
Abstract
Intracerebroventricular injection of angiotensin IV, a ligand of insulin-regulated aminopeptidase (IRAP), has been shown to improve cognitive functions in several animal models. Consequently, IRAP is considered a potential target for treatment of cognitive disorders. To identify nonpeptidic IRAP inhibitors, we adapted an established enzymatic assay based on membrane preparations from Chinese hamster ovary cells and a synthetic peptide-like substrate for high-throughput screening purposes. The 384-well microplate-based absorbance assay was used to screen a diverse set of 10,500 compounds for their inhibitory capacity of IRAP. The assay performance was robust with Z′-values ranging from 0.81 to 0.91, and the screen resulted in 23 compounds that displayed greater than 60% inhibition at a compound concentration of 10 μM. After hit confirmation experiments, purity analysis, and promiscuity investigations, three structurally different compounds were considered particularly interesting as starting points for the development of small-molecule-based IRAP inhibitors. After resynthesis, all three compounds confirmed low μM activity and were shown to be rapidly reversible. Additional characterization included activity in a fluorescence-based orthogonal assay and in the presence of a nonionic detergent and a reducing agent, respectively. Importantly, the characterized compounds also showed inhibition of the human ortholog, prompting our further interest in these novel IRAP inhibitors.
Introduction
More than 46 million people worldwide suffer from cognitive impairment or age-related cognitive decline, and this number is estimated to increase to more than 130 million by 2050. 1 Consequently, there is a strong and urgent demand to develop new treatment approaches for these disorders. A prerequisite for such activities is the identification of well-validated mechanisms and the chemical entities required to test these in suitable preclinical models of cognition. Our lab has a longstanding interest in the hexapeptide angiotensin IV (Ang IV), a degradation fragment of angiotensin II, as intracerebroventricular injections were shown to improve memory and learning in a large variety of animal models. 2 –8
High densities of binding sites for Ang IV are observed in regions of the brain associated with sensory, motor, and cognitive functions, for example, the hippocampus. 3,9 –12 At the time of the discovery, these sites of binding were referred to as the AT4 receptor, although the nature of this receptor was largely unknown. In 2001, the AT4 receptor was identified as the insulin-regulated aminopeptidase (IRAP), a single-spanning transmembrane zinc-metallopeptidase that belongs to the M1 family of aminopeptidases, and on which Ang IV acts as an inhibitor of the catalytic activity. 13 IRAP is known to play multiple functional roles, including the processing of cyclic peptides such as oxytocin and vasopressin, 14,15 the trimming of MHC peptides, 16 and the colocalization with Glut4 when the latter translocates to the cell membrane on insulin stimulation. 17
Given the potential importance of IRAP in cognitive disorders, several efforts have been undertaken to develop modulators of its enzymatic activity. The Vauquelin group focused on peptide-based inhibitors containing β-homo amino acids and constrained amino-acid analogs of Ang IV with inhibitory constants (Ki's) in the low nM range. 18 –20 Our group has utilized Ang IV and the IRAP substrate oxytocin, the latter encompassing a macrocyclic disulfide at the cleavage site, as starting points in the development of IRAP inhibitors. Truncated analogs of Ang IV, and N-terminal cyclizations thereof, provided highly potent macrocyclic IRAP inhibitors with Ki's down to 1.8 nM. 21 –25 Although this set of peptidomimetic structures was found to be metabolically relatively stable and could enhance dendritic spine density in rat hippocampal primary cultures, 26 they suffer from poor permeability and hence are of limited use as in vivo pharmacological tools to further study the role of IRAP inhibition in models of cognition. The Stratikos group have utilized transition state mimicking compounds based on a zinc-chelating phosphinic group, and representatives of these compounds show excellent activity (IC50 = 30 nM), but poor selectivity toward the closely related endoplasmic reticulum aminopeptidase (ERAP) 1 and 2. 27 A similar approach using a 3,4-diaminobenzoic acid as a scaffold led to pseudopeptides with good potency as well as to an improved selectivity profile toward ERAP1 and ERAP2. 28,29
In parallel, the first drug-like nonpeptidic IRAP inhibitors were identified and optimized by using a virtual screening approach. 30,31 In vivo efficacy was demonstrated in rodent models of cognition for one of these inhibitors, a pyridine-substituted benzopyran scaffold. The performance in both spatial working and recognition memory paradigms was improved after administration into the lateral ventricles. However, the compound suffered from short elimination half-life and high plasma clearance. 31
Since most of the available IRAP inhibitors are still peptide based, suffering from inadequate selectivity and/or poor permeability, we initiated a collaborative effort to identify less complex small-molecule-based inhibitors of IRAP. An existing enzymatic assay was used as a starting point for the development of a screen compatible assay that was adapted to 384-well microtiter plate format and automated liquid handling procedures. We hereby report on the development of the assay, the outcome of the screening of ∼10,500 lead-like and drug-like compounds, and the subsequent hit confirmation experiments in which the structure, the chemical stability, the concentration dependency, and the reversibility of the identified inhibitors were confirmed. The study resulted in the identification of three novel chemical clusters of IRAP inhibitors, with the most potent representatives in the low μM range.
Materials and Methods
Cell Culture Conditions
Chinese hamster ovary (CHO) cells were cultured in 75 cm2 cell culture flasks (430641U; Corning) or Nunc™ EasyFill™ Cell Factory™ System 2528 cm2 (140360; Nunc) in Dulbecco's modified essential medium (D6046; Sigma-Aldrich) that was supplemented with 2 mM
Overexpression of Human IRAP
Overexpression of human IRAP was achieved in Freestyle HEK293-F Cells (R790-07; Invitrogen) that were grown in 80 mL of Freestyle Expression Medium (12338-018; Invitrogen). The cell culture was grown in 250 mL Erlenmeyer flasks (431144.40; Corning) that were kept at 37°C, 8% CO2, and 70% relative humidity and with shaking at 130–135 rpm in an Infors Multitron incubator. The cells were transfected with 1.1 μg plasmid/106 cells (pCIneo containing the gene of human IRAP, obtained from Prof. M. Tsujimoto, Laboratory of Cellular Biochemistry, Saitama, Japan) in the presence of 2 μg polyethyleneimine/106 cells (Polyethyleneimine, linear, MW-25 000, Cat. No. 23966; Polysciences). Before the overexpression studies, the plasmid was transfected and propagated in Mach1 Escherichia coli (C8620-03; Invitrogen), from where the plasmid DNA was prepared by using QIAGEN Plasmid Plus Maxi Kit (12963). Forty-eight hours post-transfection, the cells were harvested by centrifugation at 130 × g for 3 min. The cells were washed once with PBS, and the cell pellets were stored at −20°C until they were used for membrane preparations.
Membrane Preparation
All experiments were based on membrane preparations from either CHO cells or HEK293T suspension cells overexpressing human IRAP. Membrane preparation was achieved essentially as previously described 32 by using the following procedure. Washed cell pellets were taken out of the −20°C freezers, quickly thawed, and placed on ice. Pellets were resuspended in ice-cold 50 mM Tris-HCl at pH 7.4 that was supplemented with 0.1 mM of phenylmethanesulfonyl fluoride (PMSF, 78830; Sigma-Aldrich) and were then sonicated for 3 × 15 s with 30 s interruption time by using a Branson Digital sonifier 250. The sonicated cells were then transferred to a Dounce homogenizer and were homogenized by a minimum of 20 strokes while keeping everything on ice. The homogenate was thereafter transferred to ice-cold tubes and centrifuged at 4°C and 30,000× g for 30 min. The supernatant was discarded; the membranes were resuspended in buffer, again homogenized by using the Dounce homogenizer (20 strokes), and centrifuged as described earlier. This procedure was repeated twice, and the final membrane pellet was then kept frozen at −20°C until it was used.
Compound Library Storage and Handling
A set of 10,500 compounds from the primary screening set at Chemical Biology Consortium Sweden (CBCS) was applied in this screening campaign. A majority of the compound library was donated by former Biovitrum AB and is a mix of commercially available and proprietary compounds. Compound stock solutions at 10 mM in dimethyl sulfoxide (DMSO) were stored and frozen at −20°C under low humidity conditions in individual capped tubes in REMP 96 Storage Tube Racks located in REMP Small-Size Store™. DMSO stock solutions for follow-up studies are retrieved directly from the REMP vials by cherry-picking. For screening purposes, the compound solutions were transferred to Labcyte 384 LDV plates (LP-0200) and then further into Labcyte 1536 HighBase plates (LP-03730) to enable dispensing by using acoustic liquid handling equipment. The compound solutions (75 nl) were dispensed directly into columns 1–22 of the assay plates (242757; Nunc) by using an Echo 555™ acoustic liquid handler (LabCyte). The Echo was also used to dispense the equivalent volume of DMSO (negative control, 0% inhibition), and a 10 mM DMSO solution of AL-11 19,33 (positive control, 100% inhibition), in columns 23 and 24, with DMSO in rows A–H and AL-11 in rows I–P. The final compound concentration used in the screen was 10 μM, with a final DMSO concentration of 0.1% in all wells.
Screening Campaign
Small-molecule library screening was accomplished in transparent 384-well plates with preplated compound solutions and controls in all wells (75 nl). A homogenized solution of IRAP-containing CHO cell membranes was prepared on ice by using a minimal of 20 strokes in a Dounce homogenizer in an ice-cold solution of 50 mM Tris-HCl, 150 mM NaCl, and 0.1 mM PMSF at pH 7.4. This solution was kept on ice until 25 μL thereof was added to each well of the assay plates by using a MultiDrop Combi (Thermo Scientific), resulting in membranes from 25,000 cells in each well. This was followed by the addition of 50 μL of a substrate solution containing 1.5 mM
IRAP Activity Assay for Concentration-Response Characterizations in 96-Well Format
Concentration-response experiments measuring the impact of identified inhibitors on IRAP activity as a function of compound concentration were conducted in several different formats. The absorbance assay, based on the
Concentration-response experiments based on solubilized enzyme were conducted by using the same setup, except for the treatment of the CHO cell membranes before addition to the plates and the presence of Triton X-100 in the assay buffer. For these experiments, aliquots of membranes were thawed, suspended in assay buffer, and homogenized by using the Dounce homogenizer (20 strokes). Triton X-100 was then added to give a final presence of 1% in the membrane containing buffer, and the suspension was subsequently rotated for at least 5 h at 4°C to solubilize the membrane proteins. After solubilization, membranes were pelleted by centrifugation at 10,000× g in a tabletop centrifuge at 4°C for 15 min. The supernatant was used as the source of IRAP activity and was applied at a volume of 50 μL per well. This procedure gave a final IRAP concentration corresponding to the content of 322,000 cells per well. Concentration-response experiments for controlling redox reactivity of the hits were conducted by using the standard absorbance-based assay with CHO cell membranes, but with the addition of dithiothreitol (DTT) to a final concentration of 1 mM in the assay buffer.
Concentration-response experiments in an orthogonal assay utilizing a fluorescence-based readout were performed in black, low-volume 96-well plates (3686; Corning). The experimental design was identical to what is described earlier, except for the substrate and the order of addition to the assay plates. Here, the substrate solution was 150 μM
Data Analysis
Raw data from the microplate readers were imported into Microsoft Excel and GraphPad Prism 6 for analysis and visualization. Determination of Z′-factor values were done based on the controls of each plate as described. 34 All data were converted into % inhibition values based on the negative (0%) and positive (100%) controls on each plate. Data from triplicate samples were averaged, and the curves were fitted to a four-parameter concentration-response model within IDBS XLfit (model 205) or GraphPad Prism 6 to obtain best-fit values for the IC50, Hill slope, and the upper and lower limits of the concentration-response curve.
Measurements of Identity and Purity of Test Compound Solutions
Assessment of identity and purity of the test solutions that were used for hit confirmation purposes, that is, those being stored in REMP vials, was done by means of reversed-phase high-performance liquid chromatography coupled to mass spectrometry (HPLC-MS). A small aliquot of each test solution (2 μL of a 10 mM solution) was placed in a deep-well plate and was diluted with 20 μL of methanol. The plate was then placed in an Agilent 1100 HPLC UV/MS with electrospray ionization (ESI+). The HPLC method was based on an ACE C8 3 μm column (3.0 × 50 mm) and a mobile phase [0.1% CF3COOH/CH3CN]/[0.1% CF3COOH/H2O]. All solvents were HPLC grade, and absorbance was monitored at 305 ± 90 and 254 nm. Compounds that did not give satisfactory data were re-analyzed by using a method based on a Waters XBridge C18 3.5 μm column (3.0 × 50 mm), 3.5 min gradient mobile phase [CH3CN]/[10 mM NH4HCO3/H2O]. The instrument software was used to integrate the UV response for each peak and provided a list of the peaks and their associated masses. The estimated purity was calculated based on the integrated area for the expected mass compared with the areas of all other peaks. The result was manually controlled and if there were deviations from the expected outcome, a meticulous investigation of the UV response and mass spectrometry was performed.
Hit Resynthesis and Characterization
General information and materials
All chemicals were purchased from Sigma-Aldrich and were used as received. Purification was performed by flash column chromatography using silica gel (60–120 mesh size) and for test compounds also, reversed-phase HPLC (UV-triggered, 254 nm) fraction collection was done with a Gilson Trilution HPLC system, using a Macherey-Nagel Nucleodur C18 column (21 × 125 mm, 5 μm particle size), and H2O/CH3CN/0.1% CF3COOH as eluent in a gradient (30%–70%, 10 mL min−1 over 15 min). Analytical HPLC-MS spectrometry was performed on a Dionex UltiMate 3000 HPLC system with a Thermo Scientific MSQ Plus mass spectrometer (ESI+) and detection by UV (diode array detector), using a Phenomenex Kinetex C18 column (50 × 30 mm, 2.6 μm particle size, 100 Å pore size). A gradient of H2O/CH3CN/0.1% HCOOH at a flow rate of 1.5 mL min−1 was used. High-resolution mass spectra (HRMS) were recorded on a Micromass Q-Tof2 mass spectrometer that was equipped with an electrospray ion source. 1H and 13C NMR spectra were recorded at 25°C on a Varian 400-MR spectrometer at 400 and 100 MHz, respectively. For rotamers, NMR experiments were performed both at room temperature and at elevated temperature; see Supplementary Data (Supplementary Data are available online at
(2-Methoxyphenyl)(pyridine-2-yl)methanamine (24)
A solution of 2-cyanopyridine (0.5 mL, 5.2 mmol) in dry toluene (25 mL) under inert atmosphere was cooled to 0°C on an ice bath. Then, 2-Methoxymagnesiumbromide in THF (1 M, 6.2 mL, 6.2 mmol) was added dropwise. The reaction mixture was stirred for 40 min at 0°C. Isobutanol was added dropwise to a clear brown solution. NaBH4 (393 mg, 10.4 mmol) was added, and the mixture was stirred at ambient temperature for 14 h. The reaction was quenched with a 1:1 mixture of MeOH and H2O, and the organic solvents were removed under reduced pressure. The remaining aqueous mixture was extracted with DCM (×3) and the combined organic phases were washed with brine, dried over MgSO4, filtered, and concentrated. The crude product was purified by flash column chromatography by using 5% MeOH in CHCl3 as eluent to afford
(2S)-tert-Butyl-2-(((2-methoxyphenyl)(pyridine-2-yl)methyl)carbamoyl)pyrrolidine-1-carboxylate (25)
To a solution of
(S)-tert-Butyl-2-(1-(2-methoxyphenyl)imidazo[1,5-α]pyridine-3-yl)pyrrolidine-1-carboxylate (26)
A solution of
(S)-1-(2-Methoxyphenyl)-3-(pyrrolidine-2-yl)imidazo[1,5-α]pyridine hydrochloride (27)
To a solution of
(S)-2-(1-(2-Methoxyphenyl)imidazo[1,5-a]pyridin-3-yl)-N-((S)-1-phenylethyl)pyrrolidine-1-carboxamide
To a solution of
Results
Assay Adaptation to a 384-Well Microplate Format
The enzymatic activity of IRAP can be studied by several different means, including HPLC analysis of the aminopeptidase activity toward endogenous substrates as well as the use of synthetic substrates for which the absorbance (
A number of changes had to be made to our standard assay format to facilitate the screening logistics. The most significant of these involved a change from 37°C to room temperature incubation to avoid plate edge effects, 40 prolonged incubation times to use less membranes on a per-well basis, and the use of automated liquid handling for compound, membrane, and substrate dispenses. After substrate Km determinations and membrane titrations in the form of time-course experiments to ensure less than 50% substrate conversion (Fig. 1a, b), we used known peptidic and small-molecule-based inhibitors to investigate whether the assay was responding pharmacologically as expected. The assay accurately ranked the metabolically stable peptidic-based inhibitors IVDE77 20 and AL-11, 19 with IC50 values of 11 and 55 nM, respectively, as well as the high μM inhibitor amastatin (Fig. 1c). We also confirmed the low μM inhibitory activity of ZnCl2 as previously reported. 37
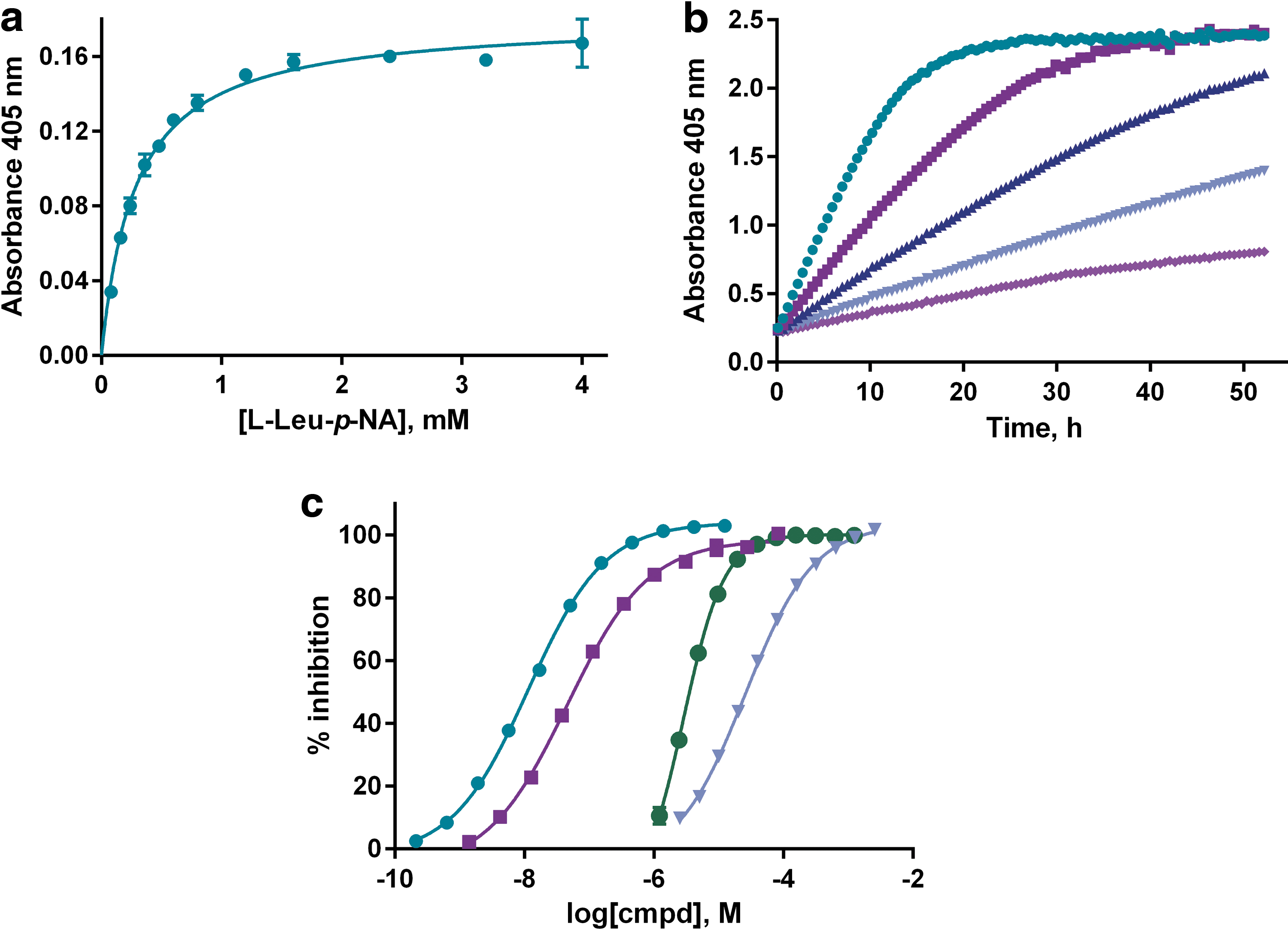
Assay characterization in the 384-well microtiter plate format.
Library Screening and Hit Confirmation
Using the microtiter plate formatted assay for IRAP, we completed a screening campaign by using the primary screening set at CBCS (
Protocol for the IRAP Inhibition Assay
1. Transparent 384-well plate, Nunc no. 242757. Echo 555™ acoustic liquid handler. Controls were added to columns 23 and 24 of the assay plates.
2. Echo 555 acoustic liquid handler. One thousand-fold assay concentration.
3. MultiDrop Combi instrument. Prepared by diluting the CHO cell membranes to 500 mL in ice-cold buffer (988,000 cells/mL). The assay buffer consists of 50 mM Tris-HCl, 150 mM NaCl, and 0.1 mM PMSF at pH 7.4. Final amount of 25,000 cells/well.
4. MultiDrop Combi Instrument. Prepared by diluting a 100 mM stock solution in 2-methoxythanol into the assay buffer. Final assay concentration is 1 mM.
5. Plates lidded and kept dark.
6. Absorbance measured.
CHO, Chinese hamster ovary; DMSO, dimethyl sulfoxide; IRAP, insulin-regulated aminopeptidase;
The increase in absorbance over time as the synthetic substrate was processed by IRAP was followed at four different time points, including a control reading immediately after the initiation of the reaction. This first reading allowed a compensation of any absorbance contributed by colored compounds in the library. However, contrary to our fears of significant compound color interference, the impact turned out to be marginal, as illustrated in Figure 2a. The small increase in absorbance from the first to the last plates represents a delay between addition and reading, and this time was longer for the later plates. Data analysis showed that the substrate conversion was optimal at 17 h incubation time as the signal increased linearly with time over the investigated period, and this time point corresponded to less than 50% substrate conversion 41,42 and a signal well within the linear range of the microplate reader (Fig. 2b). Furthermore, both the signal from the uninhibited enzymatic reaction and the background values in fully inhibited wells (16 control wells each per plate) remained constant in all assay plates, demonstrating suitable stability of the enzyme, substrate, and control inhibitor solutions (Fig. 3a). The distinction between the controls was significant throughout the screening campaign, with calculated Z′ factors for individual plates ranging from 0.85 to 0.91 (Fig. 3b), and plate edge effects were largely absent (Supplementary Fig. S1).
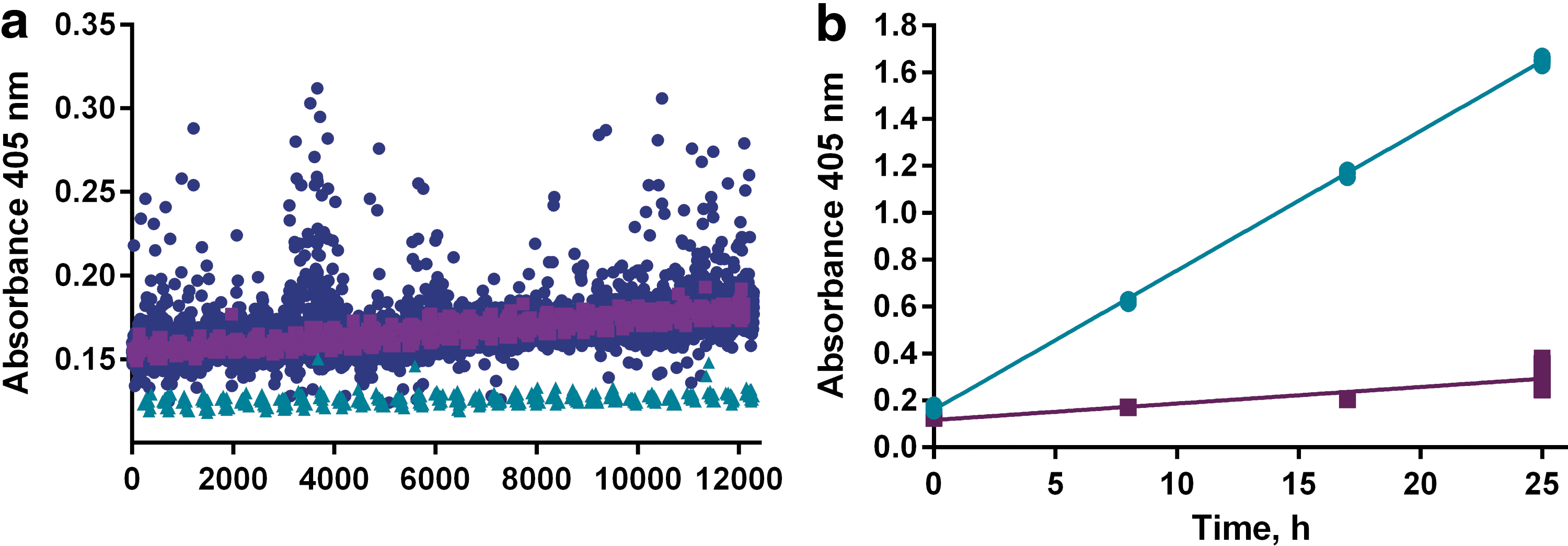
Investigation of compound interference and linearity of reaction under screen conditions.
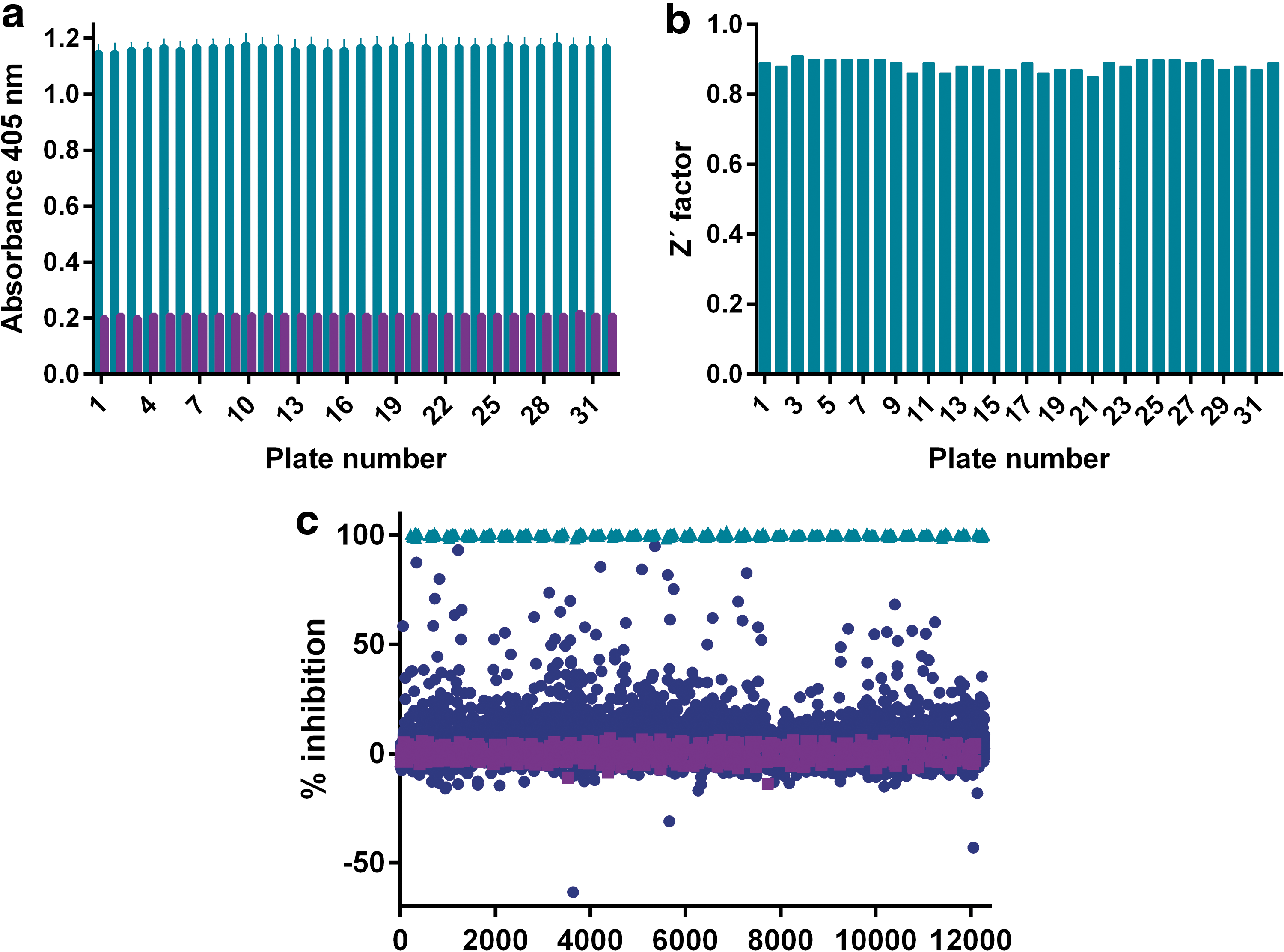
Screen performance and hit identification.
The impact of the tested compounds is summarized in the form of a scatter plot in Figure 3c. A hit limit of 25.3% was calculated based on the average plus three standard deviations of the observed inhibitory response for all library compounds, resulting in a hit list of 165 compounds (1.6% hit rate). A selection of the most potent hits, for which the apparent inhibition in the primary screen was above 60%, was pursued in concentration-response experiments for confirmation purposes. Out of these 23 compounds, 16 confirmed concentration-dependent activity in the screening assay with an IC50 value around 10 μM or below (Table 2 and Fig. 4).
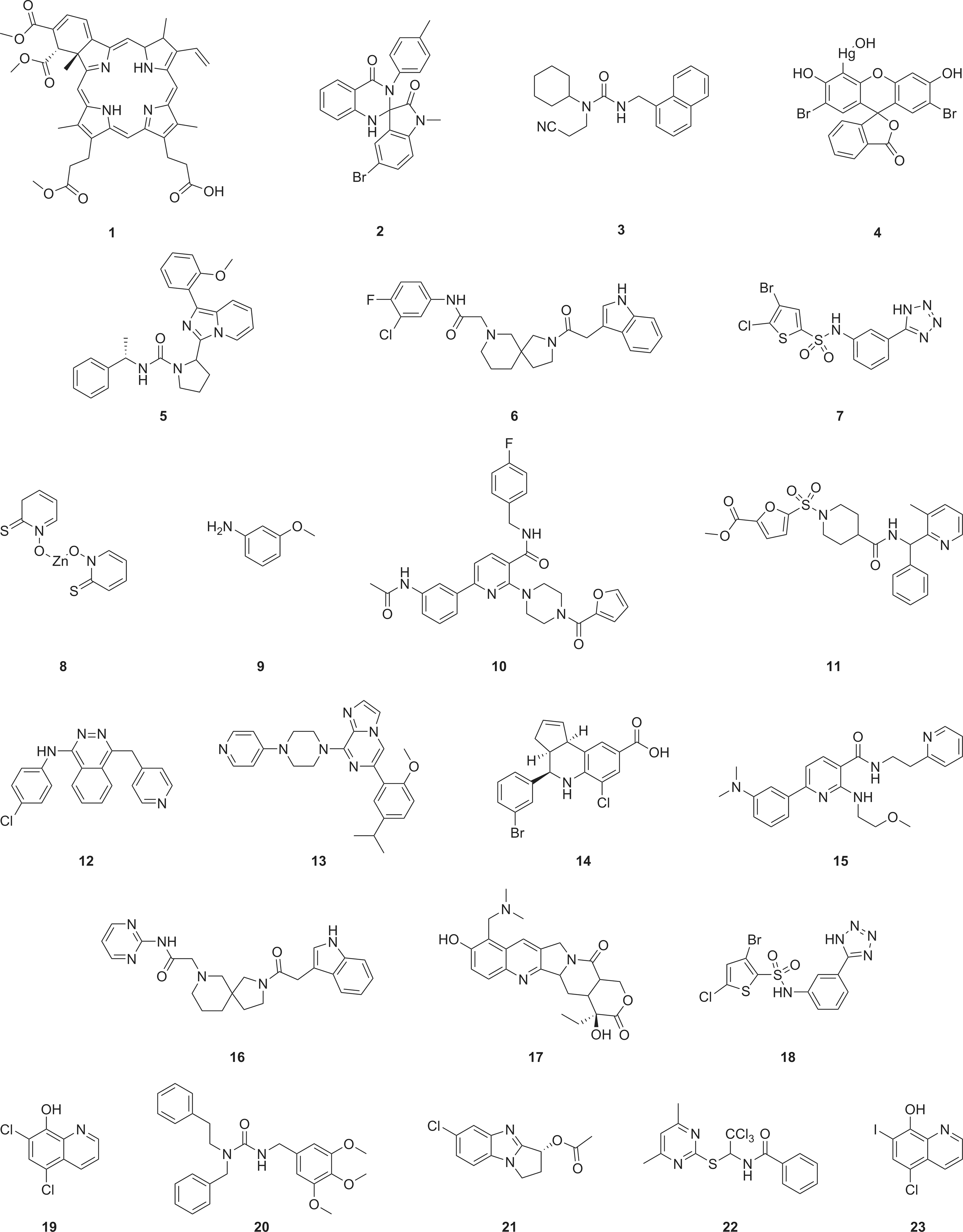
Chemical structures of the top-ranked IRAP hits.
Summary Data for Top Hits in the IRAP Screening Campaign
Membrane preparations forming CHO cells were used as the IRAP activity source.
Correct m/z ratio by LC-MS (ESI+).
Purity according to LC-UV at 305 ± 90 and 254 nm.
Numbers within brackets represent the number of test occasions on the screen batch solutions. Additional replicates were done on resynthesized compounds (see page 189, paragraph 1).
Numbers represent the amount of times the compounds have appeared as hits in internal screens. Numbers within brackets represent the number of screens of which the compounds have been included as a part.
Analog searches were based on the in-house cheminformatics program Beehive by using similarity searches with a Tanimoto coefficient >0.7. The numbers refer to available analogs in-house/tested in screen/actives above hit limit.
Reported as part of training set for hepatotoxicity.
ESI, electrospray ionization; LC-MS, liquid chromatography-mass spectrometry.
One compound (
The hit compound solutions were analyzed for purity and the appearance of the correct mass by using liquid chromatography and mass spectrometry-based detection (LC-MS) to associate the measured activities with the correct molecular structures. To prioritize among the hits, we also looked at the availability of structurally related analogs in our libraries that could be used to obtain fundamental structure-activity relationships (SAR). As shown in Table 2, these follow-up investigations identified six compounds denoted here as
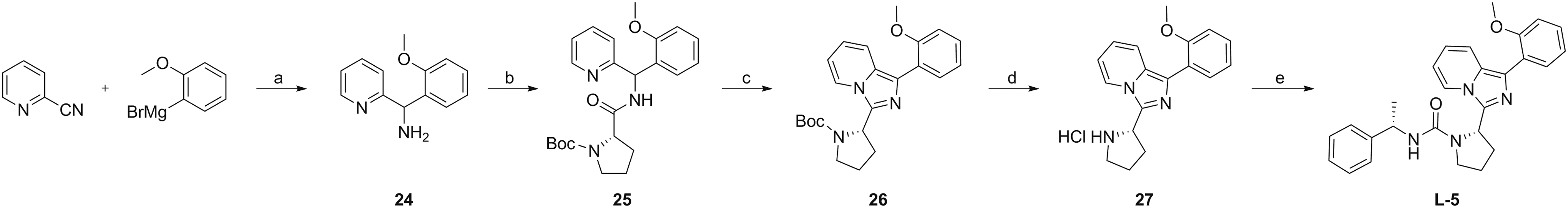
Synthesis of compound
Characterization of IRAP Inhibition Under Multiple Assay Conditions
All data were obtained on resynthesized and fully characterized material. Data are provided from triplicate samples at a minimum of two independent test occasions. CHO cell membrane preparations were used as the IRAP activity source. Cmpd
Assay performed using
Membrane preparations from human IRAP overexpressed in HEK293-F cells were used as the IRAP activity source.
Partial inhibition observed.
DTT, dithiothreitol;
Testing of the synthesized hits confirmed low μM inhibitory potency for all three compounds (Fig. 6a). Whereas the further SAR exploration for

Characterization of IRAP inhibition for resynthesized hits.
Testing Reversibility and Activity in the Human Ortholog
To motivate further investments in these novel series of IRAP inhibitors, we pursued additional activity testing, broadly following a flowchart for characterization of reversible inhibitors.
49
Before this, we ensured the compounds did not interfere with the absorbance readout (Supplementary Fig. S2), also in agreement with the observation of inhibitory activity when running the assay based on the fluorogenic substrate
With regards to the pharmacology of these compounds, we examined the Hill slopes of the concentration-response curves, which in all cases were close to unity, and the maximal inhibition obtained (Fig. 6a). Full inhibition was obtained for all these compounds, although a higher variability, sometimes resulting in partial inhibition, was observed for compound
Concentration-response experiments in the presence of a reducing agent and a nonionic detergent were employed to investigate whether any of the compounds acted by means of unwanted protein inactivation through either cysteine oxidation or protein sequestering by aggregating compounds. 50 –52 These control experiments showed that the inhibitory effect was largely independent of the presence of DTT or Triton X-100 (Table 3). The lack of evidence for these unwanted mechanisms is in agreement with the internal and external data on the absence of promiscuous appearances as hits in other screening campaigns. Furthermore, a search of the recently introduced public aggregator advisor database for these hit compounds did not show any relation to known aggregators, although a general warning was given based on the relatively high cLogP's. 52 It should also be noted that the experiments performed in the presence of Triton X-100, that is, conditions under which IRAP is known to be solubilized from the membrane, demonstrate that compound binding to IRAP is independent of the membrane anchoring.
Although CHO cells are known to abundantly express IRAP, we wanted to assert that the compounds were also active on human IRAP. Using the basic logical alignment search tool (BLAST),
53
we could conclude that IRAP from Chinese hamster (UniProtKB: A0A061IK54) and IRAP from human (UniProtKB: Q9UIQ6) have a sequence identity of 88.5%. For this purpose, human IRAP was overexpressed in HEK293-F suspension cells followed by membrane preparations to allow testing of activity of the three compounds (
Discussion
A thorough understanding of the role of pharmacological inhibition of IRAP under normal physiological as well as disease conditions requires selective and potent modulators with good pharmacokinetic properties, the effects of which can be examined in relevant animal models. We have contributed in this field by variations of Ang IV itself, driven by the beneficial pharmacological effects observed on central administration of this degradation product originating from angiotensin II. 2 –8 Based on the finding in 2001 that a central target for this effect is the aminopeptidase IRAP, 13 with activity toward cyclic peptides such as vasopressin and oxytocin, we have also worked with metabolically stable variants thereof, 21 –25 with a primary focus are primarily focus on the field of cognition impairment. However, despite the possibility to generate potent peptide-based inhibitors, the majority of these suffer from preclinical challenges when administered systemically, particularly with regards to the limited cell permeability and penetrance of the blood–brain barrier. Significant advancements were made with the identification of small-molecule-based inhibitors of IRAP that demonstrated promising effects in animal models of cognition when administered centrally. 30,31 However, pending the further development of these inhibitors into new variants that can be administered systemically, there is a need to work broadly on additional molecular scaffolds.
We were interested in the identification of additional starting points by means of a small-molecule library screen. This was accomplished by using screen formatted variants of a well-published absorbance assay based on the dipeptide analog
The screening campaign was successfully completed with excellent statistics and yielded 23 hits with an apparent inhibition above 60%. The majority of these confirmed activity in concentration-response experiments, resulting in the identification of several known drugs that interfered with IRAP activity. However, as the scope in this study was to identify new starting points that can be further developed into selective modulators of IRAP, we have not characterized these further. It is also worthwhile noting that many other biological activities are reported for a majority of these.
Instead, our primary focus was on those compounds that had not been previously active in our screening campaigns and that had no hitherto reported biological activities. Three compounds with low μM potency were of particular interest and were hence resynthesized, fully characterized, and examined for structure-activity relationships. Such efforts on compound
Footnotes
Acknowledgments
H.A., M.O., K.S., A.J.J., and T.L. acknowledge Karolinska Institutet, SciLifeLab, and the Swedish Research Council (Vetenskapsrådet), which funds Chemical Biology Consortium Sweden. M.H. and M.L acknowledge the Kjell and Märta Beijer Foundation, King Gustav V's and Queen Victoria's Freemason Foundation, and Uppsala University for financial support.
Disclosure Statement
No competing financial interests exist.