
Abstract
Astrocyte phenotypes change in a process called reactive gliosis after traumatic central nervous system (CNS) injury. Astrogliosis is characterized by expansion of the glial fibrillary acidic protein (GFAP) cytoskeleton, adoption of stellate morphologies, and differential expression of some extracellular matrix molecules. The astrocytic response immediately after injury is beneficial, but in the chronic injury phase, reactive astrocytes produce inhibitory factors (i.e., chondroitin sulfate proteoglycans [CSPGs]) that limit the regrowth of injured axons. There are no drugs that promote axon regeneration or functional recovery after CNS trauma in humans. To develop novel therapeutics for the injured CNS, we screened various libraries in a phenotypic assay to identify compounds that promote neurite outgrowth. However, the effects these compounds have on astrocytes are unknown. Specifically, we were interested in whether compounds could alter astrocytes in a manner that mimics the glial reaction to injury. To test this hypothesis, we developed cell-based phenotypic bioassays to measure changes in (1) GFAP morphology/localization and (2) CSPG expression/immunoreactivity from primary astrocyte cultures. These assays were optimized for six-point dose–response experiments in 96-well plates. The GFAP morphology assay is suitable for counter-screening with a Z-factor of 0.44±0.03 (mean±standard error of the mean; N=3 biological replicates). The CSPG assay is reproducible and informative, but does not satisfy common metrics for a “screenable” assay. As proof of principle, we tested a small set of hit compounds from our neurite outgrowth bioassay and identified one that can enhance axon growth without exacerbating the deleterious characteristics of reactive gliosis.
Introduction
Traumatic central nervous system (CNS) injury can lead to chronic disability, in part, because severed axons fail to regrow and reestablish appropriate functional connections. 1 Many factors underlie the failure of CNS repair and there are no drugs that improve functional recovery. 2 –4 In addition to neuron intrinsic factors, extrinsic factors from nonneuronal cells, including reactive astrocytes in the glial scar, also contribute to the limited capacity for axons to regrow. 5,6
Astrocytes undergo numerous changes when they react to CNS injuries. 7 –9 Glial scar astrocytes immediately adjacent to the lesion can extend long processes, and astrocytes in the lesion penumbra may also undergo hypertrophy and expansion of individual domains. 10,11 These morphological changes are accompanied by alterations in the glial fibrillary acidic protein (GFAP) cytoskeleton that give astrocytes a more stellate appearance. 12,13 Importantly, this phenotype has been used to discern reactive from quiescent astrocytes in vivo. 14 Another hallmark of reactive gliosis is the increased expression of some chondroitin sulfate proteoglycans (CSPGs). 15,16 These matrix and membrane-associated molecules play important roles in modulating inflammation as well as axon growth and guidance. 17 –19 After injury, reactive changes are required for wound healing, but negatively influence axon regeneration in the chronically injured CNS. 20,21 Accordingly, there is a need to find agents that promote neurite outgrowth without contributing to, or, exacerbating astrogliosis.
Aspects of reactive astrocytes can be modeled in vitro. 22 –24 When cultured astrocytes are subjected to physical trauma or stimulated with certain cytokines, growth factors, or ATP analogs, their volume increases as does the thickness and length of their GFAP+ processes. 25 –27 In models of the glial scar, cultured astrocytes' GFAP cytoskeletons adopt exaggerated stellate phenotypes similar to those observed in vivo after injury. 28 Like astrocytes in the injured CNS, cultured astrocytes can also be stimulated to increase their expression of axon/neurite growth inhibitory CSPGs. 28,29 Thus, stellation and CSPG expression could be exploited for screening assays.
High-content analysis (HCA) is a technology that might allow for the development of screens based on changes in astrocytes' GFAP and CSPG expression in vitro. This automated image-based approach reports about cell morphology, protein localization, and relative expression. Thus, HCA can be a powerful tool for determining how perturbagens affect cellular phenotypes. 30 Phenotypic screening has proven surprisingly successful in identification of first-in-class drugs approved by the Food and Drug Administration (FDA). 31 Although HCA can be used to assess injury-related phenotypic changes in astrocytes, few studies have done so using primary cells. 32,33
We have used HCA for in vitro studies of primary neurons that led to improved axon regeneration in vivo. 34 –38 Recently, we tested over 1,600 small-molecule kinase inhibitors (KIs) in a cell-based phenotypic screen and found many neurite growth-promoting KIs. 39,40 While these studies identified compounds that increase intrinsic axon growth programs, their effects on astrocytes are unknown. This is important because injury-induced changes in astrocytes render them less supportive of growing axons. 41 Consequently, compounds that not only promote neurite outgrowth but also induce or exacerbate gliosis may have reduced therapeutic potential.
To determine if compounds elicit changes in astrocytes in vitro that resemble their injury-induced phenotype in vivo, we developed two HCA assays. To investigate reactive-like changes in the GFAP cytoskeleton in low-density cultures, we devised a metric called the “stellation index” (ratio of the total GFAP+ process area/cell body area). In the second assay, we utilized HCA to monitor the expression of glial scar-associated CSPG epitopes in astrocyte monolayers. In these assays, reactive-like astrocytes should have an increased stellation index and/or increased CSPG expression relative to controls. As an initial proof of principle, we used these assays to identify neurite growth-promoting compounds that do not induce reactive-like astrocyte phenotypes.
Materials and Methods
All procedures involving animals were approved by and carried out in accordance with the University of Miami Miller School of Medicine Institutional Animal Care and Use Committee. Details of the spinal cord injury (SCI) experiments have been described in a metadata document, in compliance with the MIASCI reporting standard (see Supplementary Materials; Supplementary Data are available online at
Assay Protocol Table
1. GLAST-selected cells are plated on PDL coated 96-well Falcon dishes.
2. Cells are cultured at 37°C in 5% CO2.
3. Cells are washed twice with ice cold HBSS with 20 mM HEPES at room temperature.
4. After washing, 150 μL chemically defined media (DMEM with SM1 supplement) added to cells with multichannel pipette.
5. Perturbagens added at 4X. For kinase inhibitors the final concentration of DMSO is 0.2%.
6. Cells are cultured at 37°C in 5% CO2 incubator.
7. Culture media is removed and replaced with 4% PFA. After 15 minutes in PFA cells are washed with PBS and then incubated in 0.5% Triton X-100 for 10 minutes.
8. Blocking buffer contains 5% Goat serum, 1% BSA and 0.03% Triton X-100 in PBS.
9. Primary antibody incubation is performed overnight at 4°C. After primary staining, plates are washed and incubated in secondary antibodies for 1 hour at room temperature.
10. Cells are identified by their Hoechst labeled nuclei and traced based on fluorescent signals signal from 488 nm and 647 nm channels using ArrayScan VTI and Cellomics software.
Astrocyte Isolation
Enriched cultures of primary astrocytes were generated from postnatal day 6 (P6) rat cortices using the Anti-GLAST (ACSA-1) MicroBead Kit according to the manufacturer's protocol (130-095-826; Miltenyi Biotec).
43
GLAST-positive cells were eluted into Dulbecco's Modified Eagle's Medium (DMEM, 11995-065; Gibco®) containing 10% fetal bovine serum (FBS, S11050; Atlanta Biologics or 26140-079; Gibco), sodium pyruvate (1:100 dilution, 11360-070; Gibco), GlutaMAX™ (1:100 dilution, 35050-061; Gibco), and penicillin–streptomycin (1:100 dilution, 15140-122; Gibco). Live cells were counted using an automated cell counter (TC20™; Bio-Rad) by trypan blue exclusion. For low-density cultures, 1,500 live cells per well were seeded into 96-well plates (353072; Falcon) coated with poly-
KIs and Transforming Growth Factor-Beta
Individual KIs used in this article were sourced from EMD Millipore's InhibitorSelect™ Protein Kinase Inhibitor libraries (ML-7, Hydrochloride [ML-7], and ROCK Inhibitor, Y-27632) and the PKIS-I library from GlaxoSmithKline (GSK-2 and GSK-5, anonymized due to intellectual property considerations). Before screening, 10 mM KI stock solutions were prepared in dimethyl sulfoxide (DMSO, D2650; Sigma). KIs tested in astrocyte cultures were selected based on experiments from Al-Ali et al. 39 To confirm that GLAST-selected astrocytes could increase their expression of CSPGs in response to a biologically relevant perturbagen, rhTGF-β1 (10 ng/mL, 240-B; R&D Systems) was used. 29
Astrocyte Culture
Cells were cultured for 4 days in media containing 10% FBS and incubated at 37°C with 5% CO2. Three days before the addition of perturbagens, cells were washed twice with HEPES (20 mM), (15630-80; Gibco), buffered Ca2+-free HBSS (1415-095; Gibco). Immediately after washes, 150 μL of freshly prepared DMEM (11995-065; Gibco) containing the NeuroCult™ SM1 Neuronal Supplement (1:50 dilution), sodium pyruvate, GlutaMAX, and penicillin–streptomycin was added to each well.
Z-Factor Experiments
For the stellation index assay, low-density cultures were plated into the inner 60 wells of 96-well plates. For the CSPG assays, high-density cultures in the inner 54 wells of 96-well plates were used. After 7 days in vitro (DIV), compounds were prepared and equilibrated in mixing plates. Fifty microliters of compound solution (80 μM) from mixing plates was added to cultures (containing 150 μL/well) to a final volume of 200 μL/well (20 μM final concentration), as described in Al-Ali et al. 39,40 Compounds and their vehicle controls were added to plates as depicted in Supplementary Figure S1 and the workflow described in Figure 1 and Table 1.
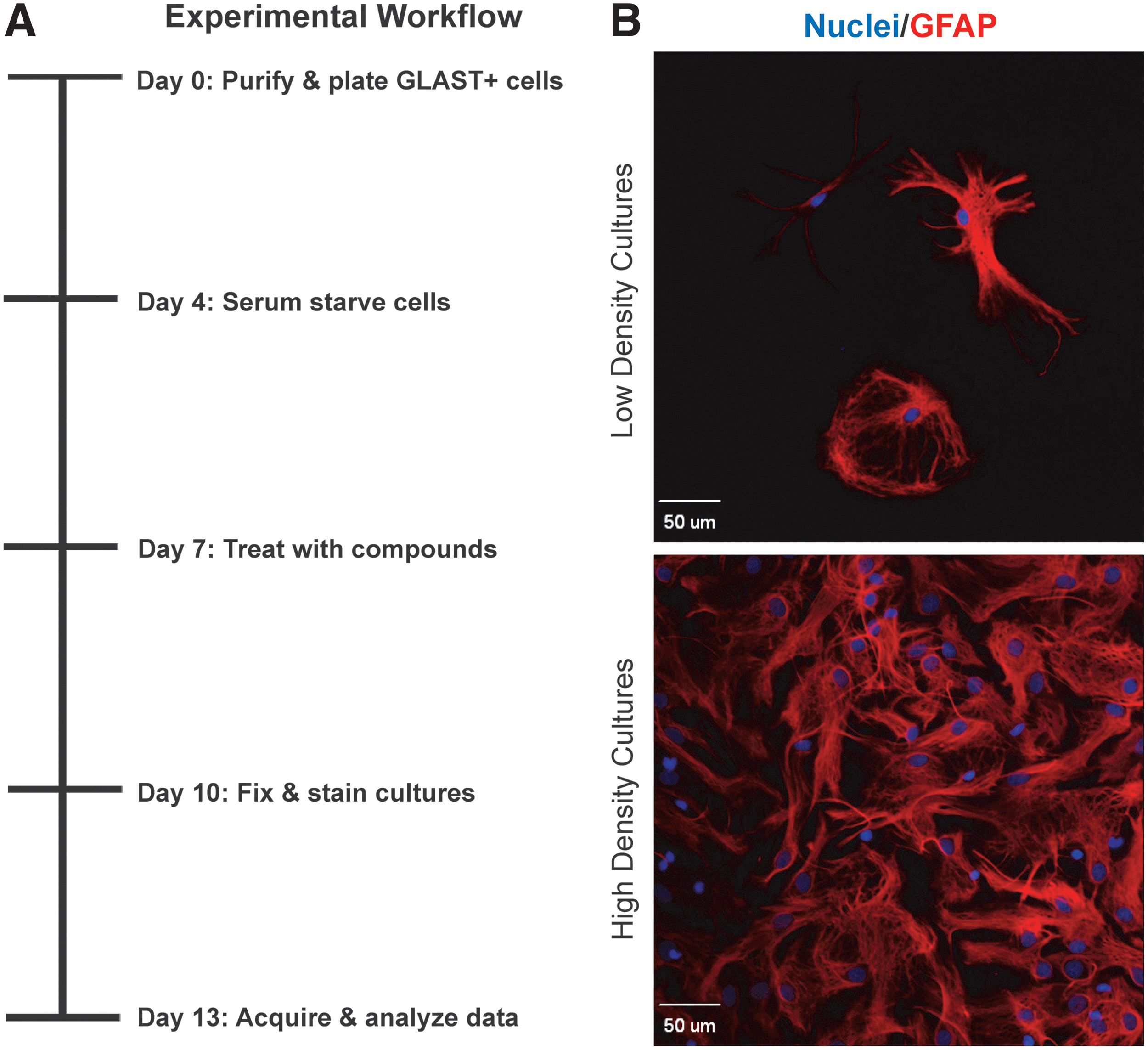
Experimental system for testing perturbagens on astrocytes.
Dose–Response Experiments with Protein KIs
After 3 days in chemically defined media (7 DIV), cells were treated with KIs in dose–response experiments as previously described with one additional concentration point (6 nM) (Table 2). 39,40 Each plate contained 12 DMSO control wells, 6 no primary antibody control wells, and 2 technical replicates per treatment condition (Supplementary Fig. S1). The concentration of DMSO was 0.2% in all wells. Biological replicates were performed on different weeks with freshly prepared primary cells and working solutions.
Dilutions for Perturbagen Treatments
SCI Experiments and Tissue Processing
Female C57BL/6 mice aged 6–8 weeks were used for SCI experiments. On the day of surgery, anesthetized (ketamine/xylazine 100 mg/15 mg/kg, intraperitoneal injection) animals received laminectomies at the T9 vertebral level and then half received 75 kdyn contusion injuries (IH-0400; Precision Systems and Instrumentation). During the first week of postoperation, animals received antibiotics (gentamicin, 2 mg/kg), analgesics (buprenorphine HCl, 0.05 mg/kg), and Lactated Ringer's. Bladders were expressed twice daily for the entire duration of the study. Fourteen days after surgery, animals were terminally anesthetized and transcardially perfused with ice-cold phosphate-buffered saline (PBS, pH 7.4, 70011-0440; prepared with Gibco) followed by 4% (w/v in 1× PBS) paraformaldehyde (PFA, 6148; Sigma). Spinal cords were removed and postfixed overnight in 4% PFA at 4°C. Tissue was cryoprotected (30% sucrose in PBS) for 72 h before embedding a 12 mm piece of spinal cord tissue (6 mm rostral and caudal to lesion/laminectomy site) in the Tissue Freezing Medium (TFM-5; Triangle Biomedical) and freezing on dry ice. Sagittal sections (20 μm) were made on a cryostat (CM 1800-3; Leica), melted onto poly-
Immunocytochemistry
Three days after perturbagen treatment (10 DIV), cultures were fixed in 110 μL of 4% PFA (w/v in PBS) for 15 min. PFA was removed and plates were washed six times with PBS dispensed from a liquid handling robot (ELx405 Select CW; BioTek). Cells were permeabilized in 0.5% Triton X-100 (9410; EMD) in PBS for 10 min and then blocked for at least 1 h in a PBS-buffered solution containing 5% goat serum (16210-064; Gibco), 1% bovine serum albumin (A9418; Sigma), and 0.03% Triton X-100. Primary and secondary antibodies were diluted in this solution (antibodies and dilutions used are listed in Table 3). Cells were incubated with primary antibodies overnight at 4°C and then washed five times to remove unbound antibodies. Alexa Fluor®-conjugated secondary antibodies and Hoechst nuclear dye (H1399; Molecular Probes) were added in a blocking buffer and incubated for 1 h at room temperature. Plates were washed five times with PBS and then stored in PBS with 0.01% (w/v) sodium azide (S8032; Sigma). Plates were covered with aluminum foil and stored in the dark at 4°C until imaged.
Immunohistochemistry
For SCI experiments, slides were defrosted and dried overnight. Sections were permeabilized, and blocked (as above in Immunocytochemistry), and then incubated with primary antibodies overnight at 4°C. After washing three times with PBS, sections were stained with Alexa Fluor-conjugated secondary antibodies for 1 h. Sections were then washed five times. VECTASHIELD® (H-1200; Vector Labs) mounting medium was applied before mounting coverslips. The antibodies and concentrations used in these experiments are listed in Table 3.
Imaging and Tracing
Image acquisition and automated tracing for HCA experiments were performed on a Thermo Scientific ArrayScan™ VTI High-Content Imager. Three-channel fluorescent images were acquired with a 5×objective using an XF93 filter set. A total of nine fields were imaged for each well. Nuclei were visualized with Hoechst 33342 nucleic acid stain (386 nm excitation). Molecules labeled with Alexa-488 and Alexa-647 secondary antibodies were visualized using 485 and 650 nm excitation, respectively. To investigate the morphology of astrocytes' GFAP cytoskeleton, GFAP-positive cells were traced using the Neuronal Profiling BioApplication in the Cellomics ArrayScan software suite. To investigate astrocyte CSPG expression, high-density cultures were traced with the Spot Detector BioApplication. In some experiments, representative images of cells were acquired using an Olympus IX81 microscope and SlideBook™ software (3I Software Company) for qualitative analysis. In all experiments, no primary antibody controls were used to set fluorescent intensity thresholds for image acquisition and image analysis. Parameters for image acquisition and tracing, and data acquired by HCA algorithms are provided in the Supplementary Materials and Methods section.
Data Acquisition and Analysis
Raw cell data were analyzed using Spotfire™ (TIBCO). The stellation index was calculated using the ratio of cell process area to cell body area. For Z-factor determination experiments, raw values were used, but in dose–response experiments, mean values for each treatment (minimum 2 technical replicates/experiment) were normalized to the respective control(s) (12 technical replicates/plate). These values are presented as the means for biological replicates (minimum N=3 per condition). We performed statistical tests and plotted data using Microsoft Excel and GraphPad Prism v5.03. For dose–response experiments comparing mean values for multiple compounds, statistical significance was determined by a two-way analysis of variance with a Bonferroni posttest. For transforming growth factor-beta (TGF-β) stimulation experiments and other experiments comparing means between two groups, Student's t-tests were used. In all experiments, the null hypothesis was rejected if P<0.05. Z-factors were calculated by Z=1−3(σStimulated+σControl)/(|μStimulated−μControl|) for each experiment with a minimum of 15 technical replicates/condition in each plate. 44 Individual wells were excluded from data analysis if pipetting errors were noted during experimentation or there were fewer than 100 Hoechst-positive cells per well.
Results
Astrocyte Cultures and Optimized Workflow for HCA Assays
Promising compounds that promoted neurite outgrowth in vitro have failed to robustly enhance functional recovery in vivo. 45,46 This may be due, in part, to their deleterious effects on astrocytes. 47,48 Given the time and cost of animal experiments, as well as the low success rate of translating hit compounds into therapeutics, our aim was to develop useful counter-screens on astrocyte phenotypes for expediting the discovery of pharmacotherapies that enhance axon regeneration beyond the glial scar.
Traditionally, neonatal astrocytes are used for in vitro studies because adult cells, including astrocytes, are more vulnerable to dissociation procedures. Despite the relatively good survival of early postnatal astrocytes, it is often necessary to culture cells in media supplemented with 10% FBS to promote their survival and differentiation, and to remove contaminating neurons and microglia by repeated passages. 49 Recently, a number of techniques have become available that deplete nonastrocyte lineage cells or select astrocytes based on cell type-specific surface markers. 43,50 These methods make it possible to obtain enriched astrocyte cultures from the postnatal cortex in as little as 1 day. We found that commercially available kits that select astrocytes on the basis of GLAST (EAAT1) expression enabled us to rapidly generate highly enriched primary cortical astrocyte cultures suitable for screening purposes (Fig. 1).
Before treatment with perturbagens, astrocyte cultures must be serum starved for at least 24 h because serum contains growth factors and cytokines that can confound interpretation and introduce unacceptable variability. 29,51,52 Accordingly, we isolated P6 astrocytes and plated cells in media supplemented with 10% FBS. Primary astrocytes were allowed to mature for 4 days and then, after washing cultures twice, changed to 150 μL/well of chemically defined media. Subsequently, astrocytes were cultured in serum-free conditions for 3 days before introducing compounds (7 DIV) and were allowed to grow for 3 more days before fixing and staining (10 DIV; Fig. 1 and Materials and Methods section). The 3-day incubation time with compounds was selected because 72 h are needed to detect changes in CSPG expression by Western blotting or ELISA, while also being suitable for detecting changes in astrocytes' GFAP cytoskeleton. 29 This system and workflow allowed us to prepare, treat, and process low- and high-density primary astrocyte cultures for two different assays on the same schedule. Each biological replicate could be cultured, treated with KIs, stained, and imaged within 2 weeks (Figure 1 and Table 1).
Stellation Index Assay Is Suitable for Screening
In low-density cultures, the stellation index was used as the metric to determine if compounds induced reactive-like changes in the astrocyte cytoskeleton (Fig. 2). To ascertain whether this assay was suitable for screening, we used the Cellomics Neuronal Profiling to compare stellation index values for control astrocytes (DMSO, 0.2%) to cells treated with a KI (GSK-5, 20 μM) (see Supplementary Fig. S1 for plate layouts and Materials and Methods section). Astrocytes stimulated with GSK-5 adopted more stellate GFAP phenotypes compared to DMSO controls (Fig. 2A, B). The effects on astrocytes' stellation index were consistent across wells from individual plates (3.54±0.28 vs. 0.69±0.12; mean and standard deviation [SD], Fig. 2C). Reproducibility among plates within a single experiment was strong as indicated by GSK-5 treatment producing an ∼5× increase in the stellation index relative to controls (3.49±0.35 vs. 0.69±0.13; mean±SD; Fig. 2D). Finally, Z-factor calculations show that this assay had excellent performance metrics (Z-factor of 0.44±0.03; mean and standard error of the mean [SEM]) (Fig. 2E) for the three biological replicates.
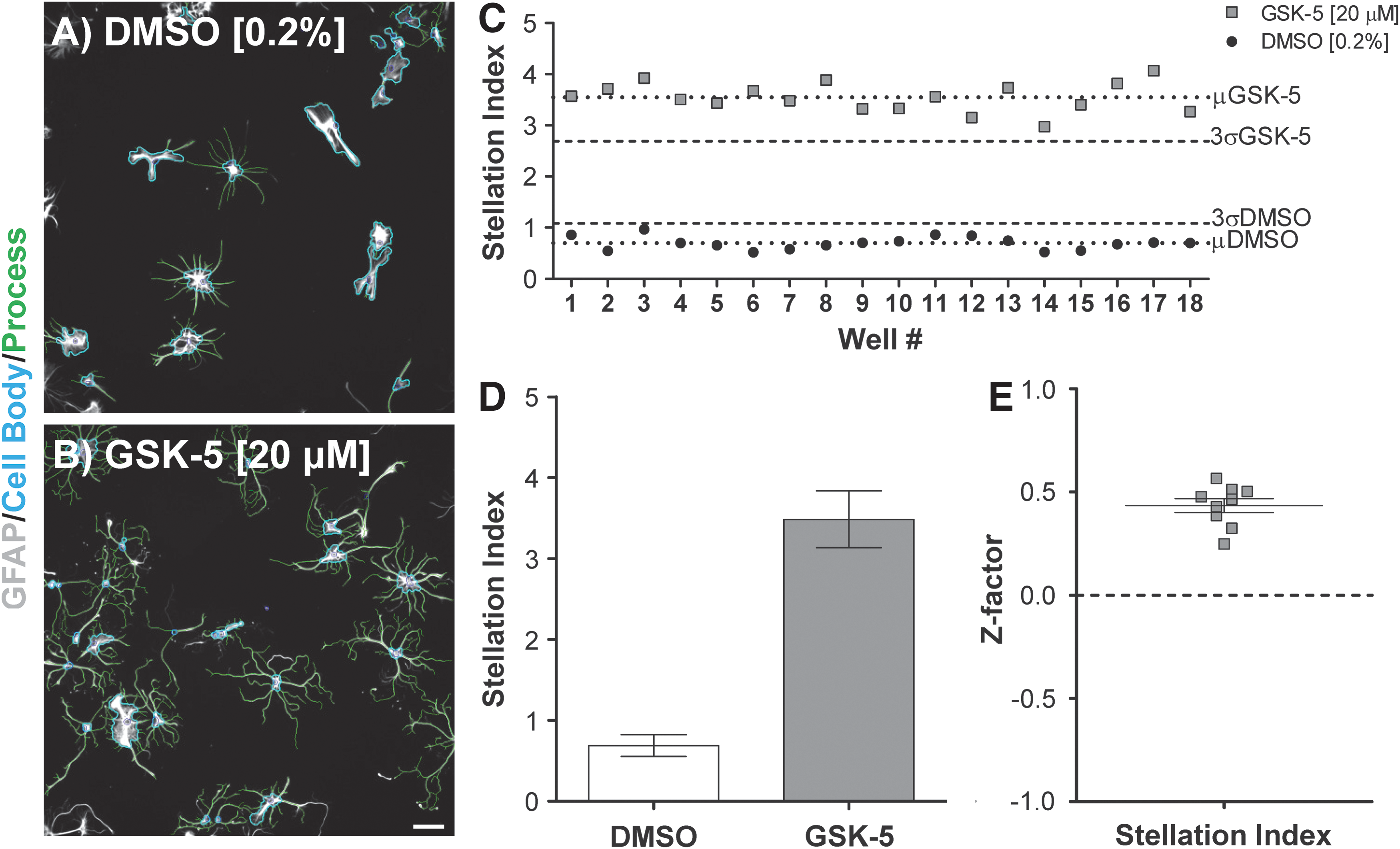
Stellation index assay is suitable for screening experiments.
CSPG Assay Is Reproducible and Informative, but Not Suitable for Screening
Given the changes in CSPGs after injury and their importance to regeneration, we wanted to determine if neurite growth-promoting KIs induce the expression of glial scar-associated CSPG epitopes from cultured astrocytes. There are numerous species of CSPGs expressed in the CNS, and the repertoire of CSPGs changes after CNS injury. 15,53,54 This is due to both differential expression of core proteins and alterations in posttranslational modifications (e.g., glycosylation and sulfation). 55,56 We tested various CSPG antibodies during assay development (data not shown), but ultimately chose the monoclonal antibody CS-56. 57 This antibody labels chondroitinase-sensitive glycosaminoglycan (GAG) moieties associated with glial scar astrocytes in vivo following traumatic CNS injuries (Supplementary Fig. S2A). 53,58,59 In addition, these CSPG species are upregulated by TGF-β-stimulated reactive-like astrocytes in vitro (Supplementary Fig. S2B–E). 29
To assess CSPG expression as an HCA target, the Cellomics Spot Detector BioApplication was utilized to measure size, number, and intensity of CSPG+ puncta in our cultures, as well as to bin data at the cell or well level (Fig. 3B). To stimulate astrocyte CSPG expression for Z-factor determination experiments, we selected the KI Y-27632 (Fig. 3). This compound was useful for these experiments because (1) it promotes neurite outgrowth and (2) it induces/exacerbates aspects of reactive gliosis in vivo and in vitro. 39,47,48 In our Z-factor determination experiments, we used conditions that were similar to our neurite outgrowth screen, where the highest concentration was 20 μM. Accordingly, Y-27632 (20 μM) was our stimulated condition and DMSO (0.2%) our control.
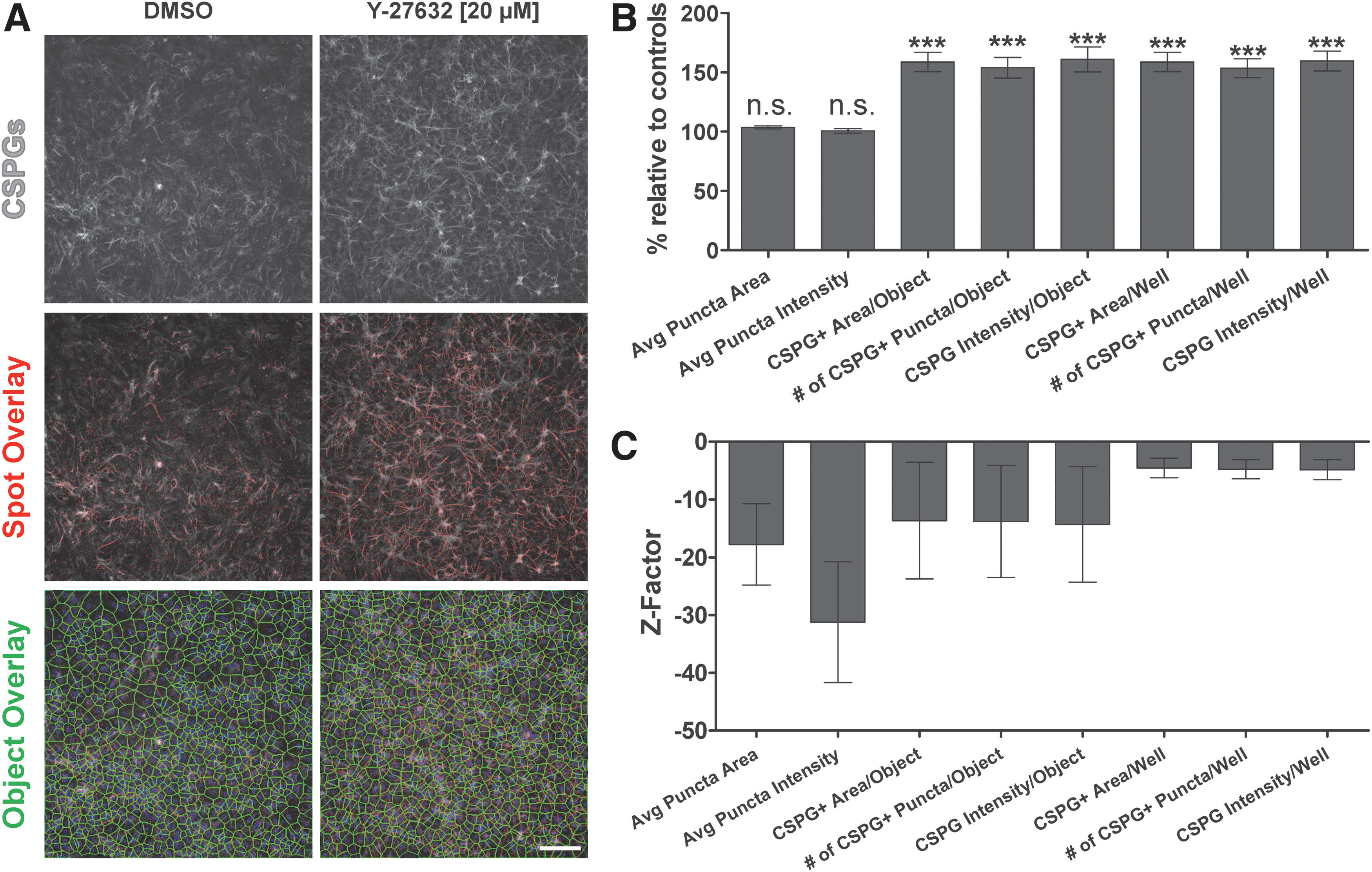
CSPG assay is reproducible.
Relative to DMSO controls, Y-27632 significantly increased (P<0.001, t-tests) the number of CSPG+ puncta per cell (153.86% of DMSO controls±8.76%; mean±SEM), the area per cell covered by CSPG+ puncta (158.74%±8.1%), and the average staining intensity per cell (160.93%±10%) (Fig. 3B). Importantly, similar changes were also observed when data were analyzed at the well level (Fig. 3B). Interestingly, Y-27632 treatment did not significantly alter the morphology of individual CSPG puncta (i.e., puncta sizes and intensities are not affected). These effects were reproducible across experiments and were of similar magnitude to those observed using biochemical or image-based approaches by other groups. 48,60 However, the parameters we analyzed produced negative Z-factors (Fig. 3C). The best performing metrics were the number of CSPG+ puncta per well (Z-factor = −3.03), CSPG+ area per well (Z-factor = −3.06), and CSPG intensity per well (Z-factor = −3.87), which suggests that our stimulation condition did not produce sufficiently large effect sizes. It is possible that, based on published results, increasing the dose of Y-27632 or extending the culture period may have improved assay performance, but we focused on the 20 μM dose, given its relevance to our neurite bioassay experiments. 39,40,47 Thus, the CSPG assay is useful for studying the expression of CSPGs in vitro, but cannot be a stand-alone readout in screening experiments.
Counter-Screening Strategy Identifies a Neurite Growth-Promoting KI That Promotes Axon Regeneration and Does Not Induce Reactive Astrocyte Phenotypes
For CNS regeneration, compounds that promote neurite outgrowth without activating astrocytes may be the most effective in vivo. Therefore, we explored whether our assays could identify neurite growth-promoting KIs that do not induce reactive-like astrocyte phenotypes. For proof-of-principle dose–response experiments with astrocytes, we selected a subset of neurite growth-promoting KIs identified as hits in our published screen. 39
In the stellation index assay, we found that Y-27632 (▼) and GSK-5 (□) induced dose-dependent reactive-like cytoskeletal changes (Fig. 4A). In contrast, GSK-2 (×) and ML-7 (○) treatment did not significantly increase the stellation index at any concentration tested (Fig. 4A). Compounds were also tested in high-density cultures for the CSPG assay, using well-level data as the primary readout (these parameters had the best performance in Z-factor experiments; Figs. 3C and 4B, C). ML-7 treatment (○) did not produce significant effects on either CSPG parameter; thus, ML-7 did not induce reactive changes from astrocytes in either assay. In contrast, Y-27632 (▼) and GSK-5 (□) increased CSPG+ area per well, as well as CSPG intensity per well. Interestingly, GSK-2 (×) treatment (20 μM) induced significant changes in CSPG intensity/well, but had no effect on the CSPG+ area per well (Fig. 4B, C).
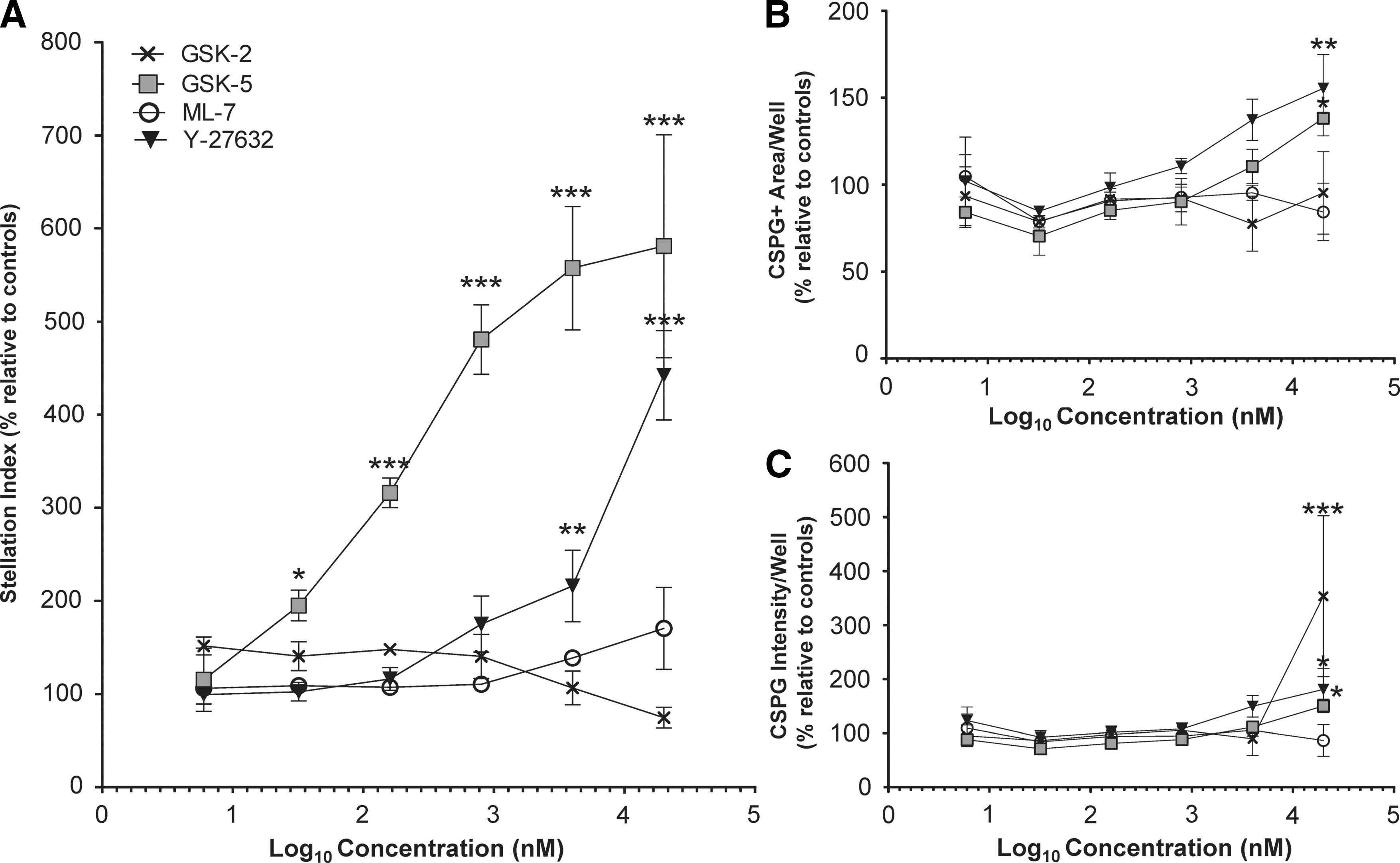
High-content analyses identify a kinase inhibitor that does not induce reactive-like changes in the GFAP cytoskeleton or CSPG expression.
Deeper investigation of GSK-2 (20 μM) data revealed that this condition produced a different CSPG phenotype compared to others (Supplementary Fig. S3). Quantification of features from individual puncta showed that they were significantly larger and more intensely stained than those observed in Y-27632 or DMSO cultures (Supplementary Fig. S3D, E). Many of these immunoreactive aggregates were associated with cell bodies (perinuclear labeling) and had limited deposition in cell-free areas of substrate (data not shown). These dense aggregates may be nascent CSPGs or GAGs stalled in the secretory pathway (e.g., endoplasmic reticulum/Golgi or vesicles). However, because all staining was performed with buffers containing detergents, antibodies had access to both intracellular and extracellular CSPGs. Thus, we cannot definitively state where molecules labeled by CS-56 are localized. Alternatively, the CSPG phenotype observed after GSK-2 (20 μM) treatment could be related to toxicity, as cell counts (data not shown) and the number of CSPG puncta appeared to be reduced compared to controls. However, neither effect reached statistical significance (Fig. 3C). These findings reinforced the fact that while the CSPG assay may not be suitable for high-throughput screening, these assays produce informative data and can detect perturbagens that induce reactive-like CSPG phenotypes, as well as deleterious effects associated with toxicity or altered processing.
Discussion
There are no FDA-approved drugs that enhance axon regeneration and functional recovery available to persons with chronic traumatic brain injuries (TBIs) or SCIs. Kinases are attractive drug targets because they are involved in many cellular processes, including neurite outgrowth, axon regeneration, and reactive gliosis. 61 –64 Until recently, there was little information about the ability of various KIs to manipulate axon/neurite growth. 39,40,65 Importantly, there is even less information about how KIs might affect the astroglial reaction to CNS injury. 48,66 This provides an exciting opportunity because identifying KIs capable of increasing axon regrowth without exacerbating reactive gliosis may ultimately lead to effective drug therapies for persons with CNS injuries.
One reason for the relative paucity of information about how KIs influence astrocytes in vivo is the fact that many KIs do not cross the blood–brain barrier (BBB) effectively. 67 As a result, extrapolating biologically relevant dosages from in vitro experiments is difficult, in part, because many KIs fail to penetrate the CNS after peripheral administration. Although it is possible to overcome BBB penetration issues through medicinal chemistry, such efforts are resource intensive and can be a significant bottleneck early in the drug development pipeline.
An approach to overcome this challenge is to directly infuse KIs into CNS tissues or cerebrospinal fluid. Unfortunately, direct injections or implanting catheters used to deliver compounds from osmotic minipumps may damage the CNS. We have observed that gliotic scar tissue can form at injection and catheter sites (data not shown). This fact confounds our ability to make discrete assessments of how compounds affect quiescent astrocytes. In addition, animal to animal variability in the severity of injury caused by surgical procedures further limits our ability to draw conclusions about KI-induced changes in astrocytes in vivo. Thus, the development of in vitro models for studying how KIs affect astrocytes represents a potentially useful advance for drug discovery in TBI and SCI.
To address this need, we developed a methodology for testing compounds on astrocytes in vitro. Enriched astrocyte cultures that were amenable to screening were obtained using a commercially available astrocyte isolation kit. Although adult astrocytes are in principle the most relevant to regeneration failure in vivo, technical limitations make these impractical for screening. 41 First, adult CNS cells survive poorly after isolation in culture. Second, our method, which relies on antibody-based positive selection of GLAST-positive cells using small-diameter columns, is inefficient when cells are obtained from myelin-rich tissues like the postnatal spinal cord and adult brain (130-095-826; Miltenyi Biotec and unpublished observations). To balance feasibility and biological relevance in our experiments, cells were prepared from postnatal day 6 rat cortices—the oldest age from which we could obtain sufficient numbers of viable cells.
We used these primary, early postnatal cortical astrocytes to develop two phenotypic assays for studying aspects of reactive gliosis in vitro. The stellation index assay, which monitors reactive-like changes in astrocytes' GFAP cytoskeleton, was robust (Z-factor=0.44) and suitable for screening experiments. The CSPG assay, which monitors the expression of glial scar-associated CSPG epitopes from astrocytes, is reproducible and useful for identifying compounds that produce an alternative aspect of reactive gliosis. However, this assay has negative Z-factors for the parameters we measured.
Finally, the assays were utilized in a small-scale pilot screen of representative neurite growth-promoting KIs. The counter-screen allowed us to identify a compound, ML-7, that did not induce reactive-like astrocyte phenotypes. Interestingly, this compound was capable of enhancing axon growth in cortical slices rich in astrocytes. 39 In light of the facts that Y-27632 exacerbated gliosis in vivo and that both GSK-2 and GSK-5 failed to enhance axon regrowth in our cortical slice model (unpublished observations), our findings suggest that these HCA assays are useful for identifying agents that produce undesirable astrocyte phenotypes. 48 Our future goals include employing these assays to screen select compound libraries as well as investigating whether our assays can be adapted for identifying agents that reverse aspects of reactive gliosis.
In conclusion, researchers have yet to discover agents that modify the injured human CNS in a manner that improves regeneration and functional recovery. A major factor that complicates drug development is the translation of findings from neurite outgrowth experiments into complex animal models, where astrocyte-derived factors can influence CNS axon regrowth. Accordingly, the tools described here can be part of a solution to this problem as scientists consider nonneuronal cells during the early phases of drug discovery. Thus, our counter-screening strategy may accelerate the development of effective drugs for CNS injury by prioritizing the most promising compounds from a primary screen for preclinical regeneration studies in small animal models.
Footnotes
Acknowledgments
We are grateful to the members of the Lemmon-Bixby laboratory, past and present. We would like to thank M. Danzi for feedback on the manuscript as well as T. Slepak and Y. Martinez for their technical assistance. We thank S. Zuchner, J. Lee, N. Ayad, P. Tsoulfas, R. Brambilla, M. Byrne, and H. Geller for constructive criticism. Finally, we appreciate Bill Zuercher and David Drewry at GlaxoSmithKline for providing some of the compounds for these experiments. S.R.B. is a Lois Pope LIFE Foundation Fellow. V.P.L. holds the Walter G. Ross Distinguished Chair in Developmental Neuroscience. This work was supported by the Miami Project to Cure Paralysis, the Buoniconti Fund, the Walter G. Ross Foundation, the Department of Defense (W81XWH-13-1-077), and the National Institutes of Health (2R01HD057632).
Authors' Contributions
S.R.B., J.L.B., and V.P.L. conceived the project. S.R.B., H.A., Y.S., J.E.J., J.L.B., and V.P.L. designed experiments, analyzed data, and wrote the manuscript. S.R.B., J.E.J., and Y.S. performed experiments.
Disclosure Statement
No competing financial interests exist.