Dominguez-Bello MG, De Jesus-Laboy KM, Shen N, Cox LM, Amir A, Gonzalez A, Bokulich NA, Song SJ, Hoashi M, Rivera-Vinas JI, Mendez K, Knight R, Clemente JC: Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer.Nat Med2016;22:250–253.
Abstract: Exposure of newborns to the maternal vaginal microbiota is interrupted with cesarean birthing. Babies delivered by cesarean section (C-section) acquire a microbiota that differs from that of vaginally delivered infants, and C-section delivery has been associated with increased risk for immune and metabolic disorders. Here we conducted a pilot study in which infants delivered by C-section were exposed to maternal vaginal fluids at birth. Similarly to vaginally delivered babies, the gut, oral and skin bacterial communities of these newborns during the first 30 d of life was enriched in vaginal bacteria—which were underrepresented in unexposed C-section–delivered infants—and the microbiome similarity to those of vaginally delivered infants was greater in oral and skin samples than in anal samples. Although the long-term health consequences of restoring the microbiota of C-section–delivered infants remain unclear, our results demonstrate that vaginal microbes can be partially restored at birth in C-section–delivered babies.
Commentary:By now, it is well established that newborn babies stand to benefit long term from exposure to commensal microorganisms during the birth process, as establishment of complete microbiota helps drive immune development and metabolic programming. Conversely, incomplete or skewed microbial colonization at birth has been associated with long-term effects on human metabolism or impaired immune development. Further, the lack of complete microbiota in babies born through a cesarean section, relative to vaginally delivered babies, has been documented. In an effort to correct for this deficiency, Dominguez-Bello and colleagues evaluated a microbiota restoration procedure, called vaginal microbial transfer, which involved incubating sterile gauze in the vagina of healthy mothers prior to them giving birth through a cesarean section and, within the first 2 min of birth, swabbing the newborns with the gauze, covering their mouth, face, and the rest of their bodies (see
figure
). The study included a total of 18 babies, of whom 7 were born vaginally and the remaining 11 through scheduled C-section, with 4 of the 11 receiving the above maternal vaginal fluid treatment. The team noted that microbial colonization of the various vaginal fluid application sites in the newborns occurred quickly, within days. The authors further noted that bacterial source tracking of the infant microbiome revealed that the microbiomes of the four C-section–delivered infants exposed to vaginal fluids resembled those of vaginally delivered infants, and that Lactobacillus species were detected in anal samples from exposed infants, matching their vaginally delivered counterparts, while those newborns that were not exposed to vaginal fluids lacked this key component of human gut microbiota. There were no reports of negative reactions associated with the procedure. The present study provides an excellent proof of principle for this extremely simple procedure. However, a much larger study size combined with monitoring the cohort for a longer period will likely be needed in order to ascertain benefit. Contributed by Anton Simeonov.
Restoring the maternal microbiota in infants born by C-section. (a) Infants born by C-section were swabbed with a gauze that was incubated in the maternal vagina 60 min before the C-section. All mothers delivering by C-section received antibiotics (ABX) as part of standard-of-care treatment (top). The gauze was extracted before the procedure, kept in a sterile (middle) container and used to swab the newborn within the first 1–3 min after birth, starting with the mouth, then the face and the rest of the body (bottom). (d) Representative bacterial taxa enriched in infants with perinatal exposure to vaginal fluids during the first month of life. S24, Bacteroidales family S24-7 members. Error bars indicate mean ± s.d.
Targeting Mutant Kinases
Duong-Ly KC, Devarajan K, Liang S, Horiuchi KY, Wang Y, Ma H, Peterson JR: Kinase inhibitor profiling reveals unexpected opportunities to inhibit disease-associated mutant kinases.Cell Rep2016;14:772–781.
Abstract: Small-molecule kinase inhibitors have typically been designed to inhibit wild-type kinases rather than the mutant forms that frequently arise in diseases such as cancer. Mutations can have serious clinical implications by increasing kinase catalytic activity or conferring therapeutic resistance. To identify opportunities to repurpose inhibitors against disease-associated mutant kinases, we conducted a large-scale functional screen of 183 known kinase inhibitors against 76 recombinant mutant kinases. The results revealed lead compounds with activity against clinically important mutant kinases, including ALK, LRRK2, RET, and EGFR, as well as unexpected opportunities for repurposing FDA-approved kinase inhibitors as leads for additional indications. Furthermore, using T674I PDGFRα as an example, we show how single-dose screening data can provide predictive structure-activity data to guide subsequent inhibitor optimization. This study provides a resource for the development of inhibitors against numerous disease-associated mutant kinases and illustrates the potential of unbiased profiling as an approach to compound-centric inhibitor development.
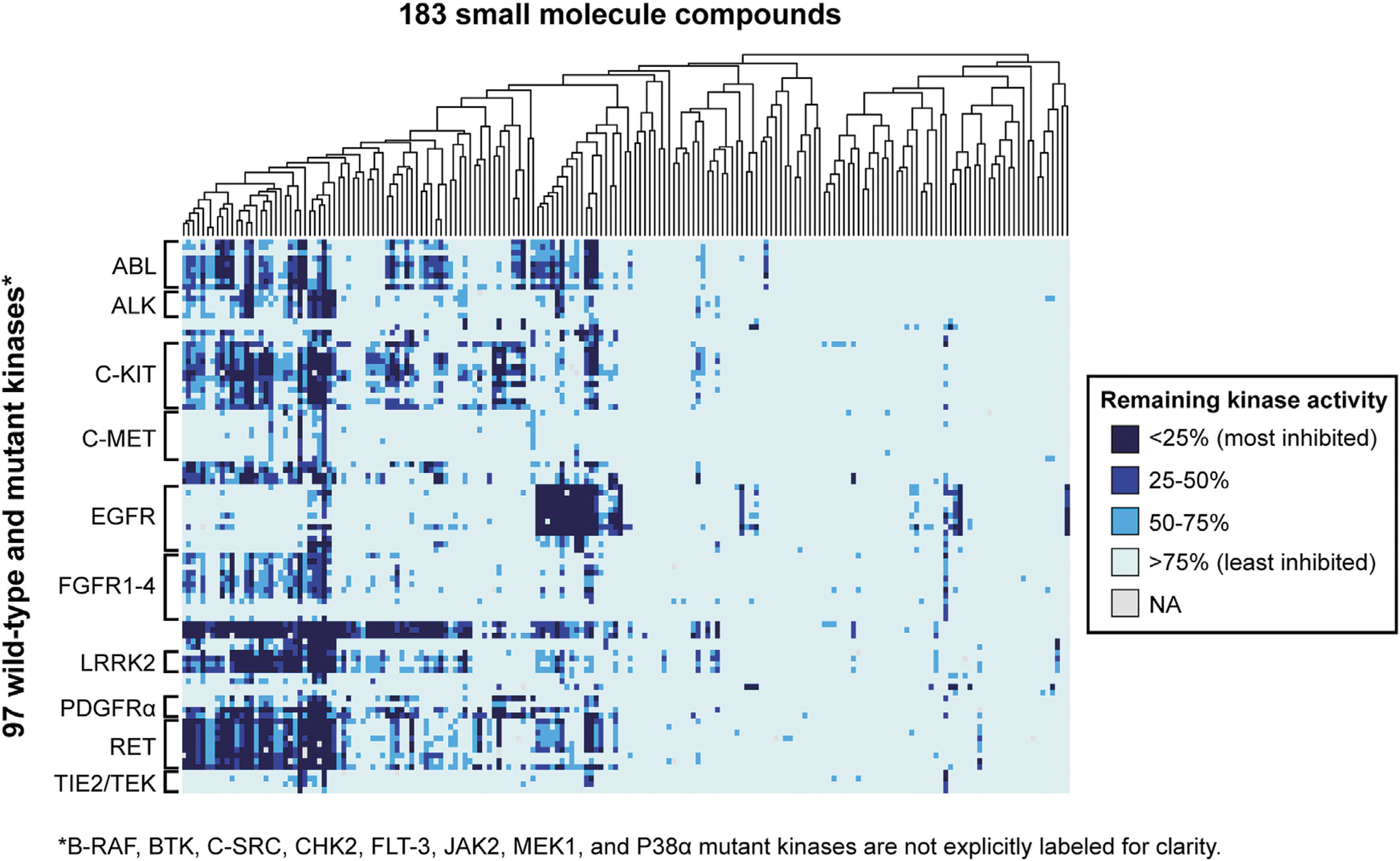
Commentary:Kinases have been the target of extensive research to identify drugs to treat a variety of diseases in which the wild-type kinase or a mutant kinase plays a crucial role. Kinase mutations frequently lead to an activated state where the kinase is always active and no longer tightly regulated. Considering resistance mutations are also important for kinase inhibitors. A frequent escape route is a mutation to the gatekeeper amino acid that blocks inhibitor binding. A variety of large screening panels have been developed that range from binding assays to enzyme assays, each with their unique pros and cons. Having a variety of assay formats allows hits to be confirmed in orthogonal assays. When a compound of interest is screened against a large kinase panel, unexpected “off-targets” can be identified. These off-targets can themselves be of interest for other uses, such as repurposing. Drug repurposing has been hotly pursued by many groups, and here the authors took 183 small molecules, including 12 FDA-approved drugs, and screened them against a panel of 76 mutant kinases (covering 21 wild-type kinases), where all but 8 of the mutations have been implicated in a human disorder. The assay utilized was a HotSpot radiometric filter-binding kinase assay. The authors screened at one concentration (500 nM) of compound and used 10 μM ATP for each kinase. Most kinases have a Km >10 μM, and most kinase inhibitors are ATP competitive, although there is much interest in developing (and some success with) inhibitors that target other pockets. It would be interesting to know the range of Km values for the mutants studied here to see how different that ratio of [ATP]/Km is across this panel given the fixed ATP concentration. It's expected that the majority of [ATP]/Km would be less than one, and thus the affinity for ATP competitive inhibitors would be quite accurately measured at this fixed concentration. The
figure
shows the % remaining kinase activity heatmap for the screen that includes data on the 21 cognate wild-type kinases from an earlier paper. It can be seen that some kinases are more frequently inhibited and that some compounds are not very selective across this panel. The authors show good concordance with their current data and previous large-scale screens, usually orthogonal assay formats. A compound known to target EGFR/ErbB2/ErbB4 was identified as having excellent selectivity for T790M mutant EGFR (gatekeeper mutation). They also identified several compounds with activity against the T641I imatinib-resistant mutation of PDGFRα. This study contains more mutant kinases than earlier large screens and helps to grow the database of compound-kinase interactions further. Contributed by Mindy I. Davis.
Large-scale screen of kinase/compound pairs. Shown is a heatmap representing the distribution of remaining kinase activities for kinase/compound pairs in this study as well as data for their wild-type cognates, as reported previously (Anastassiadis et al., 2011). The 183 compounds in the screen were subjected to hierarchical clustering, as shown by the dendrogram at the top. The 76 mutant kinases are grouped according to cognate wild-type kinase. Only kinase families with three or more members are labeled for clarity. A fully labeled version of this figure is provided in Figure S1; and the complete dataset is available in Table S2 and can be searched at the Kinase Inhibitor Resource (kir.fccc.edu).
Mitochondrial Nitric Oxide Probe
Kitamura K, Kawaguchi M, Ieda N, Miyata N, Nakagawa H: Visible light-controlled nitric oxide release from hindered nitrobenzene derivatives for specific modulation of mitochondrial dynamics.ACS Chem Biol2016 [Epub ahead of print]; DOI: 10.1021/acschembio.5b00962.
Abstract: Nitric oxide (NO) is a physiological signaling molecule, whose biological production is precisely regulated at the subcellular level. Here, we describe the design, synthesis, and evaluation of novel mitochondria-targeted NO releasers, Rol-DNB-mor and Rol-DNB-pyr, that are photocontrollable not only in the UV wavelength range but also in the biologically favorable visible wavelength range (530–590 nm). These caged NO compounds consist of a hindered nitrobenzene as the NO-releasing moiety and a rhodamine chromophore. Their NO-release properties were characterized by an electron spin resonance (ESR) spin trapping method and fluorometric analysis using NO probes, and their mitochondrial localization in live cells was confirmed by costaining. Furthermore, we demonstrated visible light control of mitochondrial fragmentation via activation of dynamin-related protein 1 (Drp1) by means of precisely controlled NO delivery into mitochondria of cultured HEK293 cells, utilizing Rol-DNB-pyr.
Commentary:The molecule nitric oxide (NO) is known as endothelium-derived relaxing factor (EDFR) due to its ability to relax blood vessels. NO also plays a key role in many cellular signaling events through nitrosylation of cysteine residues in proteins such as hemoglobin and activation soluble guanylate cyclase through binding to a heme group. This paper describes a fluorescent probe that binds to mitochondria and acts as a NO donor after brief exposure to either UV or non-toxic green-yellow light (530–590 nm). The rhodamine chromophore is known to localize to mitochondria, and the authors coupled this to a 2,6-dimethylnitrobenzene (DNB) moiety, which acts as the NO donor (see
figure
). Light absorption by rhodamine is thought to lead to a photo-induced isomerization of the sterically hindered DNB, resulting in homolytic cleavage of the phenolic oxygen–nitrogen bond and the release of NO. The authors use these probes to study mitochondrial dynamics in HEK293 cells. Mitochondria undergo both fusion and division, and this process may be dependent on the S-nitrosylation of proteins that regulate this process. Treatment of HEK293 cells expressing a GFP fusion to a mitochondrial enzyme with these probes followed by targeting the probe bound to mitochondria with a 562 nm laser resulted in increased mitochondrial fragmentation, which was prevented by an inhibitor of dynamin-related protein 1 (Drp1, a GTPase involved with mitochondria division). In addition, no mitochondria fragmentation was observed when rhodamine without the DNB group was used. This supports a role for activation of Drp1 by NO in mitochondria. The synthetic route to construct these probes is also provided in the paper. These mitochondrial-targeting NO probes should be useful tools for further studying the role of NO in mitochondrial function. Contributed by Doug Auld.
Molecular design of novel caged NO releasers. Rol-DNBmor (1) and Rol-DNB-pyr (2), which are visible-light-controllable and mitochondria-targeted.
Quantum Bret
Kumar M, Kovalski L, Broyles D, Hunt EA, Daftarian P, Dikici E, Daunert S, Deo SK: Design and development of high bioluminescent resonance energy transfer efficiency hybrid-imaging constructs.Anal Biochem2016;498:1–7.
Abstract: Here we describe the design and construction of an imaging construct with high bioluminescent resonance energy transfer (BRET) efficiency that is composed of multiple quantum dots (QDs; lem ¼ 655 nm) self-assembled onto a bioluminescent protein, Renilla luciferase (Rluc). This is facilitated by the streptavidine biotin interaction, allowing the facile formation of a hybrid-imaging construct (HIC) comprising up to six QDs (acceptor) grafted onto a light-emitting Rluc (donor) core. The resulting assembly of multiple acceptors surrounding a donor permits this construct to exhibit high resonance energy transfer efficiency (∼64.8%). The HIC was characterized using fluorescence excitation anisotropy measurements and high-resolution transmission electron microscopy. To demonstrate the application of our construct, a generation-5 (G5) polyamidoamine dendrimer (PAMAM) nanocarrier was loaded with our HIC for in vitro and in vivo imaging. We envision that this design of multiple acceptors and bioluminescent donor will lead to the development of new BRET-based systems useful in sensing, imaging, and other bioanalytical applications.
Commentary:Bioluminescent resonance energy transfer (BRET) has been applied to study protein–protein interactions in cells using Renilla luciferase (RLuc) and GFP donor/acceptor pairs. BRET can provide improved sensitivity because the excitation light arises from the enzymatic reaction of RLuc acting on a luciferin substrate therefore no external excitation light is needed, which prevents autofluorescence of cellular components. Reduced spectral overlap between RLuc and GFP emission spectra can be improved by using a blue-light shifted variant of RLuc called RLuc8. As well, the use of luciferases emitting bright blue light and red-light-emitting organic dyes as acceptors has been described (NanoBRET, Promega). In this paper, the authors describe a high efficiency BRET-based hybrid imaging construct (HIC) based on biotinylated RLuc and streptavidin linked quantum dots (QD; see
figure
). The emission of RLuc overlaps the broad excitation spectrum of the QD and results in near-infrared QD emission. The product of the RLuc enzyme reaction is coelenteramide, which remains bound to RLuc and is fluorescent. The stoichiometry of the HIC was determined to be 1:6 (RLuc:QD) by measuring the change in anisotropy of coelenteramide fluorescence in the presence of increasing concentrations QD streptavidin. This results in very high BRET efficiency. The HIC was applied as an in vivo imaging probe by encapsulating the HIC with a peptide, allowing targeting to antigen-presenting cells in the spleen of mice. Imaging on the IVIS Spectrum reader showed localization to spleen for QD emission, while emission from an RLuc targeted probe without the QD was not detectable. New imaging tools based on BRET through the application of nanostructures as described here should help in vivo imaging applications and could improve labeling of in vitro cell-based assays as well. Contributed by Doug Auld.
A simplified schematic representation of the imaging construct synthesis. Although enlarged slightly to show detail, the diameter of Rluc measures approximately half the length of the cylindrical quantum dots, with axial crystallographic dimensions of 8.92 × 5.1 × 7.4 nm for Rluc (PDB ID: 2PSJ) versus approximately 20 nm (Invitrogen) for the streptavidin-functionalized nanocrystals. Biotinylated Rluc (1 pmol) mixed with streptavidin-modified QDs (6 pmol) results in formation of the HIC.
Finding Better Drug–Drug Combinations Faster
Silva A, Lee BY, Clemens DL, Kee T, Ding X, Ho CM, Horwitz MA: Output-driven feedback system control platform optimizes combinatorial therapy of tuberculosis using a macrophage cell culture model.Proc Natl Acad Sci U S A2016, [Epub ahead of print]; DOI: 10/1073/pnas.1600812113.
Abstract: Tuberculosis (TB) remains a major global public health problem, and improved treatments are needed to shorten duration of therapy, decrease disease burden, improve compliance, and combat emergence of drug resistance. Ideally, the most effective regimen would be identified by a systematic and comprehensive combinatorial search of large numbers of TB drugs. However, optimization of regimens by standard methods is challenging, especially as the number of drugs increases, because of the extremely large number of drug–dose combinations requiring testing. Herein, we used an optimization platform, feedback system control (FSC) methodology, to identify improved drug–dose combinations for TB treatment using a fluorescence-based human macrophage cell culture model of TB, in which macrophages are infected with isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible green fluorescent protein (GFP)-expressing Mycobacterium tuberculosis (Mtb). On the basis of only a single screening test and three iterations, we identified highly efficacious three- and four-drug combinations. To verify the efficacy of these combinations, we further evaluated them using a methodologically independent assay for intramacrophage killing of Mtb; the optimized combinations showed greater efficacy than the current standard TB drug regimen. Surprisingly, all top three- and four-drug optimized regimens included the third-line drug clofazimine, and none included the first-line drugs isoniazid and rifampin, which had insignificant or antagonistic impacts on efficacy. Because top regimens also did not include a fluoroquinolone or aminoglycoside, they are potentially of use for treating many cases of multidrug- and extensively drug-resistant TB. Our study shows the power of an FSC platform to identify promising previously unidentified drug–dose combinations for treatment of TB.
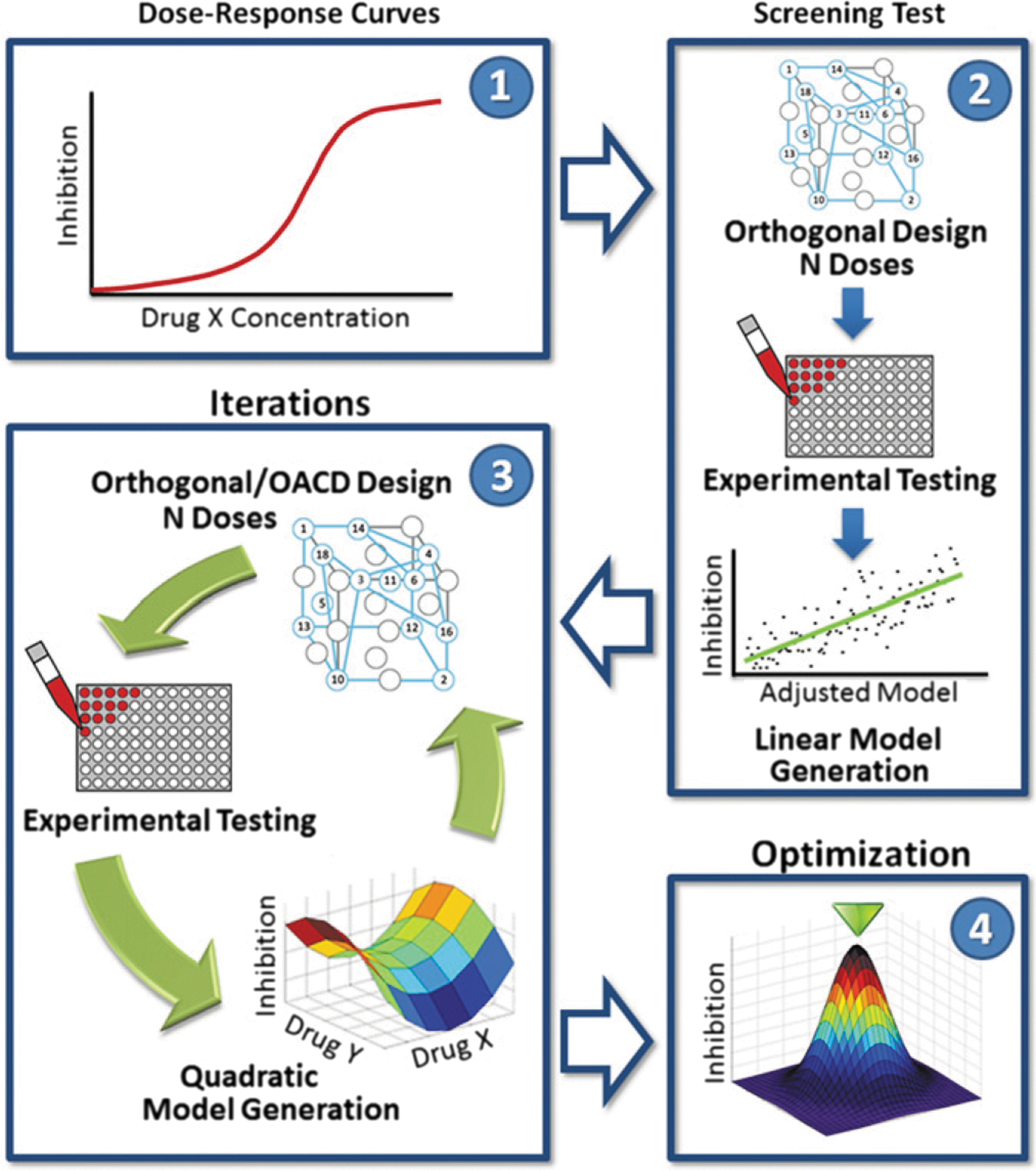
Commentary:Combinations of two or more drugs frequently offer therapeutic benefits due to improved potency and safety, and decreased possibility of developing resistance to therapy. However, it remains extremely challenging to identify potent drug combinations by trial and error because of the large number of possible candidate combinations that need to be tested. For example, even with the recent dramatic advances in screening technologies (aided, for example, by the establishment of the acoustic dispense platforms), it still remains impossible to screen a relatively modest set of a dozen drugs at half a dozen concentrations efficiently, as the total number of combinations already exceeds a billion. To tackle this problem, the team resorted to building a multiphase process (see
figure
) based on stochastic search algorithms, which used biased random walks of the input states (drug concentrations) to maximize the output (a chosen biological activity measured through an assay). First, a single-agent screen is conducted to determine the concentration range of interest for each drug. The second phase, called the screening test, uses a two-level orthogonal array experimental design to construct a first-order linear model of the relationship between drug–dose combinations and bioactivity whose application yields the drugs with the greatest impact on bioactivity. In the third phase, the selected drugs are tested in an iterative fashion to yield a quadratic model of the relationship between drug–dose combinations and bioactivity. In the last phase, in-depth analysis of the finalized second-order quadratic model is used to construct a model surface of the relationship between drug–dose combinations and bioactivity, ultimately yielding the final drug combinations and corresponding dose ratios. The team used an IPTG-inducible GFP expression system to evaluate the antimicrobial efficacy against Mycobacterium tuberculosis of drug combinations in an in vitro human macrophage culture system. The fluorescent readout reported on inhibition of Mtb metabolic activity and could be taken to a high throughput. Fourteen drugs were tested at five doses each, an experiment that if conducted using the traditional all-versus-all strategy would have required the setup of more than 6 billion tests. Using the present approach allowed the elimination of low-performing drugs very rapidly. Interestingly, both isoniazid (INH) and rifampicin (RIF) were eliminated from the optimized regimens because of a small effect on efficacy (INH) or antagonistic interactions with other drugs (RIF), in agreement with previous observations of antagonistic interactions of INH and RIF with other first-line tuberculosis drugs. While the method may seem complicated as described, automation of the steps involved offers the promise to make this platform accessible to a wide range of users. Contributed by Anton Simeonov.
FSC.II schematic. The diagram depicts the FSC.II technique loop used for drug optimization with the fluorescence-based assay. The FSC.II methodology has four phases: (i) dose–response curve established for each drug (1), (ii) screening test with two-level orthogonal array design (results used to construct a first-order linear model; 2), (iii) iterations testing drug combinations based on orthogonal or orthogonal array central composite design (OACD) design (results used to construct a second-order quadratic model and surface model; 3); and (iv) optimization of the drug combinations and drug ratios from the final surface model constructed in phase 3.