mM Tris pH 7.8, 150 mM NaCl, 2 mM DTT, and 0.05% DDM. Running buffer: Two buffer conditions were studied. They were as follows: Buffer A: 20 mM Tris pH 7.8, 150 mM NaCl, 2 mM DTT, 0.05% DDM, and 2% DMSO. Buffer B: 20 mM Tris pH 7.8, 150 mM NaCl, 2 mM DTT, 5% glycerol, 0.05% P20, and 2% DMSO.
LC/MS/MS Assay
Biochemical assay
Compounds were diluted in Tris-HCl buffer, pH 8.1 (100 mM Tris-HCl, 10 mM KCl, and 1 mM EDTA). To 10 μL of the compound dilutions, 25 μL of the reaction mix (1 mM NADPH, 3 mM glucose-6-phosphate, 500 U/mL glucose-6-phosphate dehydrogenase, 1 mM L-kynurenine in Tris-HCl buffer) was added. 9 Fifteen microliters of 0.1 mg/mL of KMO enzyme was added last to the reaction. Plates were incubated at room temperature for 2 h. The reaction was stopped by adding 10 μL of 30% trichloroacetic acid. The samples were stored at −20°C until analyzed by LC/MS/MS.
Sample preparation
Screening sample (10 μL) preparation involved dilution by fivefold in 250 μL polypropylene BASi inserts using a 0.2% acetic acid aqueous solution containing 100 ng/mL of internal standards (carbon 13 analogues of 3-HK and KYN). These samples were organized in deep 96-well plates and sealed with Easy Peel heat sealing foil from Thermo Scientific before injection onto the LC/MS/MS system.
LC/MS/MS method
A Waters ACQUITY UPLC system equipped with a YMC ODS AQ 2 × 100 mm, 3 μm particle column provided the separation of the KP analytes before detection by a Waters Quattro Premier XE triple quadrupole mass spectrometer operating in the MS/MS mode. Column and precolumn tubing were maintained at 40°C, while eluting KP metabolites with a mobile phase consisting of an aqueous component A (0.5% formic acid in milliQ water) and an organic component B (1% formic acid in acetonitrile). Gradient elution included a 0.5-min hold at 100% A followed by a gradient of 0%–90% B for 2.2 min. The column was washed using 100% B for 0.5 min before a 0.5-min re-equilibration with 100% A for a total run time of 3.9 min. The UPLC was equipped with a 5-μL loop, which was utilized in the full-loop injection mode with a 4 × overfill factor for a total sample consumption of 20 μL. The operating conditions of the triple quadrupole mass spectrometer were as follows: capillary voltage 0.7 kV, cone voltage 15 V, source temperature 150°C, desolvation temperature 500°C, cone gas flow 50 L/h, and desolvation gas flow 1,000 L/h. For each analyte (presented in the order of elution), the following transitions were monitored using the collision energy (CE) indicated: (1) 3-HK 225.12-109.8, CE 18V, (2) 13 C6-3-HK 230.0/115.80, CE 20V (3) KYN 209.05/93.7, CE 15V, and (4) 13 C6-KYN 215.12/99.95, CE 15V. The resulting LC/MS/MS chromatograms exhibited detection limits of 0.1 ng/mL (3-HK) and 0.02 ng/mL (KYN) based on a signal-to-noise ratio of three. These detection limits were more than sufficient to analyze the analytes of interest in the enzyme screen samples.
SPR Assay
Chemical biotinylation of KMO proteins and capturing of biotinylated proteins on the NeutrAvidin-coated CM5-chip
KMO proteins were first exchanged for a biotinylation buffer using a ZEBA spin column and then biotinylated by the incubation of 4 μM of KMO with 1.2 × molar excess of sulfo-NHS-LC-LC-biotin (from Thermo scientific) at 4°C for ∼18 h. The biotinylated KMO was separated from unreacted biotin by size-exclusion column (ZEBA 7K column). For every chemical biotinylation, 0.2 mg of KMO was consumed.
Biotinylated KMO was injected onto a NeutraAvidin-coated CM5 sensorchip surface at a concentration of 4 μM in an immobilization buffer at 10°C. The NeutraAvidin coated sensorchip was prepared as described previously. 14 Typical immobilization levels of GST-KMO were 2,800 RU during the buffer and protein construct selection study and 4,000 ± 1,000 RU during the titration of compounds. Unless stated otherwise, all experiments were performed at 10°C.
Affinity characterization of inhibitors by an SPR assay
The compounds identified as inhibitors of KMO by the LC/MS/MS assay were titrated into the immobilized KMO from the lowest to highest concentration in a series of two or threefold dilutions up to six concentrations. The top concentration of the titration was 5–10-fold excess over its corresponding IC50. UPF648 was used as a positive control to normalize the response to decrease in the ligand-binding capacity of the immobilized KMO during each run. Samples were injected for 60 s at 30 μL/min and their dissociation was monitored for 120 s. The data generated by Biacore T200 were analyzed using Biacore Evaluation software v2.0. The sensorgram from the flow cells in which KMO was immobilized was first referenced with the sensorgram obtained in the presence of biotin-blocked NeutrAvidin. The bulk contribution from the DMSO mismatch in the concentrations between the injected sample and the running buffer was corrected using a DMSO calibration curve. The resulting sensorgram was double referenced by the subtraction of the nearest blank buffer responses (the buffer injections acquired before the injection of ligands). The equilibrium binding responses (R eq) at 5 s before the end of the injection were extracted from the sensorgrams obtained at different concentrations. These responses were plotted against their respective concentrations and fitted to the Langmuir isotherm equation to determine both the K D and R max. 15 For compounds that displayed slow interactions with kinetic constants (k a ≤ 106 M−1s−1 and k d < 0.3 s−1), the affinity and kinetic rate constants were determined by global fitting the sensorgrams at different concentrations using a 1:1 kinetic interaction model (with or without a mass-transport term as appropriate) in Biacore evaluation software v2.0 to determine the R max, k a, k d, and K D values. 16
Based on the R
im of KMO, the R
max (theoretical maximum R
eq) for each compound was calculated using Eq. 1.
where R im is the amount of KMO immobilized, expressed in RU, and MW is the molecular weight.
SPR competition assay
Samples were injected in the order shown in Supplementary Table S1 (Supplementary Data are available online at
In the first injection, UPF648 (100 nM) was injected to saturate the substrate-binding site with an association time of 80 s. In the second injection, a mixture of UPF648 (100 nM) and a test compound was injected for 60 s. All the sensorgrams were subtracted with that of the buffer injections. The binding response was then extracted from the buffer-subtracted sensorgrams at 5 s before compound dissociation. This entire cycle was repeated twice for each compound. To monitor the integrity of immobilized KMO on the sensorchip, the affinity and binding saturation of UPF648 and RO61-8048 were determined by titration after every 96 sample injections. All experiments were performed at a flow rate of 30 μL/min with a temperature of 10°C
RT calculation
The RT (τ) of a compound for which the k d (s−1) value was available from SPR, was determined by the reciprocal of the dissociation rate (τ = 1/k d). 17
Results and Discussion
SPR Feasibility Study Using KMO With and Without GST Tag
To identify an optimal construct of protein for the analysis of small molecule binding by SPR, the ligand-binding ability of two KMO proteins with and without GST tag and various assay conditions that could improve the activity and stability of KMO in the SPR assay were investigated. KMO proteins that displayed the binding affinity of tool compounds by SPR, closely correlated to their affinity obtained by LC/MS/MS, were chosen to evaluate the assay further.
The proteins were immobilized by capturing chemically biotinylated KMO proteins on a NeutrAvidin-coated sensorchip. Because the initial observations showed poor ligand-binding activity and the stability of biotinylated KMO in SPR at 20°C, further SPR experiments were performed at 10°C on a Biacore T200 instrument. To determine the ligand-binding activity and stability of two KMO proteins constructs, two well-characterized tool compounds, RO61-8048 18 and UPF648 19 (Fig. 1), were repeatedly titrated into the immobilized KMO. The full-length KMO proteins displayed the ligand activity (Fig. 2A) and the affinity correlated with their reported IC50 only when the buffers for the chemical coupling of biotin and the SPR experiment contained a detergent DDM, a nonionic detergent often used to solubilize and stabilize membrane proteins. 20

Structures of UPF648 and RO61-8048.
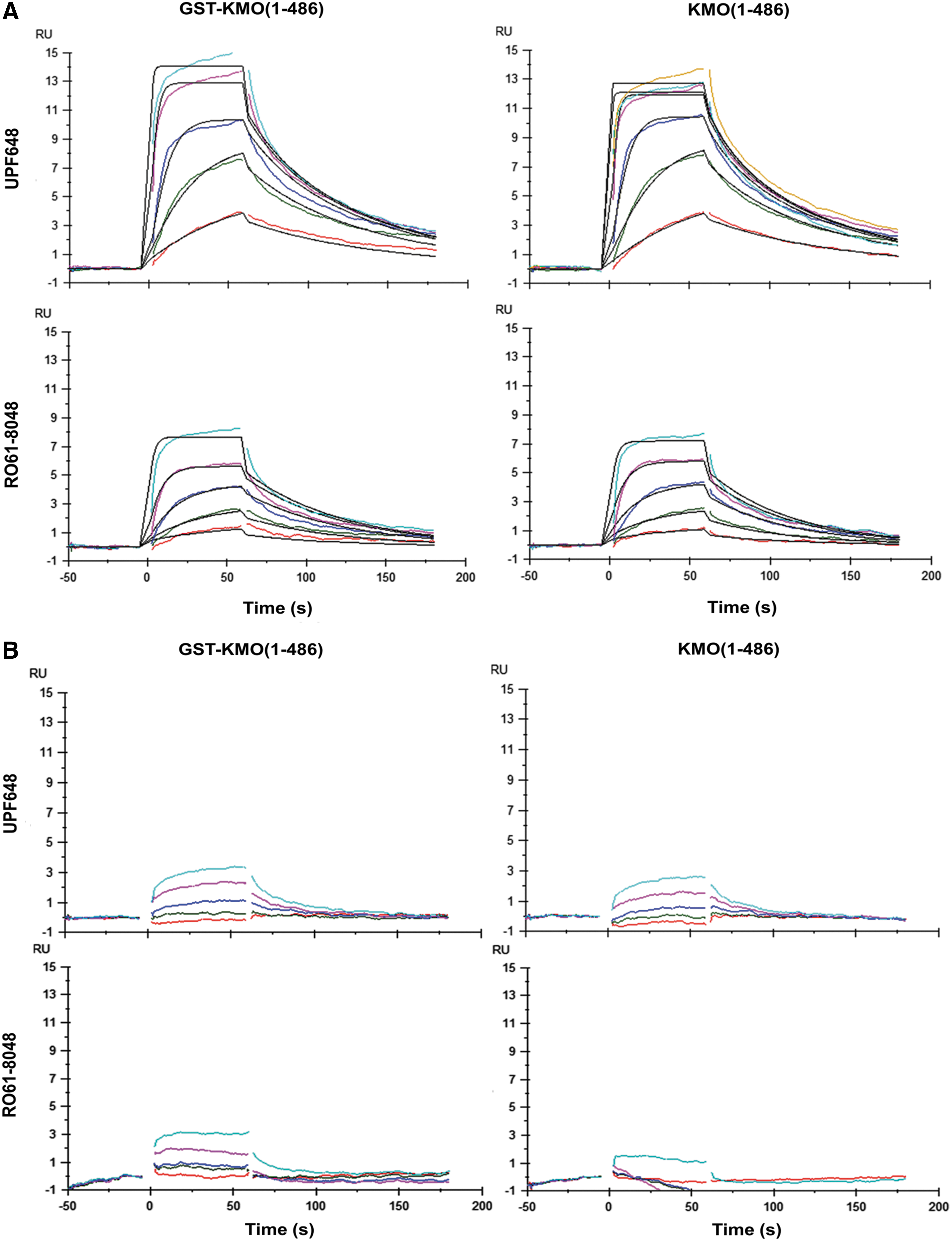
The sensorgrams of RO61-8048 and UPF648 in Buffer A
The characteristics of the two compounds binding to the full-length KMO in the two buffer conditions (A & B) are summarized in Table 1A. In the biotinylation buffer, the presence of DDM significantly improved the initial activity of KMO to >90% compared with 18% in the absence of DDM (Fig. 2B). It was observed that the binding affinity of the tool compounds for the two KMO proteins was better correlated to their reported IC50 in buffer A (+0.05% DDM) compared with buffer B (+5% glycerol and 0.05% P20) (Table 1B). Furthermore, the sensorgrams of the tool compounds were of better quality in buffer A compared with buffer B (not shown). The data obtained for the tool compounds in buffer B exhibited larger variations in the observed binding affinities. In addition, the initial activity of KMO in buffer B was less than that in buffer A. Based on these observations, buffer A was selected for assaying ligand binding to KMO. It is perhaps not so surprising that the ligand-binding activity and the stability of KMO were better in the presence of DDM since the protein bears a membrane-anchoring region at the C-terminus. The use of appropriate detergents and their concentrations are typically crucial for the correct membrane protein conformations with the highest activity and stability. 20 Our earlier investigation indicated that unlike yeast KMO, 19 enzymatic as well as ligand-binding activities were lacking in the C-terminus truncated versions of human KMO when produced in E. coli. Both observations, therefore, suggest an important role of the transmembrane region in keeping the ligand-binding activity of human KMO.
Binding Characteristics of the Tool Compounds for KMO (1-486) and GST-KMO (1-486) in Two Buffer Conditions
The affinity (K D) and the observed R max (Ob, R max) were determined by a global fit of the sensorgrams at different concentrations to 1:1 kinetic interaction model or to Langmuir isotherm equation using BiaEvaluation software. 15
KMO, kynurenine 3-monooxygenase.
Affinity, R max, and Kinetic Parameters of the Tool Compounds in Buffer A
The k a (association rate constant) and k d (dissociation rate constant) affinity (K D) were determined by a global fit of the sensorgrams at different concentrations to 1:1 kinetic interaction model using BiaEvaluation software, v.1. SPR kinetic and K D values are average of n = 3 with standard error of mean.
The average chi-square deviation of the mathematical model. It indicates the closeness of the fit.
RT is determined by the reciprocal of the dissociation rate ( = 1/k d).
LC/MS/MS IC50 values are average n = 10 with standard error mean SEM.
RT, residence time; SPR, surface plasmon resonance.
The initial activity of GST-KMO (1-486) and KMO (1-486) was similar (Table 1A) based upon the maximum level of tool compound binding. The GST-KMO (1-486) construct showed good stability over a period of 12 h in the optimized buffer A. An average decline of 32% in the GST-KMO activity was observed over 12 h (Supplementary Fig. S1). A similar stability was also seen for KMO (1-486) under identical conditions (data not shown). During these 12 h, UPF648 exhibited an average K D of 6 ± 0.6 nM, whereas RO61-8048 displayed an average K D of 17 ± 5 nM for GST-KMO in the buffer A. A good reproducibility in the K D determination of these two tool compounds for the immobilized GST-KMO at six time points was accomplished (Table 1B). Subsequently, GST-KMO (1-486) was selected as the preferred construct over the KMO (1-486) for the SPR assay, since the GST-tagged protein can be produced in larger quantities in insect cells and a higher level of GST-KMO could be captured on a sensorchip, providing a better detection window.
Based upon the findings described in this study, GST-KMO (1-486) was the best construct for an SPR assay to characterize small-molecule binding to KMO. The initial activity and stability of this protein were significantly improved when the biotinylation of protein and the SPR assay were performed in the presence of 0.05% DDM at a lower temperature. The K D values of tool compounds (RO61-8048 and UPF648) in the SPR assay were comparable to the IC50 values generated by the LC/MS/MS method. Furthermore, the SPR assay provided information on the binding kinetics of the tool compounds (Table 1B). To assess the reproducibility of the SPR assay, the titration of eight compounds with affinities ranging from 10 nM to 2 μM and of a nonbinding compound was repeated. These eight small-molecule compounds were initially shown to inhibit the enzymatic activity of hKMO in the LC/MS/MS/assay. The two affinity values from the two titrations correlated closely, which are clearly shown in the correlation plot (Spearman r = 0.94) (Fig. 3), while a nonbinding compound appeared as a nonbinder in the repeated titration. These results indicate that the reproducibility of the assay is good.
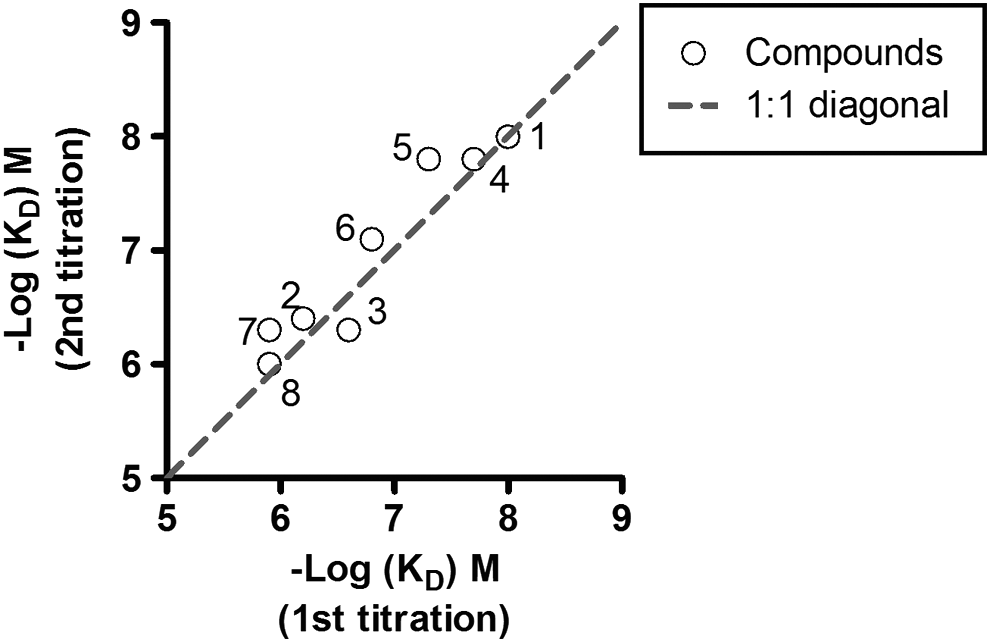
Correlation of two K D values determined from two independent titrations of eight KMO inhibitors.
Kinetic Characterization of Selective Compounds in the SPR Assay
Direct binding SPR data on affinity, kinetics, and RT have been published for a number of different drug-target systems. Drug-target RT is an emerging concept in the drug research community. Its definition, coined by Copeland in 2006, stands for an experimental measure of the lifetime of a drug–target complex, reflected by its dissociation rate (k d). 2 This is an important pharmacological feature, as a drug is only effective when bound to, and influencing, the activity of its physiological target. 2,21 Recently, several notable reviews 21 –24 have emphasized the pivotal role of RT in drug discovery. These discussions have led to an increasing recognition that drug-target RT may even be of greater importance for a drug's effect in the patient than its affinity, since the body, unlike an in vitro setting, is an open system where the concentration of free drugs fluctuates over time. 21 In this sense, optimizing drug candidates based on equilibrium-derived parameters alone, such as affinity (k d) or potency (IC50), may not be ideal. 24 Drug-target RT may thus be a useful additional parameter, as it is thought to represent a surrogate marker of drug clinical efficacy: the longer the drug occupies the receptor, the more profound the drug may exert its effect. 25 Although this reasoning most certainly is too simplistic, there is substantial evidence, as witnessed in a survey of 50 drugs on 12 different drug targets, that about 70% of long RT therapeutics display higher efficacy compared with faster dissociating drugs. 26
To assess if the SPR assay we developed can be used to differentiate the binding kinetics of selected KMO inhibitors, KMO inhibitors were titrated into GST-KMO and their binding kinetics and RTs were determined. As shown in Figure 4, although a small set of compounds identified through the LC/MS/MS HTS screen showed comparable IC50 values, some displayed a long RT compared with others, suggesting a slower dissociation constant. For example, the IC50 of compound A and B are 98 and 80 nM, respectively, but compound A has 2.5 times longer RT time than compound B. It needs to be pointed out that the criteria for long or short RT may vary for different targets and for divergent clinical indications. 27 For therapies requiring a prolonged target occupancy, a long RT drug may offer advantages, as it remains bound to the target and continuously exerts its pharmacological effect even when most of the free drug has already been eliminated from circulation. 28 Importantly, this simultaneously implies that a slowly dissociating drug will only offer a noticeably increased effect over a faster dissociating drug when its dissociation half-life (t 1/2) outlasts its pharmacokinetic half-life or when there are large fluctuations in the endogenous competitor (e.g., hormone or neurotransmitter) concentrations. 29 Further proof for the importance of RT comes from numerous examples from best-in-class drugs determined to exert slow dissociation from their target receptors. One well-known example is the Muscarinic M3 receptor antagonist Tiotropium, which is a long-acting (24-h) anticholinergic bronchodilator used in the management of chronic obstructive pulmonary disease. 30 On the contrary, there are cases where the mechanism-based side effects or toxicity outweigh the therapeutic advantages of long receptor occupancy. In this situation, a fast dissociating compound displaying short-lived intervention is preferred. This may be the case for antipsychotic D2 dopamine receptor antagonists, where a long RT is not desired because of the on-target-related extrapyramidal motor effects. In both cases, a quantitative assessment of this parameter is an important part of the overall evaluation of target–ligand interactions in drug discovery and pharmacology. Thus, the SPR assay was developed as an extremely efficient and information-rich kinetic assay to better understand the interaction of compounds acting as inhibitors of the KMO enzyme. In addition, the SPR assay was successfully used as an orthogonal assay to weed out false positives from the hits obtained by the LC/MS/MS primary screen.

The plot shows the difference in residence time of selected KMO inhibitors. The X-axis represents LC/MS/MS IC50 values and Y-axis represents residence time.
MoB Determined by Competition Assay on Biacore T200
To gain insights into the MoB of KMO ligands/inhibitors, a competition binding study was performed. 31 SPR was used to investigate whether the binding competition between UPF648 and L-kynurenine and between UPF648 and RO61-8048 could be observed. The affinity of Kynurenine for GST-KMO under this assay condition was ∼21 μM, which was similar to its K M of 15 μM in the LC/MS/MS assay.
The SPR sensorgrams from the competition study between L-kynurenine (L-KYN) and UPF648 are given in Figure 5. The study was performed by the sequential detection of ligands bound to immobilized GST-KMO in the following order: L-KYN (Fig. 5A), the competitive inhibitor UPF648 at a saturating concentration (Fig. 5B), and the mixture of L-KYN and UPF648 (Fig. 5C). If the binding sites of two ligands were different (i.e., noncompetitive binding), the level of binding response from the mixture of two ligands (Fig. 5C) would have been the sum of the responses obtained from the individual ligands (Fig. 5A, B). If the two ligands compete for the same binding site, the resulting response from the mixture of two ligands (Fig. 5C) would be similar to that of UPF648 (Fig. 5B). The observed response from the mixture of L-KYN and UPF648 was not equal to the summed responses from the respective ligands (Fig. 5D), indicative of UPF648 and L-KYN competing for the same binding site. This observation is in accordance with the X-ray structure of yeast KMO complexed with UPF648, which shows that UPF648 and kynurenine share a binding site in the KMO catalytic pocket with kynurenine. 18 The competitive inhibition exhibited by UPF648 was further confirmed in enzymatic Michaelis–Menten plots (Fig. 6), where no significant change in V max, but a shift in K m could be observed at different concentrations of the inhibitor.
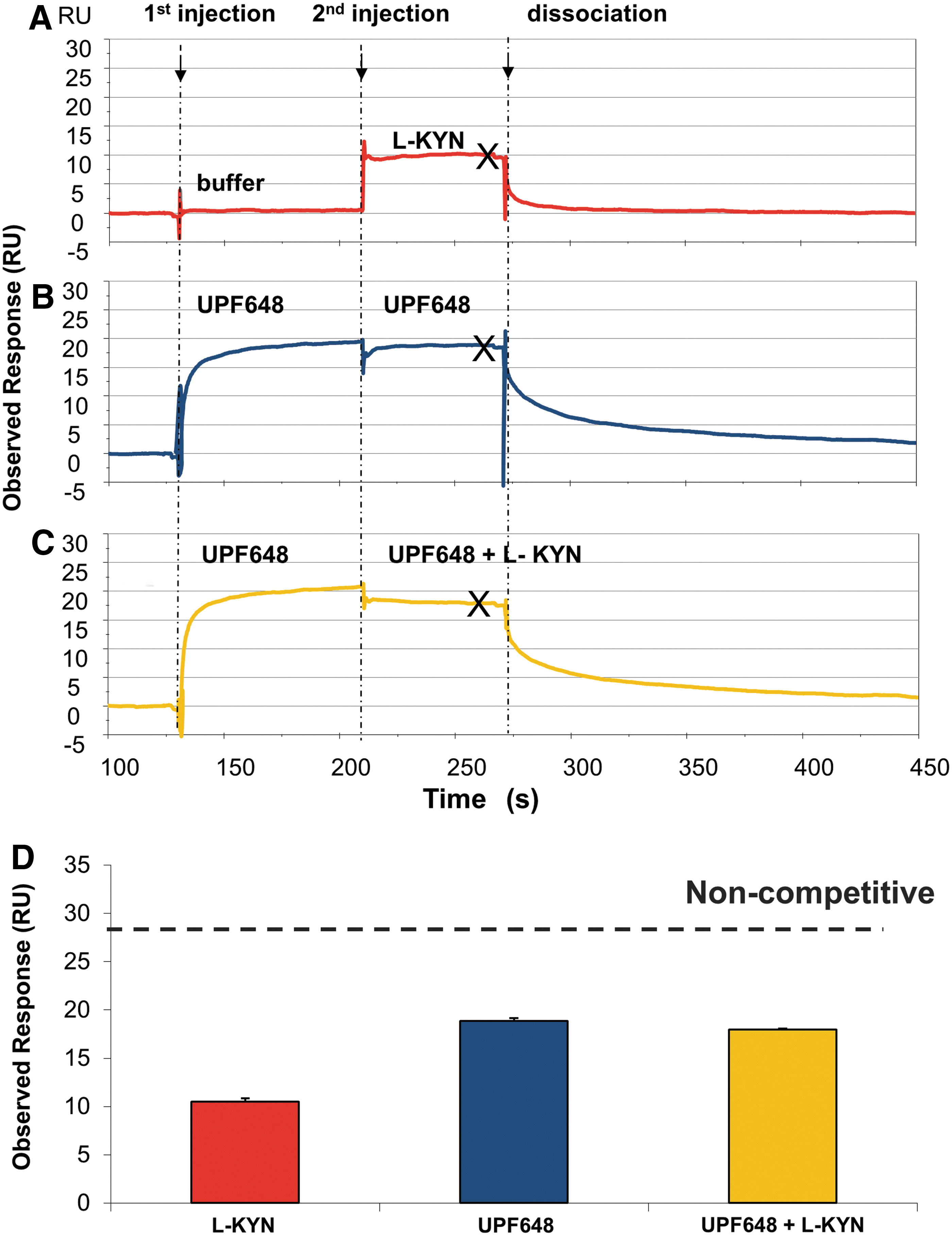
SPR sensorgrams in the presence of KMO from the injection of
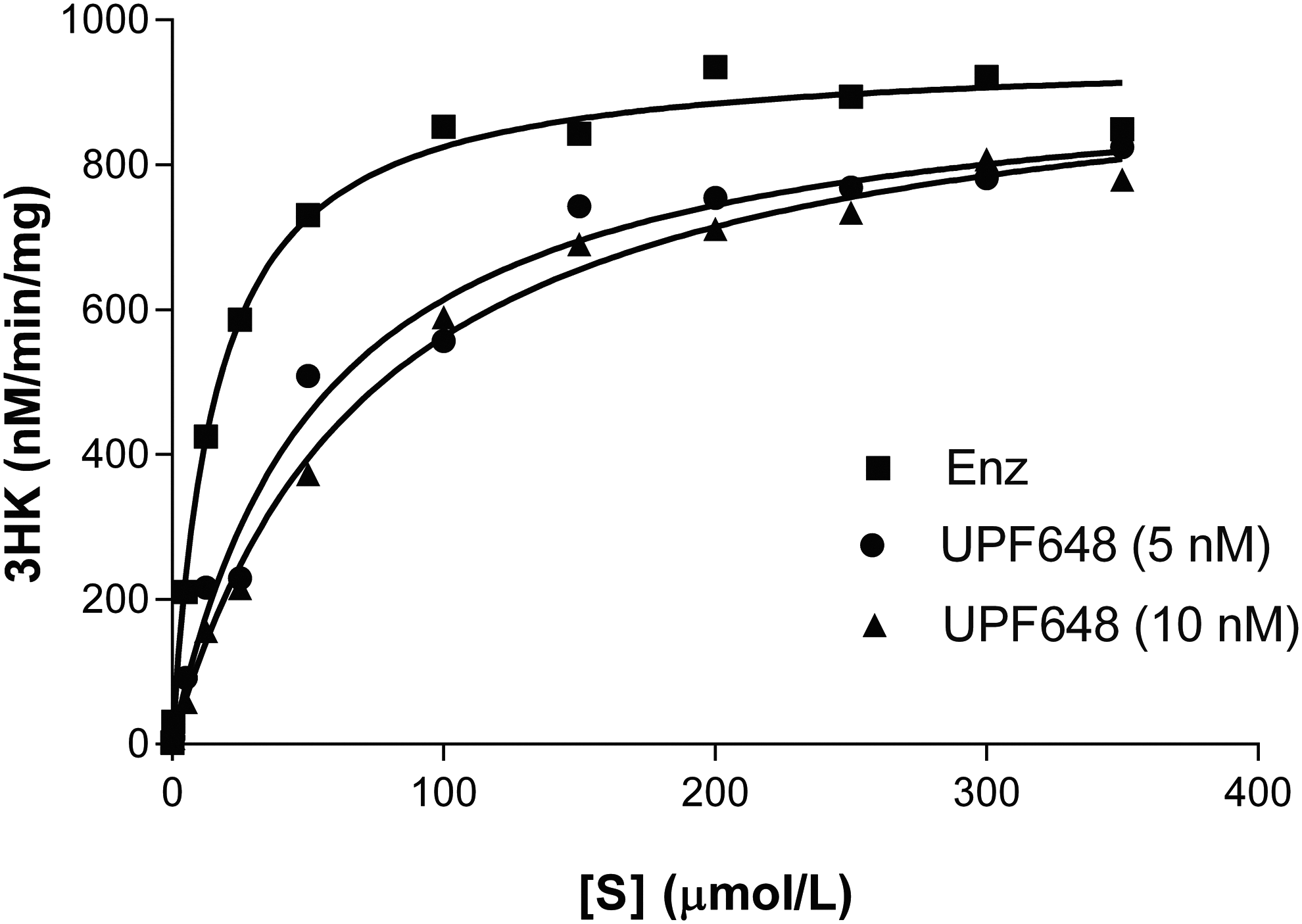
LC/MS/MS competition binding assay with KMO protein in the absence or presence of UPF648. No change in V max and right shift in K m in presence of UPF648 indicate a competitive binding of UPF648 to KMO substrate-binding site.
By using the same experimental setup above, the binding of RO61-8048 to GST-KMO was studied to validate the assay in the presence and absence of UPF648. The data suggest that RO61-8048 is able to compete with UPF648 for the substrate binding site (Fig. 7). Hence, the data presented in this paper establish that an SPR assay can be used to study the competition between two ligands binding to KMO.
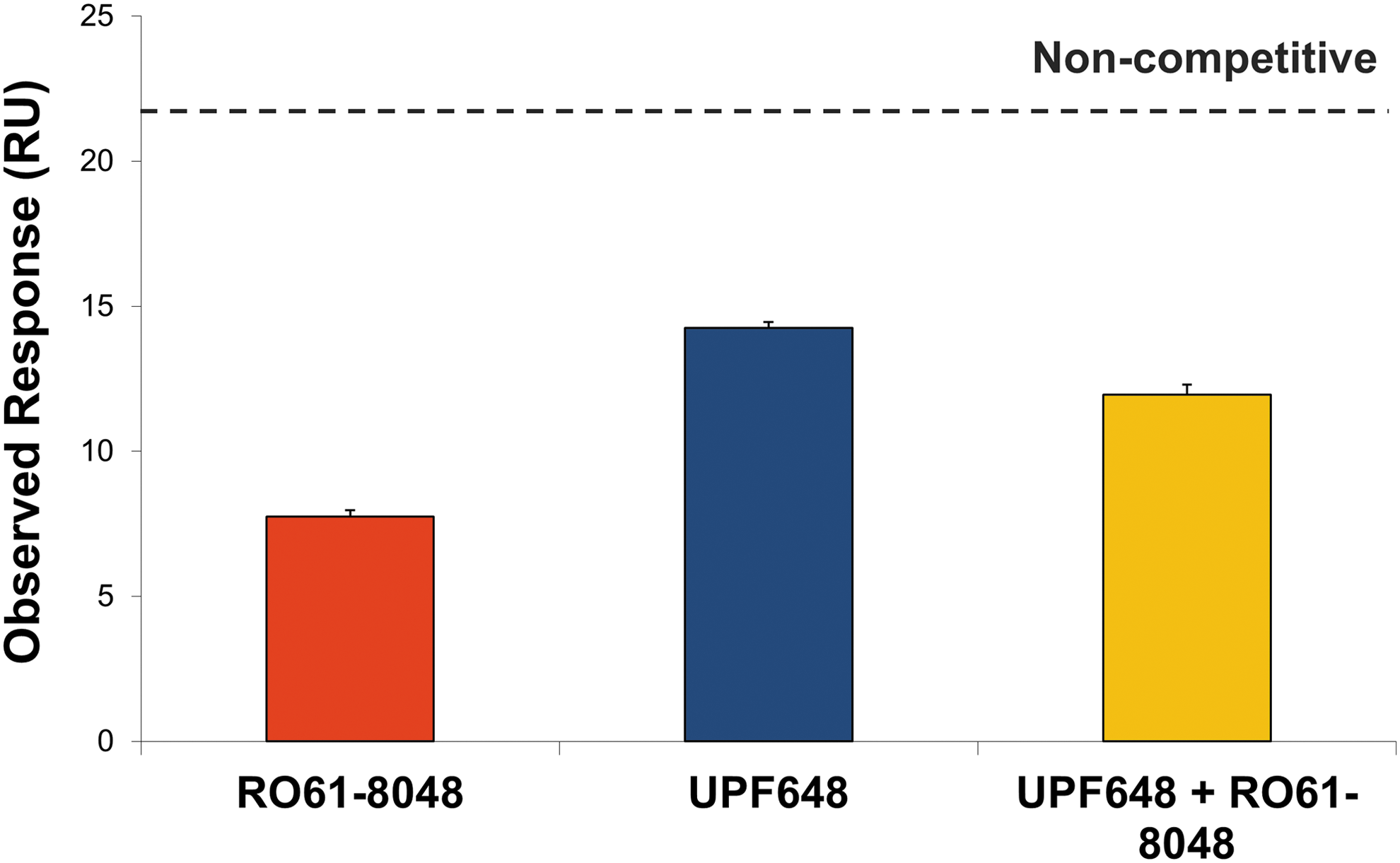
The summary of the observed responses of RO61-8048 and UPF648 and a mixture of test compound and UPF648. The expected response from noncompetitive binding of compounds and UPF648 to GST-KMO is indicated in gray. The error bar is the standard deviation from the two replicated experiments. The result indicates that RO61-8048 competes with UPF648-binding site.
Conclusions
The current results demonstrate the utility of an SPR assay to characterize KMO inhibitor binding modes and kinetic profiles. The detailed kinetic information generated using this assay will serve as a means to generate more appropriate structure–activity relationships, which could lead to the rational design of potential new drugs. Establishing binding kinetics and mechanism preclinically may help shape the clinical study design and outcomes that are important to patients and clinicians in the areas of efficacy, safety, and therapeutic differentiation.
Footnotes
Acknowledgments
The authors would like to thank Anders Damholt and Gamini Chandrasena for critical reading of the article.
Disclosure Statement
No competing financial interests exist.