Mercorelli B, Luganini A, Nannetti G, Tabarrini O, Palù G, Gribaudo G, Loregian A: Drug repurposing approach identifies inhibitors of the prototypic viral transcription factor IE2 that block human cytomegalovirus replication.Cell Chem Biol2016;23:340–351.
Abstract: New targets for antiviral strategies are needed against human cytomegalovirus (HCMV), a major human pathogen. A cell-based screen aimed at finding inhibitors of the viral transcription factor Immediate-Early 2 (IE2) was performed in HCMV-infected cells expressing EGFP under the control of an IE2-inducible viral promoter. Screening of a library of bioactive small molecules led to the identification of several compounds able to inhibit EGFP expression and also HCMV replication with potency in the low-micromolar range. Follow-up studies with four selected hits indicated that they all block viral DNA synthesis as well as viral Early and Late gene expression. Furthermore, mechanistic studies confirmed that the compounds specifically act via inhibition of IE2 transactivating activity, thus blocking viral Early gene expression and the progression of virus replication. These results provide proof of concept for identifying small molecules that modulate the activity of a microbial transcription factor to control pathogen replication.
Commentary:Human cytomegalovirus (HCMV) is a beta-herpesvirus that affects more than half of the population worldwide. The consequences of infection are greatest for HIV/AIDS patients, transplant recipients, and those who are immunocompromised. To date, the drugs to treat HCMV target the viral DNA polymerase. As resistance to the current anti-DNA polymerase drugs emerges, it is important to have new drugs available that utilize alternative mechanisms of action. In this paper, the authors utilized drug repurposing to identify inhibitors of the viral transcription factors (vTFs). vTFs are important in activating the expression of viral genes but also in reprogramming host pathways to benefit the pathogen. They developed a cell-based high-throughput screen (HTS) assay to identify inhibitors of the immediate-early 2 (IE2) TF in HCMV-infected cells that had been engineered to express the reporter EGFP stably driven by the viral promoter for the DNA polymerase. This promoter is important for the activation of both early and late genes and the progression of HCMV replication. The cell line, cell density, and multiplicity of infection were all optimized to yield a robust assay with a Z′ of 0.79. The library of 2,320 bioactive compounds (Spectrum collection) was tested at 10 μM, and 125 compounds were identified that decreased fluorescence by >50%. As general cytotoxicity would also lead to a decrease in fluorescence, compounds that lead to cell detachment prior to lysis were excluded. With this triage and following confirmation, 38 compounds were identified that inhibited EGFP expression. The assay was able to detect previously identified HCMV inhibitors, thereby providing validation to the approach. It is important to have orthogonal assays to validate initial HTS hits, and here the authors showed that four selected hits (deguelin, nitazoxanide [NTZ], thioguanosine, and alexidine dihydrochloride) were able to block viral DNA synthesis and early and late gene expression. Additionally, they developed assays with distinct detection modalities (fluorescence and luminescence), which should aid in the identification of modality-specific compound interference (see
figure
). These four compounds were tested in additional strains and shown to be broad-spectrum inhibitors of HCMV. Additionally, these compounds blocked the IE2 trans-activating activity, which leads to blocking of the viral early gene expression and viral replication. NTZ is an FDA-approved drug that had initially been developed as an anti-parasitic agent against protozoan and helminth infections. It has more recently been found to have broad antiviral activity against 20 different viruses, not including HCMV, which had not been previously reported. This drug is currently in Phase 3 clinical trials for the treatment of influenza. It will be interesting to see whether this drug can be further repurposed to treat HCMV. Contributed by Mindy I. Davis.
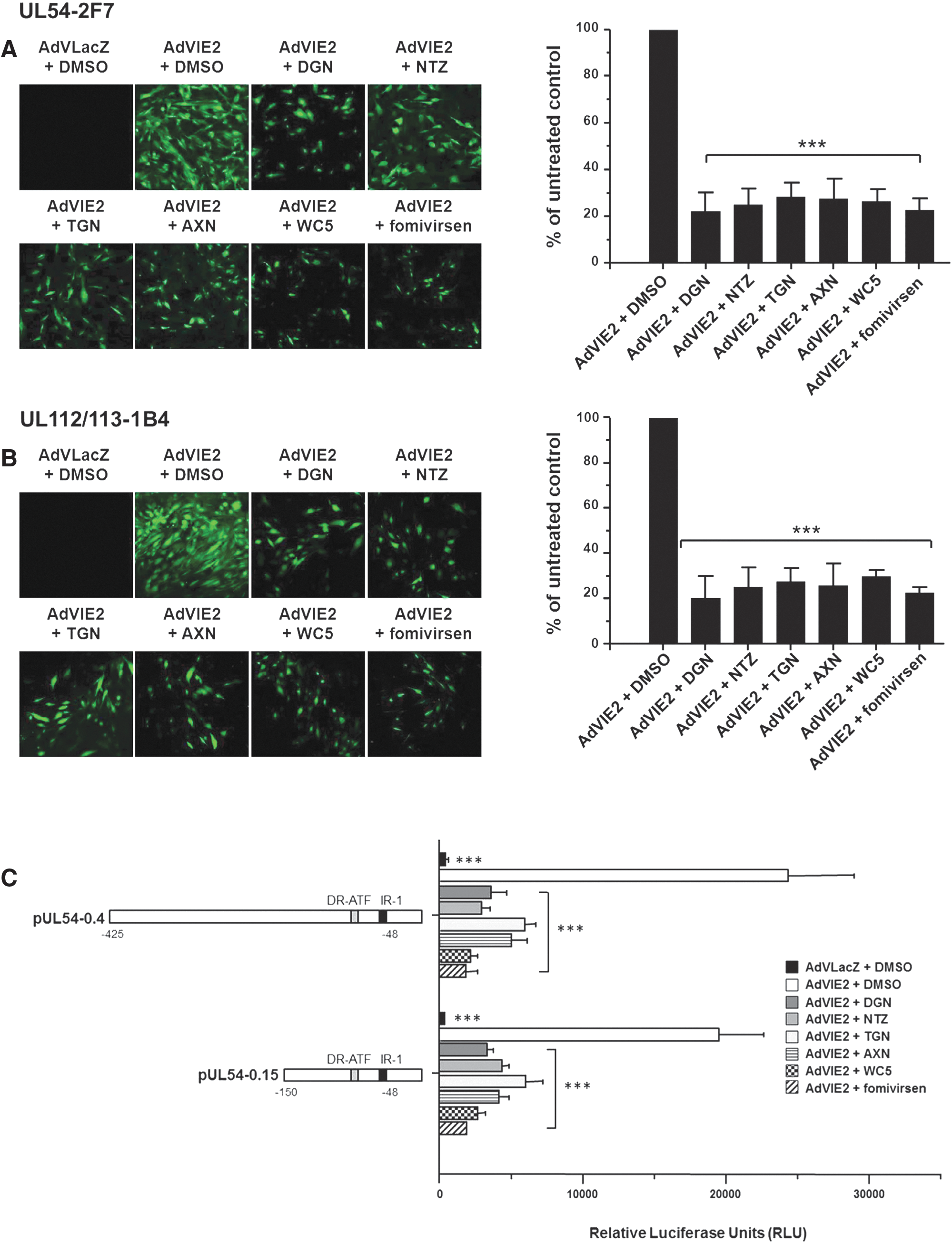
Hit compounds block the IE2-dependent transactivation of HCMV E promoters. (A and B) UL54-2F7 (A) or UL112-113-1B4 (B) cells were transduced with AdVIE2 or with AdVLacZ and then treated with test compounds or DMSO as a control. Representative confocal microscopy images (EGFP) acquired at 72 hr post-transduction (p.t.) are shown (left). At the same time, confocal microscopy was used to quantify the percentage of EGFP-expressing cells in 20 different fields. DMSO-treated transduced cells were considered 100% (right). (C) HFFs were transfected with luciferase reporter plasmids pUL54–0.4 or pUL54–0.15 and after 24 hr were transduced with AdVIE2 or AdVLacZ. Then, cells were treated with test compounds or DMSO as a control. At 48 hr p.t., luciferase expression was determined and the transcriptional activity was expressed as relative luciferase units. Data shown are the means ± SD of three experiments performed in duplicate. The results presented in all panels were analyzed with Bonferroni post-test correction for multiple comparisons. ***p < 0.001 versus calibrator sample (AdVIE2 + DMSO).
Imaging Phosphoinositides
Mondal S, Rakshit A, Pal S, Datta A: Cell permeable ratiometric fluorescent sensors for imaging phosphoinositidesACS Chem Biol2016, in press. DOI: 10.1021/acschembio.6b00067.
Abstract: Phosphoinositides are critical cell-signal mediators present on the plasma membrane. The dynamic change of phosphoinositide concentrations on the membrane including clustering and declustering mediates signal transduction. The importance of phosphoinositides is scored by the fact that they participate in almost all cell-signaling events, and a defect in phosphoinositide metabolism is linked to multiple diseases including cancer, bipolar disorder, and type-2 diabetes. Optical sensors for visualizing phosphoinositide distribution can provide information on phosphoinositide dynamics. This exercise will ultimately afford a handle into understanding and manipulating cell-signaling processes. The major requirement in phosphoinositide sensor development is a selective, cell permeable probe that can quantify phosphoinositides. To address this requirement, we have developed short peptide-based ratiometric fluorescent sensors for imaging phosphoinositides. The sensors afford a selective response toward two crucial signaling phosphoinositides, phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) and phosphatidylinositol-4-phosphate (PI4P), over other anionic membrane phospholipids and soluble inositol phosphates. Dissociation constant values indicate up to 4 times higher probe affinity toward PI(4,5)P2 when compared to PI4P. Significantly, the sensors are readily cell-permeable and enter cells within 15 min of incubation as indicated by multiphoton excitation confocal microscopy. Furthermore, the sensors light up signaling phosphoinositides present both on the cell membrane and on organelle membranes near the perinuclear space, opening avenues for quantifying and monitoring phosphoinositide signaling.
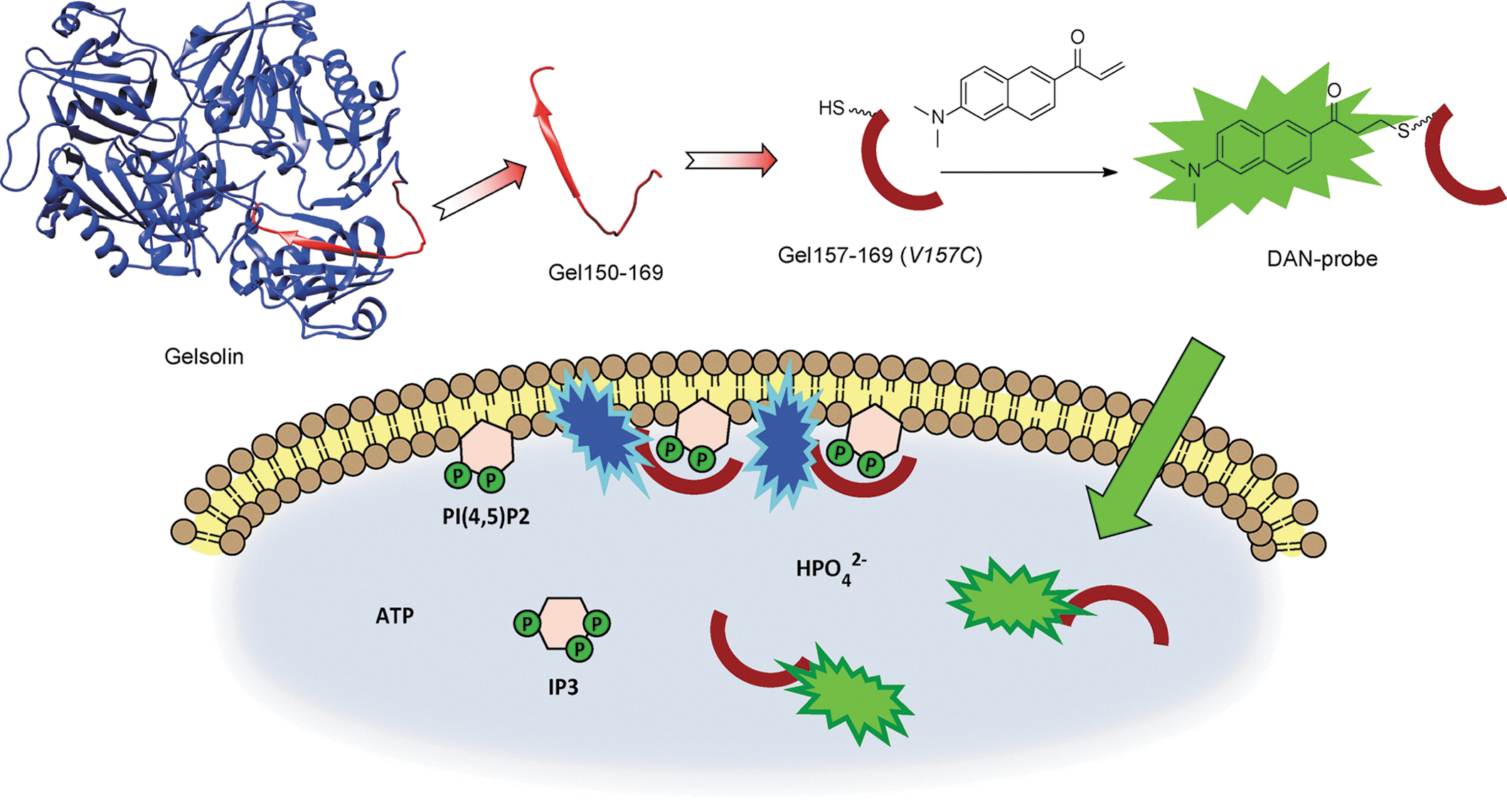
Commentary:Assays for phosphoinositides phosphates (PIP) have been constructed using PIP binding domains in cell lysates or expressed in cells. This paper describes a cell penetrating reporter peptide, which can act as cellular sensor of phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) (see
figure
). The peptide is based the protein gelsolin which has 20aa sequence known to bind PIP with particular selectivity toward PI(4,5)P2. To engineer a fluorophore into this sequence, a cysteine residue was introduced at Val59, which is a region of the peptide that interacts with membranes. A 2-dimethylamino-6-acyl-naphthalene (DAN) fluorophore was then coupled to Cys59. The DAN fluorophore undergoes a 60–70 nm shift in emission spectra (a change from green to blue emission) upon binding to membranes. Therefore, a ratiometric assay could be constructed by measuring the fluorescence of the PIP bound versus the unbound peptide (F450/F520 values were measured). Furthermore, DAN can be excited using two-photon microscopes using 780 nm light, which enables imaging of biological samples without confounding absorbance or autofluorescence from the sample. The affinity of the PIP sensor peptide was found to be in the 4–6 μM range for PI(4,5)P2. In vitro studies using small unilamellar vesicles (SUVs) were initially performed to confirm that the PIP sensor peptide responded to different concentrations of PI(4,5)P2 content in membranes. A shorter DAN-13aa sensor was found to be capable of measuring PI(4,5)P2 localization in HEK293 cells. Variation of this peptide could improve the selectivity of this probe and potentially expand to other PIP species. Contributed by Doug Auld.
Ratiometric sensing scheme for phosphoinositide imaging by using cell-permeable 2-dimethylamino-6-acyl-naphthalene (DAN)-labeled peptide-based sensors.
Antiviral Drug Resistance of Varicella-Zoster Virus
Perrier, M, Désiré N, Deback C, Agut H, Boutolleau D, Burrel S: Complementary assays for monitoring susceptibility of varicella-zoster virus resistance to antivirals.J Virol Methods2016;233:10–14.
Abstract: The emergence of varicella-zoster virus (VZV) resistance to current antivirals as acyclovir (ACV) constitutes a hindrance to antiviral treatment effectiveness of VZV infections, especially in immunocompromised patients. The molecular mechanisms of VZV resistance reported so far rely on the presence of mutations within thymidine kinase (TK, ORF36) and DNA polymerase (ORF28) viral genes. The aim of this work was to develop reliable and complementary diagnostic methods to detect VZV antiviral resistance: (i) a genotypic assay based on TK and DNA polymerase genes sequencing, (ii) a plaque reduction assay to determine antiviral 50% effective concentrations, and (iii) a functional assay to evaluate in vitro phosphorylation activity of recombinant TKs. As a whole, this study included the analysis of 21 VZV clinical isolates and 62 biological samples from patients experiencing VZV infection. Genetic analysis revealed 3 and 9 new amino acid changes that have not been previously described within the highly conserved TK and DNA polymerase, respectively. Then, VZV isolates bearing newly identified mutations considered as natural polymorphisms were characterized as susceptible to ACV using plaque-reduction assay in MeWo cells. In parallel, the impact of TK changes on ACV phosphorylation activity was examined using a nonradioactive in vitro enzymatic assay.
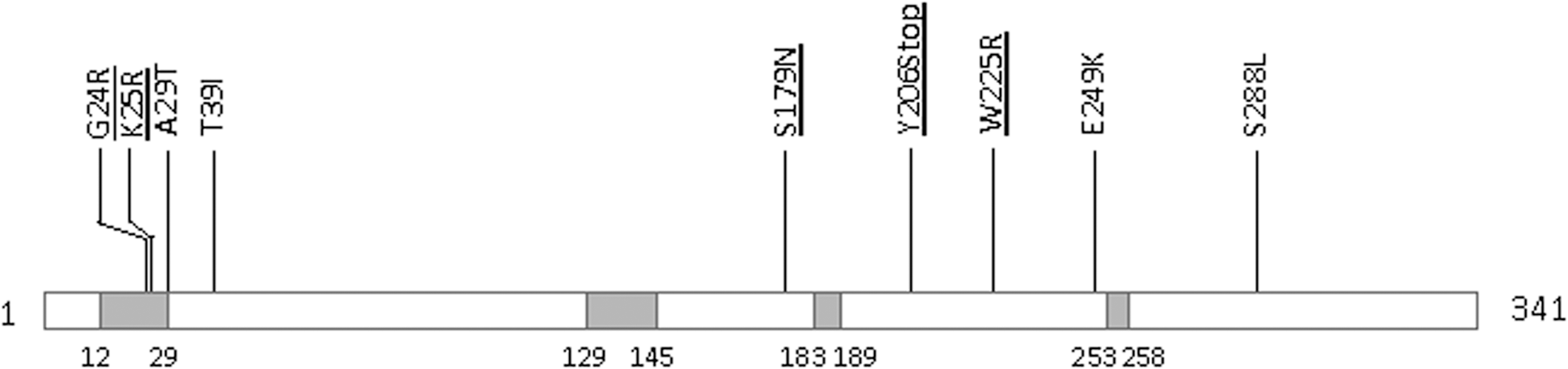
Commentary:Varicella-zoster virus (VZV) is a common herpesvirus that has the greatest impact on HIV/AIDS patients, transplant recipients, and individuals who are immunocompromised. Resistance to current antiviral drugs, such as acyclovir, is emerging, so it is important to develop assays to identify resistance as well as next-generation antivirals. The authors developed assays to monitor VZV resistance to antivirals, identified new polymorphisms within the thymidine kinase (TK) and DNA polymerase, and evaluated the in vitro kinase activity of TK. TK and DNA polymerase have been identified as the sites of mutations in antiviral resistant VZV. The antiviral acyclovir needs to be activated by TK in the cell prior to targeting the viral DNA polymerase, leading to chain termination. The authors developed three main orthogonal assay types: genotypic, plaque reduction, and functional assays. The authors were able to identify novel natural polymorphisms that are important to distinguish from mutations that confer resistance. Their system allowed sequencing of the entire TK and DNA polymerase genes. The genotypic assay utilized sequencing viral DNA (84 strains) that was extracted from clinical samples and viral stocks. The mutations to date that have been identified within VZV TK are shown in the
figure
. The plaque reduction assay involved plating MeWo cells (melanoma cell line) in 24-well dishes, infecting them with VZV isolates. After 1 h, the compounds were added and incubated for 5 days, at which time the cells were fixed and stained with crystal violet, allowing for plaque counting. The functional assay involved utilizing ADP-Glo to couple the ADP produced to a luminescent readout. The protocol used herein for VZV was from a protocol for HSV published previously with a modification of the reaction time to 60 min. This assay confirmed that three mutated TKs lead to a decrease in TK activity and the natural polymorphism S179N did not. This decrease in TK activity in the three mutants would be expected to be associated with resistance to acyclovir.One of the TK mutants (A29T) is outside of the ATP-binding pocket and therefore may not impact the kinase activity. Indeed, it was sensitive to acyclovir and is likely a natural polymorphism. It is important to have tests ready both to identify mutations and then to determine their functional significance, if any. The assays described herein provide a good starting point for assessing VZV antiviral resistance. Contributed by Mindy I. Davis.
Map of mutation identified within VZV TK. Conserved regions are represented by gray boxes. The positions (codon numbers) of these conserved regions are indicated under each box. VZV TK is a 341 amino acid protein, encoded by ORF36, containing two functional domains: an ATP binding site (codons 12–29) and a nucleoside binding site (codons 129–145) (Morfin et al., 1999). Investigated mutations using the TK functional assay described in this study are underlined.
Screening with Broccoli
Svensen N, Jaffrey SR: Fluorescent RNA aptamers as a tool to study RNA-modifying enzymes.Cell Chem Biol2016;23:415–425.
Abstract: RNA-modifying enzymes are difficult to assay due to the absence of fluorometric substrates. Here we show that the Broccoli, a previously reported fluorescent RNA-dye complex, can be modified to contain N6-methyladenosine, a prevalent mRNA base modification. Methylated Broccoli is nonfluorescent but, upon demethylation by the RNA demethylases fat mass and obesity-associated protein (FTO) or ALKBH5, it binds and activates the fluorescence of its cognate fluorophore. We describe a high-throughput screen (HTS) for FTO inhibitors using the fluorogenic methylated Broccoli substrate HTS assay, which performs robustly with a Z′ factor >0.8 in the LOPAC1280 library. This allowed the identification of novel high-affinity FTO inhibitors. Several of these compounds were selective for FTO over the related demethylase, ALKBH5, and increase methylation of endogenous FTO target mRNAs in cells. Lastly, we show that Broccoli can be modified to contain other base modifications, suggesting that this approach could be generally applicable for assaying diverse RNA-modifying enzymes.
Commentary:High-throughput assays for RNA/DNA modifying enzymes are rare due to the difficulty in developing sensitive reporters for this class of enzymes. This paper describes an assay for an RNA demethylase known as fat mass and obesity-associated protein (FTO). The assay is based on 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI), which can bind and become fluorescent upon demethylation of a m6A-containing RNA. The RNA is based on a so-called “Broccoli” RNA aptamer, which binds and activates fluorescence of DFHBI only when the RNA is present in a demethylated form. DFHBI binds within a pocket formed by a G-quadruplex and a UAU base triple (see
figure
). The paper employed a modified DFHBI (DFHBI-1T), which has fluorescent properties that are more easily accommodated on standard microtiter plate readers. Nucleotide base modifications would be expected to disrupt either the G-qaudruplex or the base triple, and prevent binding of DFHBI. Modified bases were tested at every position, which identified a RNA with nine m6A residues as reporter of FTO activity (m6A9 Broccoli). To obtain an RNA requiring fewer turnovers and thus act as a more sensitive reporter of FTO activity, further work was performed to examine the critical m6A residues in the m6A9 Broccoli reporter. This showed that substitution of adenosine below the G-quadruplex was not allowed, and identified a RNA with two less adenosine substitutions, which showed efficient activation of DFHBI fluorescence (m6A7 Broccoli). One issue with this class of enzymes is the low turnover rate, and the authors determined a kcat = 0.30 min–1 FTO acting on m6A7 Broccoli. The assay used for HTS of the LOPAC1280 library employed 7.5 μM m6A7 Broccoli substrate and 260 nM enzyme, which yielded a Z′ > 0.8. Several classes of compounds were identified, which were tested in counterscreens employing both methylated and unmethylated m6A7 Broccoli RNA alone to identify compounds that may directly displace the dye. Furthermore, compounds were tested against a different demethylase, ALKBH5, to identify selective compounds. One series included rhein analogs, a known class of FTO inhibitors. Compounds were also tested for inhibition of target transcripts in cells and for cytotoxicity. The m6A7 Broccoli reporter and the methods described in this paper should be a useful resource for those studying RNA/protein interactions. Contributed by Doug Auld.
Application of m6A-modified Broccoli as a fluorometric substrate in the FTO assay. (A) Core structure of the Broccoli-DFHBI-1T complex. DFHBI-1T (green) binds between the G-quadruplex (blue squares) and a base triple (purple squares). Grey squares indicate a tetrad below the G-quadruplexes. (B) Effect of RNA modifications on Broccoli-DFHBI-1T fluorescence. To test whether RNA modifications affect the ability of Broccoli to bind and activate the fluorescence of DFHBI-1T, Broccoli was synthesized with the modified nucleotide in place of the wild-type base at every position. Data were normalized by background subtraction (DFHBI-1T buffer). mn denotes base methylation in the n position of the ribonucleobase. 2Om denotes 20-O-methylation of the ribose. (C) Scheme of FTO-catalyzed N6-methyladenosine (m6A) to adenosine-RNA conversion. m6A is highlighted in red. (D) m6A-Broccoli is a fluorometric substrate for the FTO enzyme. The extent of FTO-catalyzedm6A-Broccoli demethylation was measured using the fluorescence resulting from the A-Broccoli that is generated.m6A9-Broccoli (red line) orm6A7-Broccoli (blue line) were treated with FTO in a standard FTO assay. The amount of A9-Broccoli-DFHBI-1T (purple line) and A7-Broccoli-DFHBI-1T (orange line) fluorescence generated was measured following incubation with DFHBI-1T. Data were normalized by background subtraction (DFHBI-1T buffer). (E) m6A7-Broccoli titrated with FTO. The extent of m6A7-Broccoli demethylation as a result of increasing FTO concentration was measured using the fluorescence resulting from A7-Broccoli generation as readout. This assay was carried out as in (D) with increasing FTO concentration. Data were normalized to A7-Broccoli control. (F) FTO demethylation time course. The extent of FTO-catalyzed m6A7-Broccoli demethylation as a result of increasing time was measured using the fluorescence resulting from A7-Broccoli generation as the readout carried out as in (E) measuring the A7-Broccoli-DFHBI-1T fluorescence generated over time. (G) Inhibition of FTO in the m6A7-Broccoli assay. Titration the m6A7-Broccoli FTO reaction with four known inhibitors: rhein (red points), meclofenamic acid (Meclofen; blue points), FG4592 (purple points) and 2-4-pyridinedicarboxylate (2-4-pyr; orange points). This assay was carried out as in (E) with increasing inhibitor concentration. Data were normalized to the negative control (vehicle). n = 3. Error bars = SD.
Going after Tumor-Specific Idiotypes with Small Peptides
Torchia J, Weiskopf K, Levy R: Targeting lymphoma with precision using semisynthetic anti-idiotype peptibodies. Proc Natl Acad Sci U S A2016, in press. DOI: 10/1073/pnas.1603335113.
Abstract: B-cell lymphomas express a functionally active and truly tumor-specific cell-surface product, the variable region of the B-cell receptor (BCR), otherwise known as idiotype. The tumor idiotype differs, however, from patient to patient, making it a technical challenge to exploit for therapy. We have developed a method of targeting idiotype by using a semisynthetic personalized therapeutic that is more practical to produce on a patient-by-patient basis than monoclonal antibodies. In this method, a small peptide with affinity for a tumor idiotype is identified by screening a library, chemically synthesized, and then affixed to the amino terminus of a premade IgG Fc protein. We demonstrate that the resultant semisynthetic anti-idiotype peptibodies kill tumor cells in vitro with specificity, trigger tumor cell phagocytosis by macrophages, and efficiently clear human lymphoma in a murine xenograft model. This method could be used to target tumor with true precision on a personalized basis.
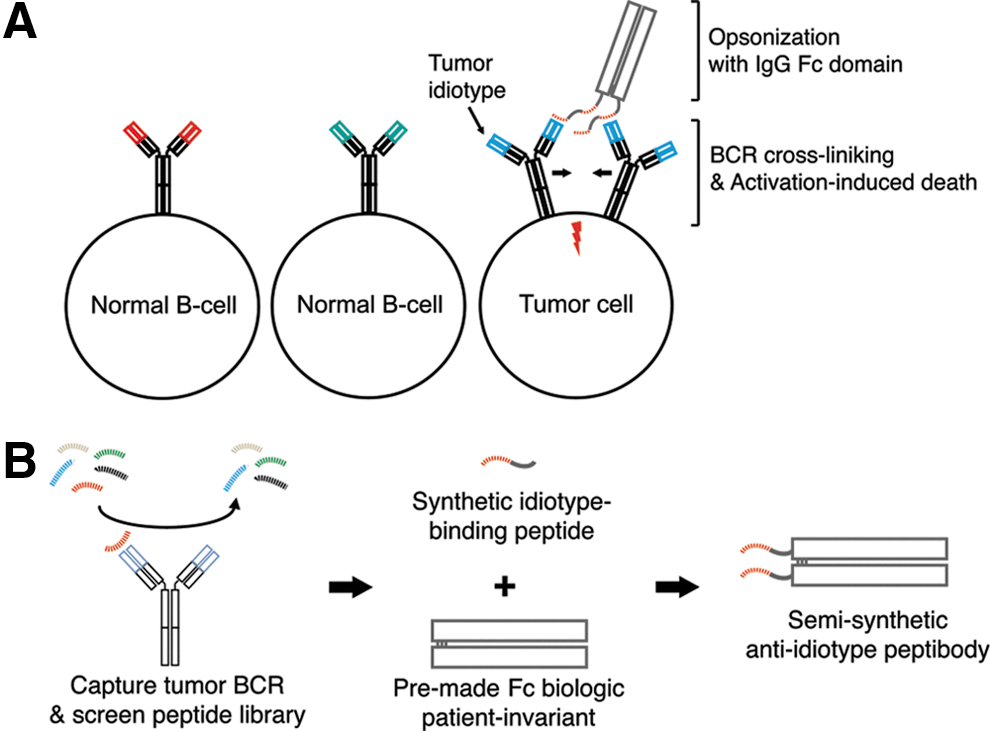
Commentary:Treatment of B-cell lymphomas with monoclonal antibodies, though promising in principle, has met with low success due to development of resistance over time. The tumor's resistance to the antibody is traced to the fact that individual tumors present unique and changing sets of epitopes associated with the malignancies in B-cell lymphomas, making anti-idiotypic therapy with traditional immunoglobulin G (IgG)-type antibodies highly impractical and ultimately unsuccessful, as such a traditional therapy would require a custom generation of anti-idiotype antibodies one patient at a time. Here, the team has provided a method of targeting lymphoma idiotype on a patient-specific basis by using semisynthetic anti-idiotype peptibodies (see
figure
). First, a recombinant murine IgG2a Fc domain was modified to carry an aminoterminal cysteine, which in turn served as a handle for a native chemical ligation later used to attach idiotype-binding peptides site selectively. The idiotype-binding peptides could in turn be easily identified through high-throughput screens of patient tumor cells against peptide libraries. In this manner, the peptibody combining an immunoglobulin Fc domain and malignant B-cell-specific peptide binder represents a therapeutic modality that is easier to scale up and personalize than a traditional monoclonal. Further, given that a single peptibody molecule can incorporate more than two antigen-binding domains, a higher avidity and cell-killing efficacy can be engineered. Finally, the peptibodies developed here were shown to kill tumor cells in vitro and to clear human lymphoma in a mouse xenograft model, thus offering a proof-of-principle for this novel therapeutic modality. Contributed by Anton Simeonov.
Concept overview. (A) Tumor idiotype is the variable region of the BCR on the surface of lymphoma cells. It is unique to the tumor clone and distinct from the idiotype on other B-cell clones. Peptibodies cross-link idiotype, triggering BCR signaling, resulting in activation-induced cell death, and opsonize tumor cells with an IgG Fc domain, promoting tumor clearance mediated by innate immune effector cells. (B) Schematic of semisynthetic anti-idiotype peptibody production. A random peptide library is screened to identify peptide sequences with idiotype-specific binding, which are synthesized and affixed to the amino terminus of a premade recombinant IgG Fc domain.
A Fast-Onset, Long-Acting, Nonaddictive Antidepressant … Based on Ketamine
Zanos P, Moaddel R, Morris, PJ,et al.: NMDAR inhibition-independent antidepressant actions of ketamine metabolites.Nature2016, in press.DOI: 10.1038/nature17998.
Abstract: Major depressive disorder affects around 16 per cent of the world population at some point in their lives. Despite the availability of numerous monoaminergic-based antidepressants, most patients require several weeks, if not months, to respond to these treatments, and many patients never attain sustained remission of their symptoms. The non-competitive, glutamatergic NMDAR (N-methyl-d-aspartate receptor) antagonist (R,S)-ketamine exerts rapid and sustained antidepressant effects after a single dose in patients with depression, but its use is associated with undesirable side effects. Here we show that the metabolism of (R,S)-ketamine to (2S,6S;2R,6R)-hydroxynorketamine (HNK) is essential for its antidepressant effects, and that the (2R,6R)-HNK enantiomer exerts behavioral, electroencephalographic, electrophysiological and cellular antidepressant actions in mice. These antidepressant actions are independent of NMDAR inhibition but involve early and sustained activation of AMPARs (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors). We also establish that (2R,6R)-HNK lacks ketamine-related side effects. Our data implicate a novel mechanism underlying the antidepressant properties of (R,S)-ketamine and have relevance for the development of next-generation, rapid-acting antidepressants.
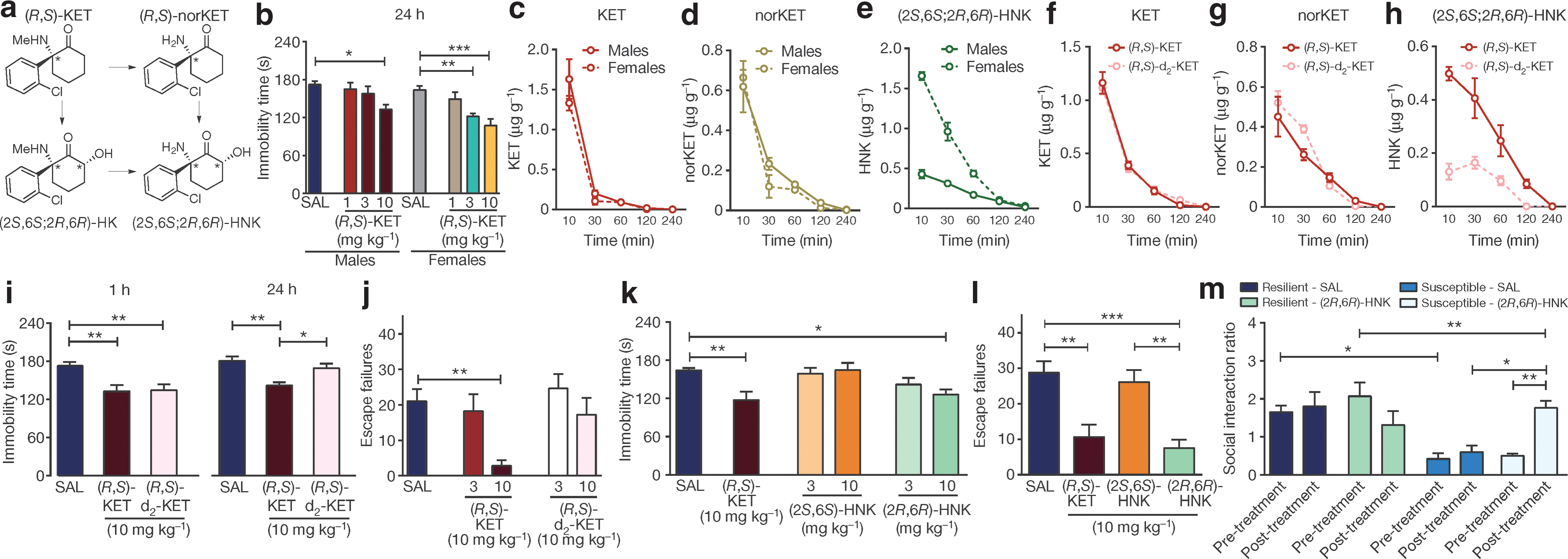
Commentary:Ketamine is an old drug, primarily used as an anesthetic. However, the potential use of ketamine as an antidepressant has also been highlighted recently. While the onset of ketamine's antidepressant action is fast, notable side effects include euphoric and dissociative properties (earning its place as a street drug by the name of Special-K), as well as the development of addiction. Ketamine's mechanism of action as antidepressant had been thought to involve N-methyl-D-aspartic acid (NMDA) glutamate receptors. However, attempts to replace ketamine with NMDA receptor blockers with a superior safety profile have thus far been unsuccessful. The present study represents a major chemical detective work to unravel the basis of ketamine's action and provide a safer, more efficacious antidepressant. Ketamine has been known to be metabolized in vivo to a number of products, and the team's painstaking work determined that a single enantiomer—(2R, 6R)-hydroxynorketamine (2R,6R)-HNK)—was the key metabolite responsible for the beneficial action of ketamine (see
figure
). In mice, (2R, 6R)-HNK produced rapid antidepressant effects and, even when delivered at high doses relative to ketamine, failed to display the negative effects associated with ketamine, such as dissociative properties and propensity to cause addiction (as seen by a complete lack of self-administration of intravenous (2R, 6R)-HNK, in contrast to ketamine which was readily self-administered). To the team's surprise, (2R,6R)-HNK did not show signs of NMDA-receptor engagement as determined by tritiated tracer displacement in a biochemical test and functional inhibition test of NMDA-receptors in stratum radiatum interneurons in hippocampal slices; rather, (2R,6R)-HNK's action appears to be associated with activation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors. The superior drug profile of (2R, 6R)-HNK, as established in murine models, should form the basis for testing this unique metabolite for toxicity and efficacy in humans. Contributed by Anton Simeonov.
Metabolism of ketamine to (2R,6R)-HNK is necessary and sufficient to exert ketamine-related antidepressant actions. (a), Simplified diagram of (R,S)-KET metabolism. (b–e), Greater antidepressant-like actions of ketamine in female mice compared to males (b) are associated with higher brain levels of (2S,6S;2R,6R)-HNK (e), but not KET (c) or norketamine (norKET) (c). (f–h), Brain levels of KET (f), norKET (g) and (2S,6S;2R,6R)-HNK (h) after administration of (R,S)-KET and 6,6-dideuteroketamine ((R,S)-d2-KET). (i, j), Effects of (R,S)-KET and (R,S)-d2-KET in the 1-h and 24-h FST (i) and the learned helplessness test (j). (k, l), Compared to (2S,6S)-HNK, (2R,6R)-HNK manifested greater potency and longer-lasting antidepressant-like effects in the FST (k) and learned helplessness test (l). (m), (2R,6R)-HNK reversed chronic social defeat-induced social interaction deficits. Data are mean ± s.e.m.*P < 0.05, **P < 0.01, ***P < 0.001 (see Supplementary Table 1 for statistical analyses and n numbers).