
Abstract
Current tuberculosis (TB) treatment suffers from complexity of the dosage regimens, length of treatment, and toxicity risks. Many natural products have shown activity against drug-susceptible, drug-resistant, and latent/dormant Mycobacterium tuberculosis, the pathogen responsible for TB infections. Natural sources, including plants, fungi, and bacteria, provide a rich source of chemically diverse compounds equipped with unique pharmacological, pharmacokinetic, and pharmacodynamic properties. This review focuses on natural products as starting points for the discovery and development of novel anti-TB chemotherapy and classifies them based on their chemical nature. The classes discussed are divided into alkaloids, chalcones, flavonoids, peptides, polyketides, steroids, and terpenes. This review also highlights the importance of collaboration between phytochemistry, medicinal chemistry, and physical chemistry, which is very important for the development of these natural compounds.
Introduction
In 1882, Robert Koch first identified Mycobacterium tuberculosis as the causative pathogen responsible for tuberculosis (TB). A little over a century later, the World Health Organization declared TB a global epidemic, due to the poor success of treatment and the high mortality rates associated with TB infections. 1 Moreover, one-third of the global population carries the asymptomatic latent or dormant bacterial form, further increasing the risk for disease spreading among the population by increasing the risk of TB reactivation. 1,2 In 2013, TB incidences were reported to be ∼9 million, where TB infections claimed 1.5 million lives. 3 The current, short-course frontline TB therapy consists of rifampicin, isoniazid, ethambutol, and pyrazinamide cocktail for a 2-month “intensive phase,” followed by rifampicin and isoniazid administration for a 4-month “continuation phase” to eradicate the latent bacilli. Except for bedaquiline, the Food and Drug Administration has not approved a new anti-TB drug in the last 40 years despite the current treatment suffering from significant shortcomings such as length of the dosage regimen, toxicity of antitubercular agents, and resistance development to currently used agents. 4 Therefore, the need for novel anti-TB drugs is crucial.
Mutations in at least 10 genes in the M. tuberculosis genome can cause the emergence of drug-resistant TB strains. 5 Drug resistance has developed, in part, by patient noncompliance and drug shortages, which has forced the use of more toxic, less effective, and more expensive drugs for a longer treatment duration. Multidrug-resistant TB (MDR-TB) strains are resistant to two of the first-line antitubercular drugs, rifampicin and isoniazid. Extensively, drug-resistant TB (XDR-TB) strains are associated with high levels of mortality because these strains are resistant to the first-line antitubercular drugs, rifampicin and isoniazid, along with any of the fluoroquinolones and at least one of the injectable second-line antitubercular agents (capreomycin, kanamycin, and amikacin). 6 Among the new TB cases and previously treated cases between 1997 and 2012, 15% and 45%, respectively, were patients infected with MDR strains. Among the drug-resistant cases, 11% were infected with the XDR strains. 7 A meta-analysis study comprising 91,538 TB patients revealed that only 54% of TB treatments were successful, where 15% of that patient population died and 20% suffered from adverse effects, 8 such as hepatic dysfunction, ocular complications, ototoxicity, and gastrointestinal side effects. 9 The previous data emphasize the need for new treatment and diagnostic tools, as well as a programmatic follow-up for patients to identify infected patients and treat them in a timely manner while reducing the spread of the disease among the population. 10
The ideal anti-TB drug must be potent, safe, easily administered, active against resistant strains, active against latent and replicating bacteria, and it should have a low incidence of drug/drug interactions. 1 Table 1 describes the ideal attributes of an anti-TB drug.
Desired Properties of a New Antituberculosis Drug
MDR, multidrug resistant; XDR, extensively drug resistant.
Several compounds in preclinical and clinical studies have been identified during the last few years for developing new TB treatment. These approaches include discovery of novel chemical entities, such as bedaquiline, 11 –13 PA-824, 14 –16 and delamanid, 17,18 and the optimization and repurposing of old drugs, such as riminophenazines, 19 –21 β-lactams, 22,23 and oxazolidinones 24,25 for anti-TB activity. These approaches have been successful in introducing many promising lead compounds in preclinical and clinical studies. However, none of these compounds meet the requirements for an ideal anti-TB drug and they all have limitations to be successful antitubercular agents. 26
Natural Products: A Potential Solution for TB Challenges
With the low output of lead compounds from high-throughput screening efforts, focus has shifted toward the exploration and development of natural products as effective antimicrobial drugs. 27 Drugs from natural origin comprise ∼75% of all the antibacterial agents discovered between 1981 and 2010. 28 The exploration of nature as a source of novel antimicrobial agents is expanding and they offer a remarkable opportunity to identify interesting chemical scaffolds for drug discovery by providing chemically diverse compounds. Many natural products have multiple activities against multiple targets, which may reduce drug-resistance rates since it is likely that multiple genetic mutations would be required. However, promiscuous compounds, whether natural or synthetic, have tendencies to be cytotoxic. Curcumin is an example of a natural product that has multiple direct molecular targets and many pharmacological activities ranging from anticancer to anti-inflammatory. 29 However, long-term consumption in mice increases the risk of ulcers, cecum and colon inflammation, and thyroid gland follicular cell hyperplasia. Thus, promiscuity is a challenge for the development of natural products as drug leads. 30 Recently, significant efforts have been made to explore and optimize various natural compounds for their antimycobacterial activities. 31
Alkaloids
Many anti-TB scaffolds are inspired by those of naturally occurring alkaloids. 32 Tiliacorinine, 2′-nortiliacorinine, and tiliacorine are bisbenzylisoquinoline alkaloids isolated from Tiliacora triandra roots (compounds 1, 2, 3, and 4, respectively, Fig. 1). They were found to exhibit activity against isolated MDR-TB strains with minimum inhibitory concentration (MIC) values in the range of 3.1–6.2 μg/mL. 33 Screening of the murine natural products library resulted in the identification of the pyridoacridone alkaloid ascididemin, which also blocks M. tuberculosis growth (MIC <0.35 μg/mL). Ascididemin was found to exhibit its cytotoxic activity by DNA topoisomerase II inhibition. However, its low selectivity (Vero cells IC50 < 0.14 μM) prohibits its use as an antitubercular agent. Decreasing the bulkiness of the pentacyclic core of ascididemin leads to the thioethyl analogue (compound 5, Fig. 1), which possessed comparable activity with a MIC value of 0.34 μg/mL and has a significantly higher selectivity (Vero cells IC50 of 6.8 μM). It can be concluded that size reduction of the ascididemin scaffold is a promising approach for TB drug discovery. 34

Tiliacorinine
The need to deliver a drug that acts on both active and dormant M. tuberculosis states focuses on the screening of active leads in hypoxic conditions that induce the dormant state. Halicyclamine A (compound 6, Fig. 1) was isolated from the Indonesian marine sponge Haliclona sp. and showed a higher activity against latent M. tuberculosis (MIC of 5.0 μg/mL) compared to isoniazid (MIC of 25 μg/mL). 35 Marine sponge Aplysina caissara extracts were found to be toxic and moderately active against M. tuberculosis with a MIC range of 200–400 μg/mL. The components of the extract and the mechanism responsible for this activity are currently unknown. 36
Chalcones and flavonoids
Chalcones isolated from Helichrysum melanacme (compounds 7 and 8, Fig. 2) are found to have a relatively low antitubercular activity (MIC of 50 μg/mL) compared to the frontline drug rifampicin (MIC of 2 μg/mL). 37 Four new flavonoids were isolated from the stems and roots of Derris indica. A peltogynan derivative (compound 9, Fig. 2) was found to be most active against M. tuberculosis H37Rv with a MIC value of 6.25 μg/mL. Unfortunately, the structure–activity relationship (SAR) was not established among these active compounds. 38
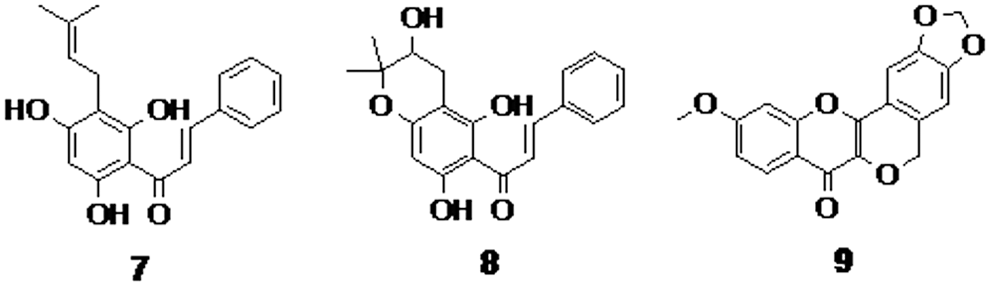
Chalcones
Peptides
Trichoderin A, Trichoderin A1, and Trichoderin B (compounds 10, 11, and 12, respectively, Fig. 3) are three aminolipopeptides isolated from the fungus Trichoderma sp. They showed significant activity against a panel of dormant Mycobacteria (MIC values of 0.12, 2.0, and 0.13 μg/mL, respectively) in comparison to isoniazid, which is inactive against hypoxic strains. Thus, aminolipopeptides can be promising agents for latent TB treatment. 39
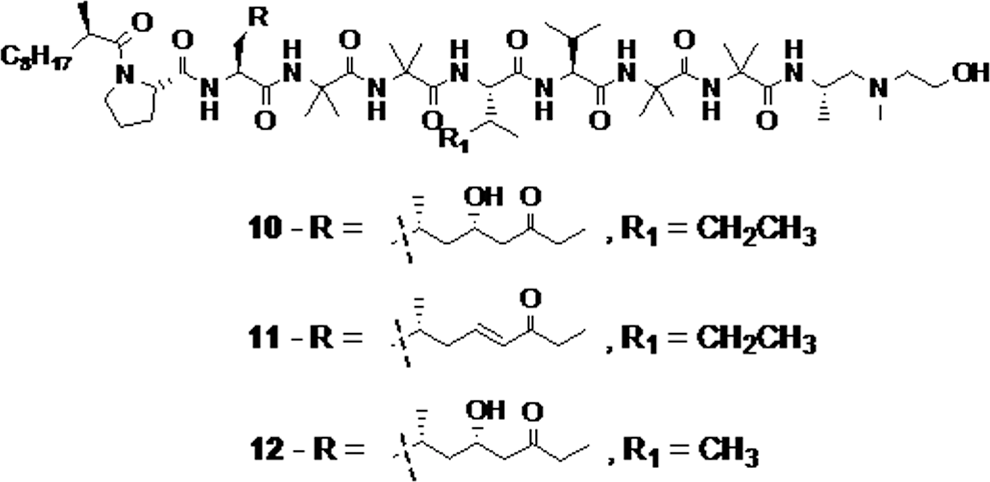
The aminolipopeptides trichoderin A
Human neutrophil peptides 1 (HNP1), isolated from azurophilic granules of humans, exhibit potent activity against TB. HNP1 have bactericidal activity and an ability to increase the permeation of rifampicin and isoniazid, resulting in a synergistic action with 16-fold lower MIC values at 50% dose reduction. The use of HNP1 as adjunct TB therapy has a potential to decrease the side effects and enhance the adherence for the treatment. 40 However, the use of an endogenous defense mechanism might lead to resistance that would diminish the human's innate host defenses, which might leave the society more susceptible to infections.
Polyketides
Urdamycinone E, urdamycinone G, dehydroxyaquayamycin, and urdamycin E (compounds 13, 14, 15, and 16, respectively, Fig. 4) are four C-glycosylated benz[a]anthraquinone derivatives isolated from Streptomyces sp. These four compounds have shown moderate antitubercular activity with a MIC range of 3.13–12.50 μg/mL. However, the lack of selectivity results in the cytotoxicity with an IC50 range of 0.092–10.07 μg/mL. 41
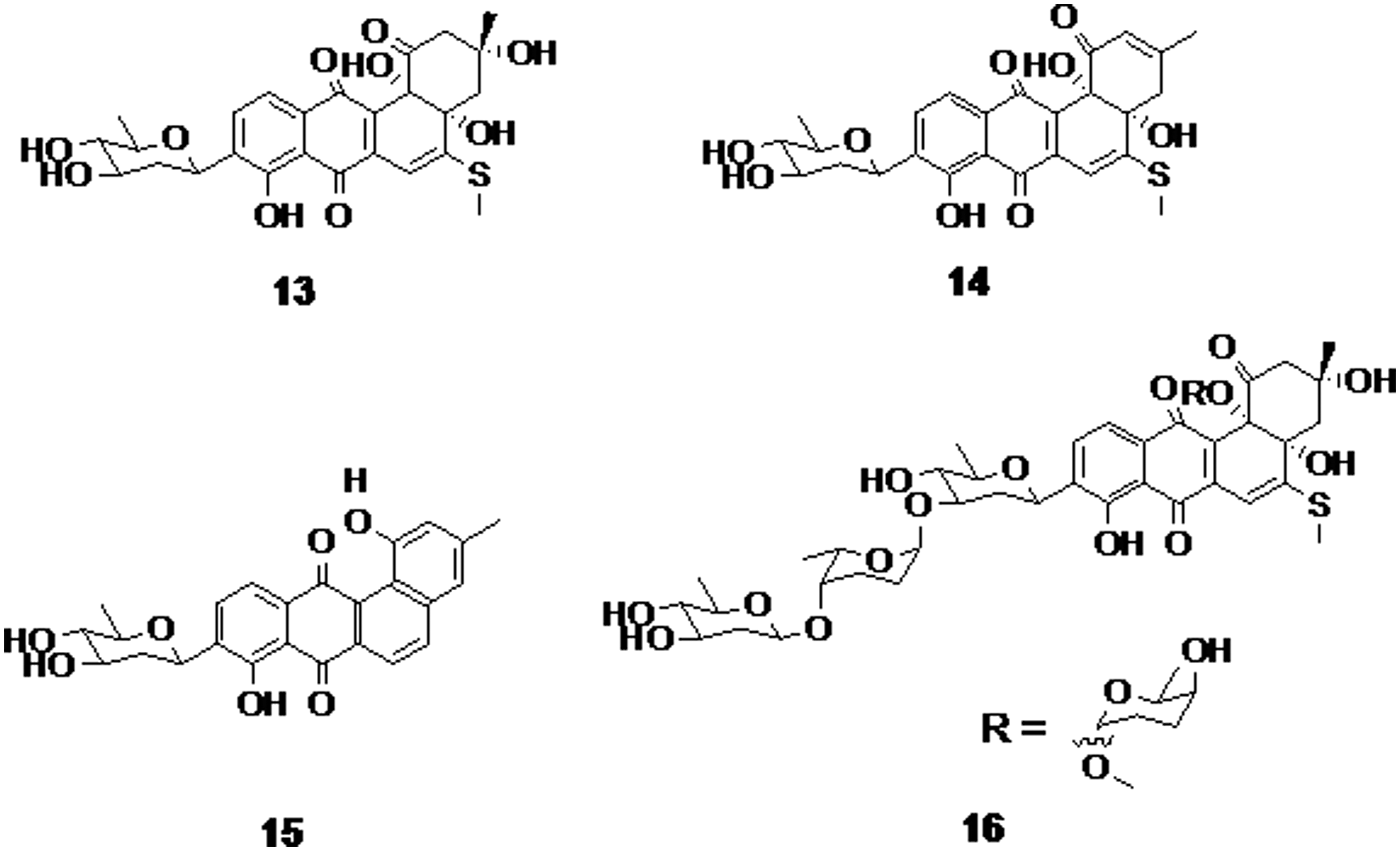
The C-glycosylated benz[a]anthraquinone derivatives urdamycinone E
Steroids
Extraction of the Caribbean Sea sponge Svenzea zeai resulted in the isolation of parguesterols A and B (compounds 17 and 18, respectively, Fig. 5). Natural parguesterols were found to have potential activity against Mycobacteria spp. and their synthetic derivatives exhibited high selectivity and potent activity with a low MIC value of 3.8 μg/mL. 42,43
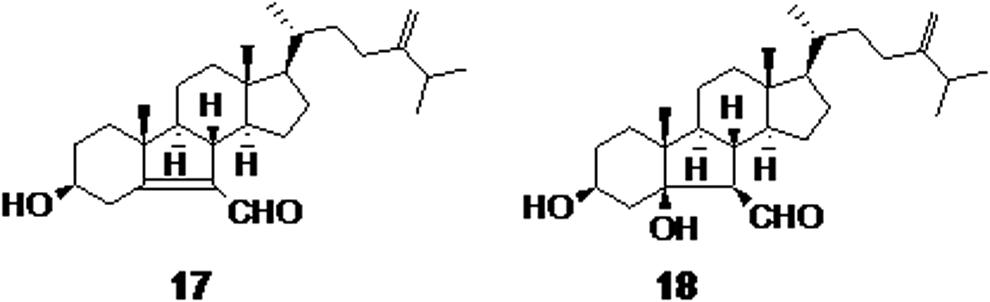
Parguesterols A
Terpenes
In a study, 20 sesquiterpene natural compounds were tested against M. tuberculosis strains. 1a-Acetoxy-6b,9b-dibenzoyloxy-dihydro-b-agarofuran (compound 19, Fig. 6) found to be more potent than conventional anti-TB drug rifampicin and having a MIC value of 6.2 μg/mL. 44 Extracts from Artemisia ludoviciana, Chamaedora tepejilote, Lantana hispida, Juniperus communis, and Malva parviflora have shown potent activity against MDR-TB strains. The fifth fraction of L. hispida hexane extract that contains sesquiterpenes has the most potent activity in this group with a MIC value of 12.5 μg/mL. However, step by step structure purification and elucidation are required to identify and develop the most efficacious active component. 45
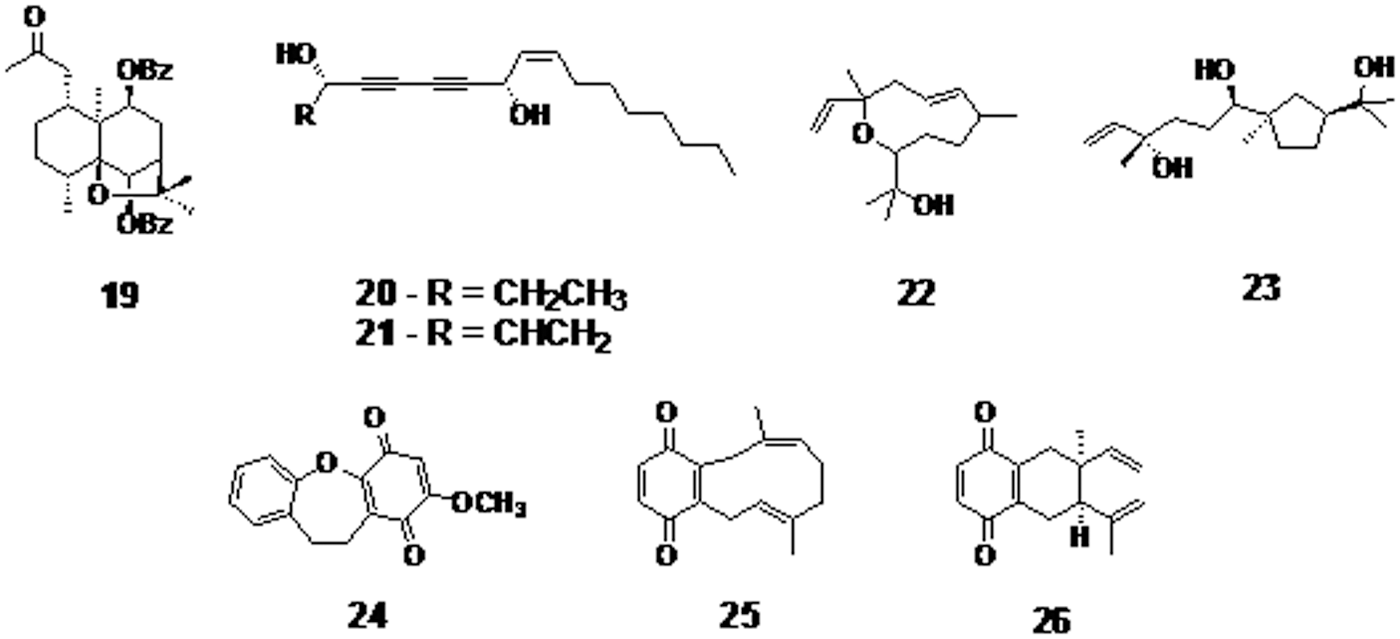
1a-Acetoxy-6b,9b-dibenzoyloxy-dihydro-b-agarofuran
Natural product isolation from Microtropis japonica, a small shrub, has been reported to yield fourteen compounds. Celahin C and salasol A were the most potent terpenes out of the isolated compounds with a MIC value of 15 μg/mL. 46 Extracts from another small shrub, Microtropis fokienensis, contained nine different compounds. 8-Acetoxymutangin was found to be the most potent extracted compound with a MIC value of 10 μg/mL. 47 Five promising compounds were isolated from the inner stem bark of Oplopanax horridus. Oplopandiol and falcarindiol (compounds 20 and 21, respectively, Fig. 6) have shown the highest activity with MIC values of 61.5 and 6.2 μg/mL, respectively. They were also found to act synergistically in the presence of neroplomacrol and neroplofurol (compounds 22 and 23, respectively, Fig. 6). 48 Artemisia afra isolates showed activity against replicating and virulent M. tuberculosis. Dichloromethane soluble extracts resulted in the most potent sesquiterpene-containing fractions with a MIC value of 10 μg/mL. Interestingly, aqueous extracts were the only extracts that showed in vivo activity. 49
Purification of Bauhinia purpurea extract yielded 18 different compounds. All of the compounds showed activity against M. tuberculosis, where bauhinoxepin (compound 24, Fig. 6) was the most potent with a MIC value of 24.4 μM. 50
Globiferin (compound 25, Fig. 6), a terpenoid isolated from the plant Cordia globifera, showed a promising activity against M. tuberculosis with a MIC of 1.5 μg/mL. However, the cope rearrangement of globiferin results in the production of cordiachrome (compound 26, Fig. 6), which is more potent and cytotoxic than its precursor. 51
Table 2 summarizes the efficacy profiles of the natural products. Currently, among all the natural compounds, ascididemin alkaloids were most active against susceptible strains, bisbenzylisoquinoline alkaloids have shown remarkable activity against the resistant strains, and the aminolipopeptides had a promising activity against the latent form.
The MICs of Different Natural Products Against Drug-Susceptible, Drug-Resistant, and Latent Mycobacterium tuberculosis
MIC, minimum inhibitory concentration.
Natural Products As Scaffolds for Synthetic Antitubercular Agents
Low concentrations and difficulty of extraction of pure active constituents from natural sources have significantly limited their potential. Furthermore, the complexity of their structures and the presence of complex mixtures of sterioisomers make the formulation and the approval process problematic. Moreover, it is difficult for medicinal chemists to characterize and purify these molecules.
However, another intelligent use of natural products is that they can provide the needed scaffolds for novel anti-TB compounds, and ideally, these efforts must be concentrated on medicinal chemistry studies to optimize their use. 52 –54 The focus now is shifting to use natural product scaffolds as leads for synthesis of natural product-like compounds. Structural modifications are important for the improvement of pharmacodynamic and pharmacokinetic properties of natural products. 55,56
Chalcones
Numerous studies have confirmed the activity of naturally occurring chalcones isolated from several plants such as H. melanacme against M. tuberculosis without prior knowledge of their mechanism of action. 37,57 A recent study has proposed that protein phosphatase inhibition is chalcone's bactericidal mechanism. 58 Thirty-eight different chalcone analogues were synthesized based on the chalcone natural scaffold and tested for their activity against M. tuberculosis phosphatase enzyme. The SAR revealed that compounds with the most hydrophobicity and the highest electronic density are able to inhibit the enzyme activity up to 70% (compound 27, Fig. 7). Compound 28 (Fig. 7) had the lowest reported IC50 of 8.3 μM. 58 Enzyme inhibition is found to be responsible for intracellular bacterial inhibition, without affecting the viability of human-derived macrophages. 59 Twenty-seven aryloxy-azolyl chalcones were synthesized and tested for their in vivo and in vitro activity. Among them, compound 29 (Fig. 7) was the most potent with a MIC value of 1.56 μg/mL and 71% intracellular killing. 60

Compounds
Flavonoids
Natural biflavonoid (isolated from Rhus succedanea L. and Garcinia multiflora Champ.) and synthetic biflavonoid containing methoxy- and nitro-substituents have shown remarkable in vitro activity against M. tuberculosis, with bacterial growth inhibition of up to 96% at 12.5 μg/mL concentration. 61 One year later, the same group tested a series of flavonoids and chalcones. Of the tested flavonoids and chalcones, five compounds showed promising activity with a MIC range of 6.8–48.3 μg/mL. 62 Quantitative SAR (QSAR) analysis correlated the importance of the hydrogen bond donor ability, and an increase in lipophilicity significantly increases anti-TB activity. 63 Other studies confirmed these results by showing that the substitution of biflavonoids with halogens, -OCH3, -NO2, and C2-indolyl significantly increased their antitubercular activity. 64,65
Coumarins
Coumarin is a natural product produced by many sources, including the tonka bean, and has been synthetically modified to achieve anti-TB coumarin-based compounds. 66 Recently, a series of coumarin derivatives were synthesized and tested. Compound 30 (Fig. 7) has shown significant activity with IC90 of 0.4 μM by targeting the essential mycobacterial fatty acid degradation protein D32 (FadD32). The synthesized compounds have also shown excellent oral bioavailability of 61.95%. 66,67 SAR studies revealed a relationship between the activities of coumarin derivatives and their lipophilicity. 68
Peptides
The remarkable activity shown by natural peptides isolated from Streptomyces sp. promoted the synthesis of sansanmycin derivatives. These derivatives were tested against both susceptible and drug-resistant TB strains. Compound 31 (Fig. 8) was found to be the most active derivative with a MIC value of 8 μg/mL in both susceptible and drug-resistant strains. 69 Calpinactam (compound 32, Fig. 8), a fungal antimycobacterial metabolite, was synthesized and derivatized with five different amino acids. This compound that has ALA instead of GLU in position IV was found to be most potent with a MIC value of 6.25 μg/mL. 70 The development of peptides as anti-TB drugs will be challenging as peptides are generally metabolically labile and tend to have short half-lives. However, bioisosteric replacement of peptide bonds has been achieved (as seen in many HIV protease inhibitors) and, therefore, provides an opportunity for synthetic modifications of this class.
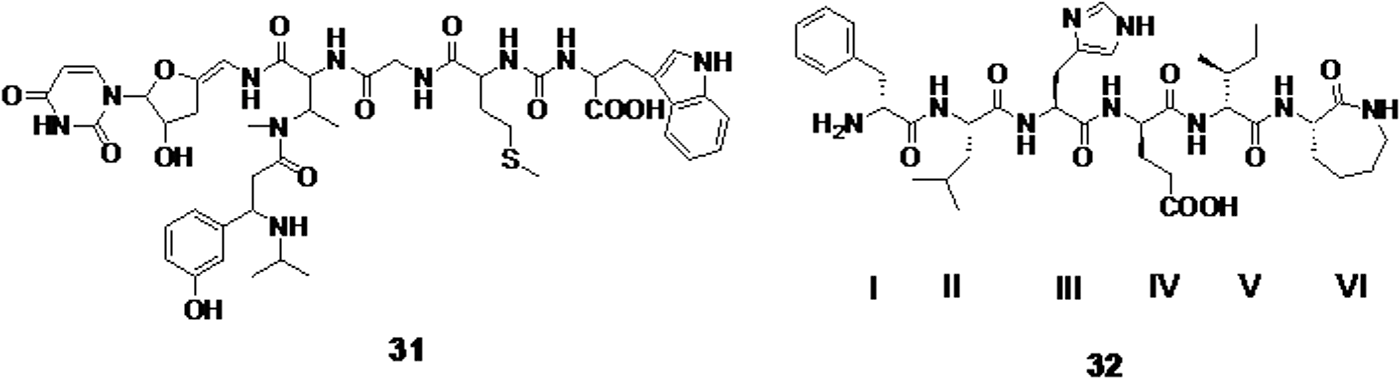
Compound
Polyketides
The natural thiolactone antibiotic isolated from a soil Nocardia spp. thiolactomycin is active against M. tuberculosis. The 4-acetyl-phenyl analogue (compound 33, Fig. 9) was able to inhibit mycobacterial mycolic acid biosynthesis with an IC50 of 4 μM. 71 Ten 3-aracylphthalide analogues were synthesized and tested against TB strains and furan substitution of the aryl methyl ketone resulted in the highest activity with an IC50 of 0.81 μg/mL. 72 Triazole fused spirochromone conjugates showed activity against M. tuberculosis H37Rv strains. Compound 34 (Fig. 9), with a 4-pentylphenyl at position 4 of the triazole ring and a cyclohexane ring on opposite side, was found to be most potent with a MIC of 0.78 μg/mL. 73 Fourteen quinone derivatives were recently synthesized and screened for their anti-TB activity. Among them, Plumbagin (compound 35, Fig. 9) was the most active with a MIC value of 400 μg/mL. 74 Furthermore, modified quinoline and naphthalene series were tested against drug-susceptible M. tuberculosis. Among them, compound 36 (Fig. 9) was found to be the least toxic with 99% inhibition at 6.25 μg/mL. Its activity is comparable to isoniazid, suggesting that the methoxy-phenyl piperazine derivatives are the most potent. 75
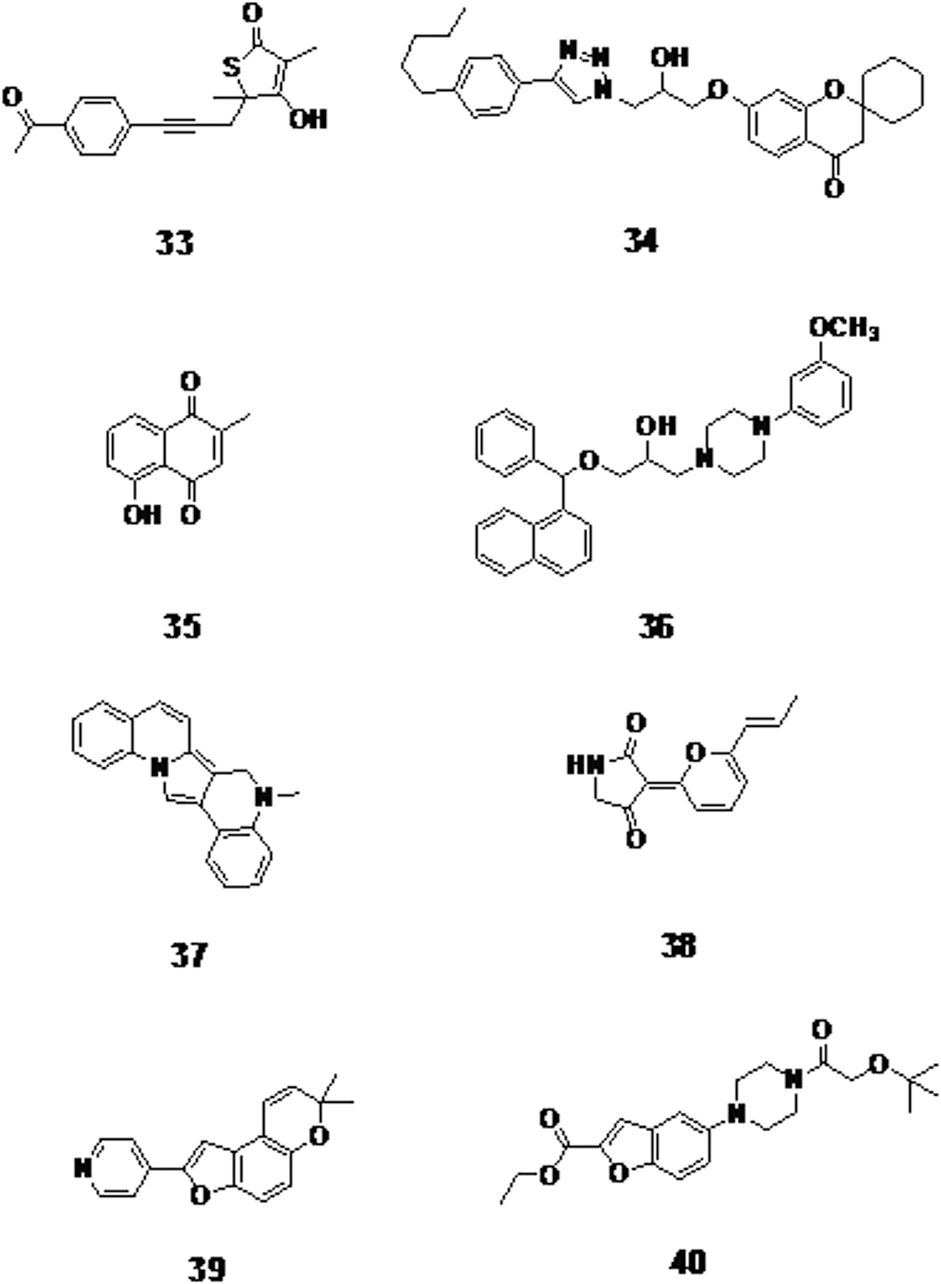
The 4-acetyl-phenyl analogue
Pyrrolodiquinoline (compound 37, Fig. 9), a compound synthesized based on vermelhotin's structure (compound 38, Fig. 9), exhibited a higher potency than vermelhotin against MDR-TB strains with a MIC range of 0.3–6.2 μg/mL. 76 4-Pyridyl derivatization of furo[3,2-f]chromene (compound 39, Fig. 9) showed increased activity against M. tuberculosis strains by fourfold with a MIC value of 2.5 μg/mL. 77 Benzofuran modified with various substituents exhibited a bactericidal effect by inhibiting mycobacterial DNA gyrase B. Compound 40 (Fig. 9) containing a piperidine linker has the most activity with low M. tuberculosis DNA gyrase activity (IC50 0.81 μM, MIC 9.18 μM). 78
Terpenes
Inhibition of class II diterpene cyclase of M. tuberculosis strains using transition state analogues of diterpenes (compound 41, Fig. 10) showed a remarkable activity with a strong binding. 79 Edaxadiene (compound 42, Fig. 10), a mycobacterial diterpene, which is important for endosomal progression inhibition, has potential for targeting the enzymes included in its synthetic pathway. 80,81 Targeting the diterpene biosynthetic pathway is a promising approach for discovery against novel bacterial synthases for anti-TB chemotherapy. 82
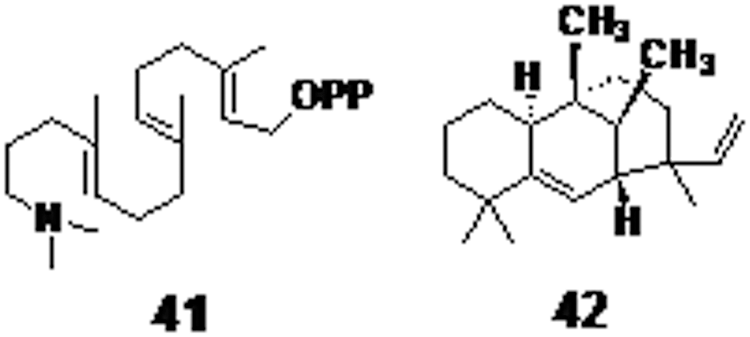
A transition-state analogue diterpene
Miscellaneous
The methylerythritol phosphate (MEP) pathway, which is essential for isoprenoid synthesis, is a promising approach for antitubercular activity, since it is vital for mycobacterial survival and humans lack some enzymes in this pathway. Fosmidomycin is a novel drug and acts by inhibition of the second enzyme involved in MEP synthesis, and had reached clinical trials. 83,84 3,6-Dialkyl-4-hydroxy-pyran-2-one is a hepatotoxic constituent of Kava extract and its analogues were synthesized and tested against M. tuberculosis. Compound 43 (Fig. 11) was found to have modest activity against M. tuberculosis with a MIC value of 170 μM. 85,86
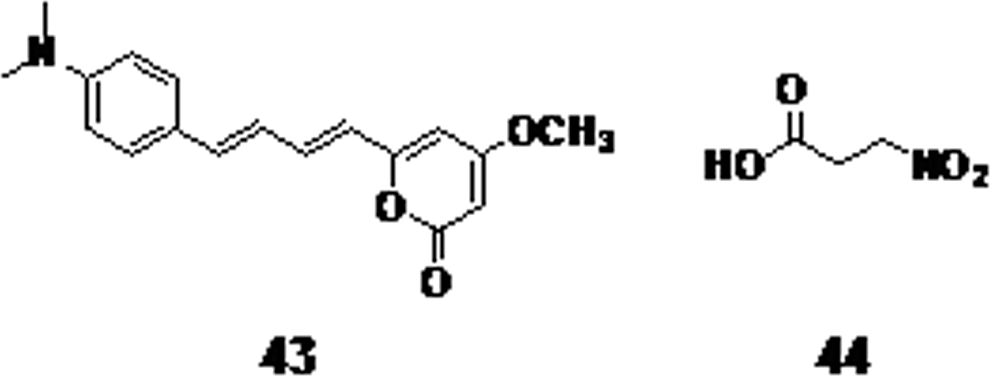
The Kava extract constituent 3,6-dialkyl-4-hydroxy-pyran-2-one
The metabolite from endophytic fungi, 3-nitropropionic acid (compound 44, Fig. 11), exhibited potent antimycobacterial activity with a MIC value of 3.3 μM. 87 The neurotoxic agent 3-nitropropionic acid has low cytotoxic activity against the Vero cell line, which makes it a potential lead for TB therapy.
Conclusion
TB is a leading cause of death among the infectious diseases. The current anti-TB therapy suffers from several pitfalls, including lengthy and complex dosing regimens that lead to patient noncompliance, which has no doubt contributed to the emergence of drug-resistant strains. Nature, since the beginning of time, has evolved to produce natural products from many sources, including bacteria, plants, and marine life. However, natural products need optimization to overcome the difficulties associated with their use. 28,88 The generation of semisynthetic analogues derived from natural product scaffolds has been a successful approach to optimize pharmacological properties along with reducing negative liabilities. 28,88 Moreover, recent advances in drug delivery have substantially improved the chances of developing natural products as antitubercular drugs. Clinical use of natural products has been limited by delivery difficulties since many of them are complex lipophilic molecules and have poor bioavailability. Development of novel drug delivery techniques like nanoparticles, liposomes, and amorphous dispersions can provide a suitable delivery platform for these natural products showing potent anti-TB activity. 89 –95 Therefore, optimization of specific natural product scaffolds using medicinal chemistry and formulation development should produce lead compounds with clinical utility as anti-TB agents.
Footnotes
Acknowledgment
The author acknowledges the School of Pharmacy and Health Professions, and the School of Medicine, Creighton University.
Disclosure Statement
No competing financial interests exist.