
Abstract
Drug design by methods such as fragment screening requires effective solubilization of millimolar concentrations of small organic compounds while maintaining the properties of the biological target. We investigate four organic solvents and three 1-butyl-3-methylimidazolium (BMIm)-based ionic liquids (ILs) as cosolvents to establish conditions for screening two structurally unrelated dihydrofolate reductases (DHFRs) that are prime drug targets. Moderate concentrations (10%–15%) of cosolvents had little effect on inhibition of the microbial type II R67 DHFR and of human DHFR (hDHFR), while higher concentrations of organic cosolvents generally decreased activity of both DHFRs. In contrast, a specific IL conserved the activity of one DHFR, while severely reducing the activity of the other, and vice versa, illustrating the differing effect of ILs on distinct protein folds. Most of the cosolvents investigated preserved the fold of R67 DHFR and had little effect on binding of the cofactor NADPH, but reduced the productive affinity for its substrate. In contrast, cosolvents resulted in modest structural destabilization of hDHFR with little effect on productive affinity. We conclude that the organic cosolvents, methanol, dimethylformamide, and dimethylsulfoxide, offer the most balanced conditions for early-stage compound screening as they maintain sufficient biological activity of both DHFRs while allowing for compound dissolution in the millimolar range. However, IL cosolvents showed poor capacity to solubilize organic compounds at millimolar concentrations, mitigating their utility in early-stage screening. Nonetheless, ILs could provide an alternative to classical organic cosolvents when low concentrations of inhibitors are used, as when characterizing higher affinity inhibitors.
Introduction
Screening for inhibitors can be challenging when the biological target is water soluble and the test compounds are hydrophobic. A decisive factor in choosing a cosolvent for inhibitor screening is its capacity to solubilize compounds at the mM concentrations required to make up stock solutions. This is particularly important during early-stage screening where hits are likely to be weak inhibitors with IC50 in the high μM to mM range, thus requiring that high compound concentrations be assayed. This was the case in our recent report of the first effort toward the identification of inhibitors against the plasmid-encoded type II R67 dihydrofolate reductase (R67 DHFR), an emerging source of microbial trimethoprim (TMP) resistance. 1,2
We identified bisbenzimidazole-type compounds with low mM to low μM IC50 for R67 DHFR, the latter of which are exemplified by 2,2′-[1,5-pentanediylbis(4-oxyphenylene)]-bis-1H-benzimidazole-5-carboxylic acid (molecule

Structures of R67 DHFR and hDHFR inhibitors. Methotrexate (MTX) is a tight binding inhibitor of hDHFR. Pemetrexed (PMTX) is not selective and inhibits both DHFRs.
DHFRs are ubiquitous targets for fighting proliferative diseases such as cancer and infectious diseases. Indeed, methotrexate (MTX) and pemetrexed (PMTX) (Fig. 1) are among the most commonly used anticancer drugs where they inhibit human DHFR (hDHFR), 5 and the antibiotic TMP is used worldwide in the treatment of bacterial infections, selectively targeting bacterial chromosomal DHFRs. 2 As a result, DHFRs are long-standing targets for inhibitor development, including recent development of multitarget inhibitors of hDHFR and further key enzymes to improve cancer chemotherapy regimens by obtaining synergetic toxicities and simplifying pharmacokinetic profiles. 6 –9
Chromosomal DHFRs share tertiary structure conservation among all species. 5,10 In contrast, R67 DHFR catalyzes the same reduction reaction of DHF using NADPH, but is structurally and evolutionarily distinct from chromosomal DHFRs, sharing no homology with respect to primary amino acid sequence or tertiary structure. 2 While structural differences form the basis for the resistance of the microbial R67 DHFR to the antibiotic TMP, they also highlight the potential in the creation of inhibitors that are selective to R67 DHFR.
To broaden our screening effort for discovery of more potent selective inhibitors of R67 DHFR, we investigate screening conditions that best satisfy the requirements of maintaining the activity of two structurally unrelated enzymes (the target, R67 DHFR, and the control, hDHFR) while providing adequate compound dissolution. While organic cosolvents increase the aqueous solubility of nonpolar molecules, they often lead to enzyme inactivation, as we observed upon exposing the DHFRs to DMSO. 1,11 As potential alternatives to DMSO for screening these DHFRs, we examine the use of dimethylformamide (DMF): it has a high capacity for dissolution of organic compounds and has been used as a water-mimicking solvent through H-bond networks with some enzymes. 12,13 Acetonitrile (ACN) and methanol (MeOH), routinely used as analytical cosolvents and for preparation and storage of compound stock solutions, were also assessed. 14
We also examine the use of ionic liquids (ILs) as they have been proposed as alternative cosolvents in various biocatalytic transformations. 15 –18 In general, enzymes dissolved in pure ILs show reduced catalytic activity, but the addition of ILs to water can help improve enzyme stability and activity, 19 –21 making ILs viable alternatives for use in compound screening. However, their effect on specific enzyme systems must be determined empirically.
To this end, we determined the effect on R67 DHFR and hDHFR enzyme activity of those organic cosolvents as well as 1-butyl-3-methylimidazolium ILs ([BMIm]) bearing different anions, previously shown to be compatible with function of certain oxidoreductases. 15,16,19,22,23 We further considered the maintenance of thermodynamic, kinetic, and thermostability properties by determining IC50 values for known inhibitors as well as k cat , KM, and Tm under various cosolvent conditions to verify whether the cosolvents affected fold or function. Finally, we considered the effectiveness of the cosolvents in aiding dissolution of high concentrations of small organic compounds such as those typically used in inhibitor design and screening.
Materials and Methods
Materials
ACN was purchased from EMD Chemicals (Gibbstown, NJ), DMF from A&C American Chemicals LTD. (Montreal, QC, Canada), isopropyl β-D-1-thiogalactopyranoside from Bioshop (Burlington, ON, Canada), and DMSO and MeOH from Fisher Scientific (Ottawa, ON, Canada). The IL, BMIm octylsulfate, was purchased from Alfa Aesar (Haverhill, MA), while the ILs, BMIm hexafluorophosphate ([BMIm][PF6]) and BMIm tetrafluoroborate ([BMIm][BF4]), were synthesized as described previously
23,24
(
1
H NMR data for [BMIm][PF6] and [BMIm][BF4] are provided as Supporting Information). MTX was bought from Sigma-Aldrich. PMTX (ALIMTA) from Eli Lilly (Toronto, ON, Canada) was supplied as a 1:1 mixture with D-mannitol as a stabilizer. Molecules
Enzyme Purification
Recombinant human chromosomal DHFR (hDHFR) was overexpressed in Escherichia coli BL21 (DE3) and purified as described. 25 Recombinant type II R67 DHFR was overexpressed in E. coli BL21 containing the plasmid pREP4 (Qiagen, Mississauga, ON, Canada) as described 26 with the following modifications. For a 1 L expression culture, the cell pellet was resuspended in 30 mL of lysis buffer (0.1 M potassium phosphate, 5 mM imidazole, pH 8.0) and the cells were disrupted by one passage through a cell disrupter (Constant Systems, Kennesaw, GA) adjusted to 27 kpsi. An additional 10 mL of buffer washed residual lysate through. Following centrifugation (30 min, 23,500 g, 4°C) and filtration through a 0.22-μm filter, the supernatant was injected onto a 5-mL His-Trap HP cartridge at a flow rate of 1 mL/min using an Äkta FPLC (GE Healthcare, Piscataway, NJ). The column was washed with 12 column volumes (CV) of lysis buffer. A linear gradient (6 CV) to a plateau (6 CV) at 30 mM imidazole was followed by a step to 300 mM imidazole for elution. Fractions containing R67 DHFR were identified according to activity assay and analysis on tricine sodium dodecyl sulfate–polyacrylamide gel electrophoresis 27 and pooled for dialysis at 4°C into 0.1 M phosphate buffer, pH 8.0, using 3,500 Da molecular weight cutoff dialysis tubing (Spectrum Laboratories, Inc., Rancho Dominguez, CA). Protein concentration was determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA) with bovine serum albumin (Bio-Rad) as a protein standard.
Determination of Enzyme Activity in the Presence of Cosolvents
Substrate concentrations were quantified spectrophotometrically in 50 mM potassium phosphate, pH 8.0 (ɛ340nm = 6,200 M−1 cm−1 for NADPH and ɛ282nm = 28,400 M−1 cm−1 for DHF). Reactions were initiated by the addition of 0.6 mU of either R67 DHFR or hDHFR in the presence of 100 μM each of DHF and NADPH, where 1 U represents the quantity of enzyme required to consume 1 μmol of substrate per minute. The reactions were in 50 mM potassium phosphate buffer at pH 7.0 for R67 DHFR and pH 8.0 for hDHFR in 384- or 96-well polystyrene plates. Enzyme activity was determined by monitoring the initial rate (within the first 10% of substrate consumption) of depletion of NADPH and DHF at 340 nm (Δɛ340nm = 12,300 M−1 cm−1). 28 Liquid handling was mainly carried out using a BioMek NX automated workstation (Beckman Coulter, Brea, CA) and data collected with the integrated Beckman DTX 880 plate reader.
Determination of KM and kcat of R67 DHFR and hDHFR in the Presence of Cosolvents
Kinetic assays were performed with 10% DMSO, 10% DMF, 10% MeOH, 15% ACN, 10% [BMIm][BF4], or 10% [BMIm][OctSO4]. All kinetic assays were performed using 1-cm cuvettes at 300K in 50 mM potassium phosphate except for [BMIm][BF4], which tended to form a precipitate due to a salting-out effect frequently observed upon mixing ILs with ionic buffers such as the phosphate buffer used here. 29 Thus, kinetic assays in 1-cm cuvette using [BMIm][BF4] were performed in MATS buffer, pH 7.0 (25 mM MES, 25 mM acetate, 50 mM Tris, 100 mM sodium acetate).
For the determination of KM NADPH and KM DHF, the concentration range of the variable substrate bracketed the reported KM values (NADPH: 1–50 μM; DHF: 1.56–100 μM) except when saturation was attained at concentrations too low for accurate activity determination. 26 The second substrate was kept at a saturating concentration of 50 μM. Initial rates were measured where <20% substrate conversion to product had occurred. The reaction was initiated by adding 0.1–4 mU of R67 DHFR or 0.1–0.6 mU of hDHFR to the reaction mix. The measurements were carried out using a Cary 100 Bio spectrophotometer (Varian Canada, Inc., ON, Canada). Data were fit to the Michaelis-Menten equation using nonlinear regression analysis with GraphPad Prism version 6.04 for Windows (GraphPad Software, San Diego, CA).
Concentration Effect in the Two-Phase Aqueous/[BMIm][PF6] System
To mimic the reduced volume of the aqueous phase upon increasing the concentration of the water-immiscible [BMIm][PF6] (to 10%, 20%, 40%, 60%, and 80% of [BMIm][PF6]), the concentration of DHF, NADPH, and enzyme was increased by a factor of 1.14, 1.15, 1.7, 2.5, and 5, respectively. For example, to mimic 60% of [BMIm][PF6], the concentration of DHF and NADPH was increased from 100 to 250 μM and the concentration of enzyme increased from 200 to 500 nM (R67 DHFR) or from 30 to 75 nM (hDHFR). Assays were carried out in 384- or 96-well plates as described above. When absorbance surpassed the linear detection range of the plate reader (1.5 absorbance units), the volume of the assay was decreased to diminish the path length.
Inhibition of hDHFR and R67 DHFR
MTX was dissolved in 0.05 M KOH and quantified by spectrophotometry in 0.1 M NaOH using ɛ258nm = 22,100 M−1 cm−1.
30
PMTX was dissolved and quantified in 0.9% NaCl (154 mM) (ɛ226nm = 31,200 M−1 cm−1).
30
Dilutions were made in the appropriate pure cosolvent except for R67 DHFR inhibition where dilutions of PMTX were made in 0.9% NaCl (154 mM) for assays with organic solvent or in 50 mM potassium phosphate, pH 7.0, for assays with IL. Molecules
Thermal Scanning Fluorimetry Shift Assays
Differential scanning fluorimetry (DSF) can be used to determine the thermal stability of a protein or a protein–ligand complex. It exploits the difference in quenching of a fluorescent dye when in a polar environment such as aqueous solution relative to a hydrophobic region such as the exposed hydrophobic core of a denatured protein. 31 DSF assays were performed as previously described. 32 For hDHFR, 2 or 4 μM of enzyme was assayed with 2.5 × and 3.33 × SYPRO Orange (5000 × solution in DMSO from Invitrogen). For R67 DHFR, 5 or 10 μM of enzyme was assayed with 2.5 × , 3.33 × , and 5 × SYPRO-Orange. Assays were conducted in 50 mM potassium phosphate, pH 8, with 1% DMSO as the carrier of SYPRO Orange in a final volume of 20 μL per well. Control reactions were performed in the absence of enzyme or cosolvent. The 96-well plates were sealed with optically clear sealing tape (Sarstedt). The plate was heated from 20°C to 95°C in a LightCycler 480 apparatus (Roche) with a ramp speed of 0.02°C s−1. Twenty acquisitions/°C were obtained with 1-s exposure time. A CCD camera measured the fluorescence using λexc = 483 nm and λem = 568 nm.
Results and Discussion
Cosolvents Affect the Activity of R67 DHFR and hDHFR in Distinct Ways
To perform fragment screening, cosolvents are required to solubilize the test compounds. At early-stage screens for hits, assays should be performed in the millimolar range to increase the likelihood of observing even weak inhibition as in the case of fragment-based design, thus requiring effective solubilization without denaturing the target enzyme. While screening for inhibitors of R67 DHFR, inhibitor selectivity was verified by assaying hDHFR in parallel. Their structural differences are such that the effect of a cosolvent on one DHFR may not be predictive of its effect on the other DHFR. To this effect, we investigated the impact of the commonly used organic cosolvents, MeOH, ACN, DMF, and DMSO, on the activity of both DHFRs in concentrations ranging up to 25%. We note that the high volatility of MeOH required that it be handled with special consideration (Table 1).
Protocol Table for Cosolvent Analysis and Final Choice for Early- and Late-Stage Screening
1. Appropriate buffer for each enzyme assayed.
2. Freshly prepared stock solution: fivefold the assay concentration (100 μM of each, final).
3. Choose cosolvent that minimally affects enzyme activity: DMSO, DMF to dissolve high concentrations for early screening, and MeOH for late screening using lower compound concentrations; ensure no visible precipitation. Note that compounds should be dissolved immediately before use unless it is known that compounds remain stable upon storage in each given cosolvent.
4. Careful mixing.
5. Same cosolvent as in Step 3. Careful observation should be made to ensure that automated pipetting of MeOH volumes is reproducibly aspirated and dispensed rapidly to prevent loss (dripping) due to low surface tension. If loss is observed, it can be circumvented by conditioning (prewetting) tips, by aspirating, dispensing, and aspirating the desired volume again to prevent leaking. Finally, enzyme assays were conducted at a smaller scale (for example, one single row or column of a multiwell plate) in the presence of MeOH to prevent significant evaporative loss.
6. Same buffer as the enzyme solution.
7. Analysis of the initial rate: ≤10% substrate consumption; visual inspection of assay wells to confirm no precipitation has occurred.
DHF, Dihydrofolate; DHFRs, dihydrofolate reductases; hDHFR, human DHFR; DMSO, dimethylsulfoxide; DMF, dimethylformamide; MeOH, methanol.
In the case of R67 DHFR, increasing concentrations of each of the tested cosolvents resulted in a gradual decrease of the initial reaction rate (Fig. 2A). While DMSO was the least disruptive toward activity of R67 DHFR at 5% cosolvent (85% activity remaining, compared with ≈50%–60% for all others), 10% DMSO resulted in a drop in activity to 30%, as did 10% DMF. Higher concentrations of DMSO and DMF dropped activity to below 10% and were not further considered. MeOH and ACN both retained nearly 60% of the enzyme activity at 10% cosolvent and 30% activity at 15% cosolvent.
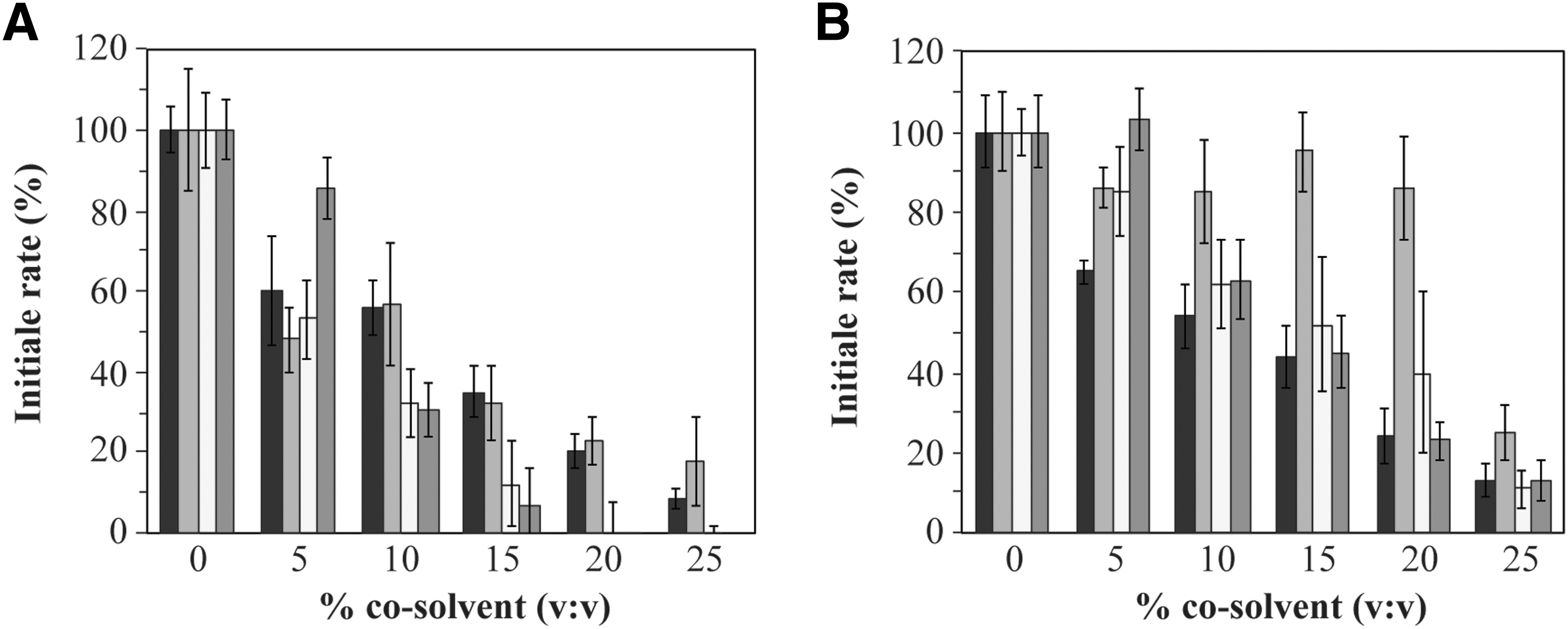
Initial rate of the enzymatic reaction for
In the case of hDHFR (Fig. 2B), a similar effect of DMSO was observed, where 5% DMSO had no effect on activity, while 10% DMSO reduced activity to 60%. MeOH was the most disruptive to activity when used at 5% (≈60% activity remaining), although its effect at higher concentrations was similar to that of DMF and DMSO (≈50%–60% activity remaining at 10% cosolvent and ≈45%–55% activity remaining at 15% cosolvent). Strikingly, the addition of 5%–20% ACN scarcely reduced hDHFR activity (≈90% activity remaining).
Because all organic solvents tested significantly reduced DHFR activity, ILs were tested as alternative cosolvents. The most widely studied ILs in biocatalysis using oxidoreductases such as dehydrogenases and peroxidases are the imidazolium-based ILs, bearing different counterions, including PF6 −, BF4 −, OctylSO4 −, MeSO4 −, and NTf2 −. 16,18,33 –37 While we previously demonstrated that hDHFR is active when surface-immobilized using an imidazolium bromide IL self-assembled monolayer, 38 to our knowledge, no study has yet reported on the effect of ILs on any DHFR in solution. In this study, we tested two water-miscible ILs, [BMIm][OctSO4] and [BMIm][BF4], and water-immiscible IL, [BMIm][PF6], as cosolvents for R67 DHFR and hDHFR.
Activity of R67 DHFR decreased gradually with increasing concentrations of [BMIm][OctSO4] (Fig. 3A). At 10% and 40% of [BMIm][OctSO4], ≈75% and 50% of R67 DHFR activity remained, respectively. In contrast, hDHFR lost all activity even at the lowest concentration of [BMIm][OctSO4] tested (5%). Changing the counteranion to procure [BMIm][BF4] produced very different results: hDHFR was fully active up to 10% [BMIm][BF4], while only 5% [BMIm][BF4] reduced R67 DHFR activity to 65% (Fig. 3B). Tests at higher concentrations of [BMIm][BF4] were limited by increased solution acidity, 21 which is crucial for maintenance of the quaternary structure of R67 DHFR 39 and to maintain chemical integrity of the substrates. Accordingly, increasing the concentration of [BMIm][BF4] to 10% and 15% decreased the activity of R67 DHFR drastically to 17% and 5%, respectively. Approximately 85% inactivation of hDHFR was observed at 20% [BMIm][BF4].

Initial rate of the enzymatic reaction for R67 DHFR (light gray) and hDHFR (black) with a water miscible IL as cosolvents.
We also investigated the water-immiscible IL [BMIm][PF6]. Water-immiscible cosolvents provide an organic phase with the potential for better dissolution of hydrophobic compounds. Partition of the components through the interface of the biphasic system has been investigated in a number of systems. 40,41 However, water-immiscible organic solvents may result in enzyme denaturation at the aqueous interface; less is known about the effect of biphasic ILs on enzymes, particularly with respect to water-soluble enzymes.
Enzyme activity of both DHFRs increased with increasing concentrations of [BMIm][PF6] (Fig. 4). Indeed, at 70%–80% [BMIm][PF6], the relative activity of both enzymes was ≥200% of the initial activity. For these enzyme assays, the initial quantity of the substrate DHF, the cofactor NADPH, and the DHFR enzyme was held constant, while the variables were the volumes of IL and aqueous buffer (with final volume held constant). Our results suggest that water-soluble DHFRs and reagents become more concentrated as the volume of the aqueous phase shrinks, with poor partitioning into the increasing volume of nonmiscible IL. To take this into account, the concentrations of substrate, cofactor, and enzyme were recalculated as a function of only the volume of aqueous phase. For example, at 80% volume of IL, the aqueous phase (20% volume) will contain a five times greater concentration of enzyme and reagents.
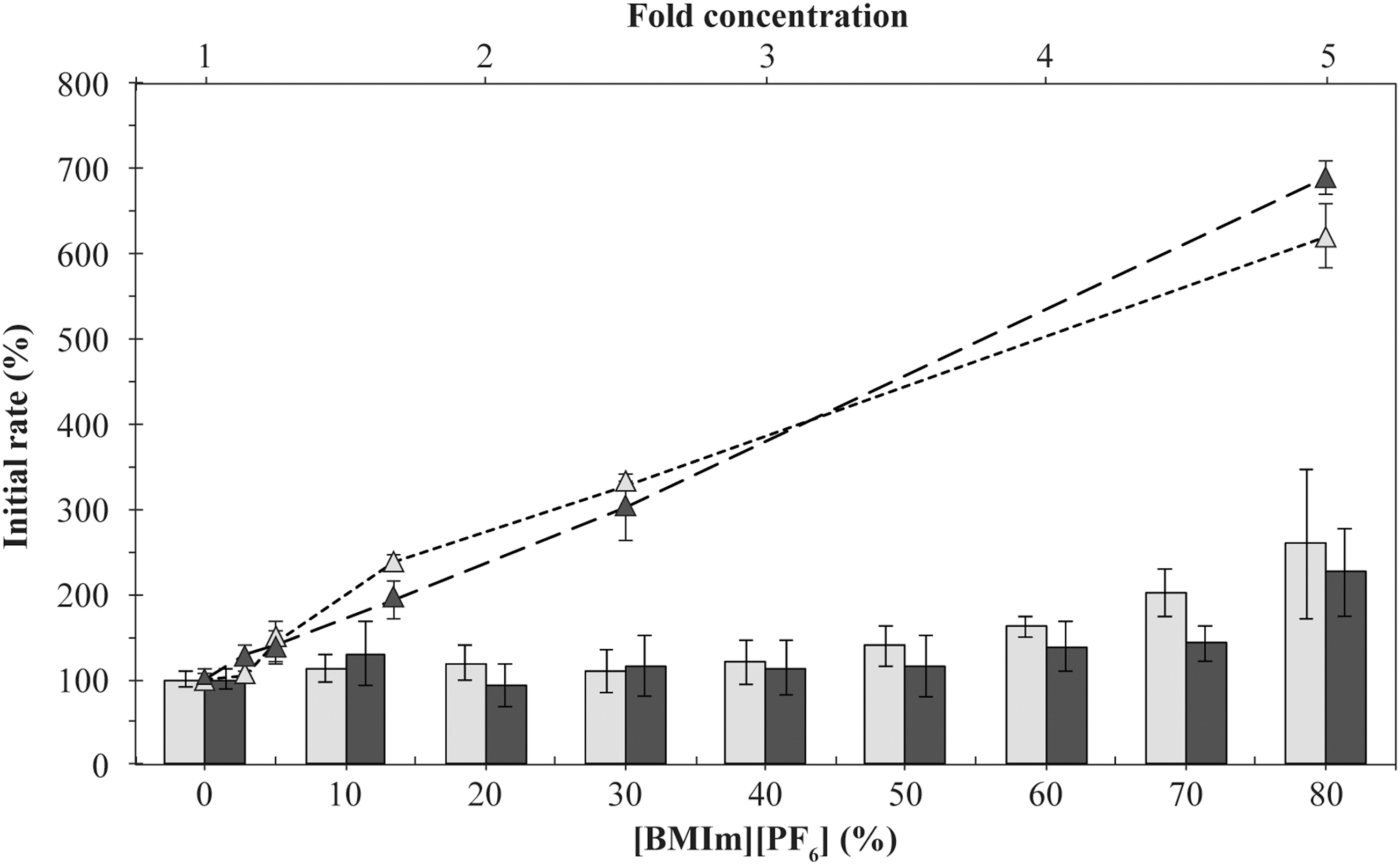
Initial rate of the enzymatic reaction for R67 DHFR and hDHFR with the water-immiscible IL [BMIm][PF6], and expected increase in enzyme activity if reactants and enzyme partition fully into the aqueous phase. The bar chart plots the R67 DHFR (light gray) and hDHFR (black) activities with increasing concentrations of [BMIm][PF6]. Enzyme activity in absence of cosolvent was the reference value. The scatterplot displays the effect of increasing concentrations of reaction components (DHF, NADPH and DHFR) on activity of R67 DHFR (light gray triangles) and hDHFR (black triangles), as described under Results. The fold-concentrations of 1.0, 1.1, 1.3, 1.7, 2.5 and 5.0 mimic the residual aqueous volume at 0, 10, 20, 40, 60 and 80% [BMIm][PF6], respectively, assuming full partitioning of the enzyme and reactants in the aqueous phase. Values are given as the mean ± standard deviation for the results of three independent experiments performed in triplicate. BMIm, 1-butyl-3-methylimidazolium.
Upon applying the calculated concentrations to simple enzyme assays (in the absence of IL), enzyme activity increased with increasing concentration of enzyme and reagents, as expected (Fig. 4, dashed lines). However, the increase was greater than that observed in the presence of [BMIm][PF6], reaching 600%–700% of the control (standard reagent concentrations) under conditions mimicking 80% [BMIm][PF6]. The increased activity observed at high concentrations of [BMIm][PF6] is thus less than expected if the enzyme and reactants remained entirely in the aqueous phase. Analysis of the protein concentration remaining in the aqueous phase after incubation with 80% [BMIm][PF6] indicates that only ∼30% of R67 DHFR remained (Supplementary Fig. S1; Supplementary Data are available online at
Overall, the cosolvent system offering the highest conservation of enzyme activity for R67 DHFR was 10%–15% of [BMIm][OctSO4], MeOH, or ACN and for hDHFR was [BMIm][BF4], MeOH, ACN, DMSO, or DMF. The differences in cosolvent preference of both DHFRs underscore the empirical nature of choice of cosolvent for a given enzyme. The concentrations of organic cosolvents chosen for further experiments were 15% ACN, 10% DMF, 10% MeOH, and 10% DMSO, as well as 10% [BMIm][OctSO4] for R67 DHFR and 10% [BMIm][BF4] for hDHFR. It is noteworthy that activity of both DHFRs was not significantly affected by 10%–20% [BMIm][PF6]. While it could be considered a potential cosolvent system compatible with activity of both DHFRs, the unknown partitioning of reagents and, crucially, of the various compounds being screened led us to discount the biphasic IL [BMIm][PF6] as a potential screening cosolvent.
Effect of Cosolvents on Inhibition of R67 DHFR and of hDHFR
To verify whether the cosolvent systems promote native-like inhibitor binding, we investigated the reproducibility of R67 DHFR inhibition with inhibitors
Molecules
IC50 and LE of MTX and PMTX Against hDHFR and IC50 of 1, 2, and PMTX Against R67 DHFR in Organic Cosolvents and ILs
Values are given as the mean ± standard deviation for the results of two independent experiments performed in triplicate.
IC50 values are in μM and LE in kcal/(mol × number of heavy atoms).
—, Is not an inhibitor of that DHFR.
MTX, methotrexate; PMTX, pemetrexed; BMIm, 1-butyl-3-methylimidazolium; NS, not soluble; ND, not determined, enzyme is inactive in that cosolvent; IL, ionic liquid.
The same trend for PMTX inhibition was observed for hDHFR, with three- to fourfold increases in IC50 PMTX and with 0.02–0.03 difference in LE, except in the presence of ACN (>20-fold increase and 0.06 difference, respectively) and [BMIm][BF4] (nearly 30-fold increase and 0.07 difference, respectively). MTX inhibition of hDHFR showed yet greater maintenance of IC50 MTX, remaining within twofold except a threefold increase with ACN (LE values varying by 0.01–0.02).
While ACN effectively maintained high activity of both DHFRs (Fig. 2), it was the most disruptive with respect to IC50 values and may thus bias inhibitor-screening results. Similarly, [BMIm][BF4] procured high activity of hDHFR, but poor conservation of IC50 PMTX for both DHFRs. Notably, [BMIm][BF4] did not alter MTX inhibition. Globally, our results indicate that the cosolvents investigated generally maintained inhibitor binding and may thus be considered for inhibitor screening.
Efficiency of Cosolvents in Solubilizing Compounds for Inhibitor Discovery
For fragment screening purposes, stock solutions of compounds can be made in pure solvent, then diluted in aqueous buffer to 10% cosolvent. 43 We thus verified the capacity of pure solvents to solubilize mM concentrations of a set of small organic molecules that we had previously screened against both DHFRs (Supplementary Table S1). 1 Those molecules are often nitrogen-containing heterocycles, chosen in loose analogy to folates, and thus represent a moderate chemical diversity. DMSO and DMF performed equally well and were more efficient at solubilizing the compounds of interest than were MeOH and ACN. However, as shown above, both 10% DMSO and 10% DMF resulted in a loss of ≥70% of activity for R67 DHFR and, as such, do not represent ideal screening conditions.
The two water-miscible ILs, [BMIm][OctSO4] and [BMIm][BF4], were only moderately effective in solubilizing mM concentrations of the small hydrophobic test compounds (Supplementary Table S1). [BMIm][OctSO4] dissolved slightly more than half of the compounds, while [BMIm][BF4] dissolved less than half of the compounds. The high viscosity of pure [BMIm][OctSO4] made it a difficult medium in which to work. Thus, despite procuring good retention of enzyme activity, the narrow capacity of these ILs for dissolution of small organic compounds indicates that they are not ideal cosolvents for screening hydrophobic compounds in high concentrations, such as required for early-stage screening.
We have thus shown that 10% of MeOH, DMSO, DMF, [BMIm][BF4], [BMIm][OctSO4], and 15% ACN could maintain sufficient activity of both DHFRs to conduct fragment screening assays with confidence. DMSO and DMF were shown to be most efficient in solubilizing hydrophobic compound-based libraries in the high mM range and could thus be choice cosolvents for early-stage screening, whereas MeOH, [BMIm][BF4], and [BMIm][OctSO4] could be suited to screening more hydrophilic compound-based libraries and/or lower concentrations of compounds when screening more advanced compound generations. In all these studies, compounds were dissolved immediately before use. The cosolvents resulted in modest increases in IC50 for the inhibitors tested, generally within threefold. A protocol table reporting guidelines for setting up fragment screening is presented in Table 1. To clarify the effects of the cosolvents on both DHFRs, we investigated their effects on enzyme kinetics and thermostability in more detail.
Effect of Cosolvents on Catalytic Parameters and Protein Thermostability
The decreased enzyme activity and altered inhibition may result, among other causes, from direct enzyme inhibition by the cosolvent or from partial enzyme denaturation. To distinguish between these possibilities, we investigated the kinetic parameters k cat (turnover number) and KM (productive affinity) using the natural substrates (Table 3) and determined Tm values (melting temperatures) through DSF analysis to verify whether cosolvents induce enzyme denaturation or destabilization (Table 4). Thus, through kinetic and thermostability studies, we sought to understand if cosolvents act not only as solvent molecules but also as ligands or inhibitors.
Kinetic Constants for the Reaction of DHF and NAPDH by R67 DHFR and hDHFR
Values are given as the average ± standard deviation from the mean of experiments performed in triplicate.
ND, not determined; enzyme activity was too low.
KM and k cat/KM estimated; the lowest substrate concentration used was saturating.
KM not determined; substrate saturation was not achieved at 100 μM.
ND, not determined; Vmax was not reached.
ACN, acetonitrile.
Tm (°C) Values of R67 DHFR and of hDHFR from Thermal Scanning Fluorimetry Shift Assays
DMF was used at 5% because thermal denaturation curves performed in 10% DMF were not interpretable.
ND, activity of the enzyme being too low or undetectable in that cosolvent.
ND, even at 5% of [BMIm][OctSO4], the fluorescence signal decreased in a sigmoidal-shaped curve, while temperature increased.
The fluorescence signal decreased as the temperature increased.
The cosolvents generally resulted in a reduction of k cat/KM NADPH and k cat/KM DHF. This was mainly due to variation of KM (up to 35-fold) rather than k cat (no more than fivefold). This implies that cosolvents disrupt productive binding of the substrates to a greater extent than the rate of product formation. In the case of R67 DHFR, the reference kinetic values determined in the absence of cosolvent were consistent with those previously reported (Table 3). 26 Marginal decreases were observed for k cat/KM NADPH (two- to fourfold) in organic cosolvents, whereas significant reductions were observed for k cat/KM DHF (6- to 170-fold). According to those results, the catalytic efficiency of R67 DHFR was the highest in MeOH, followed by DMSO, DMF, and [BMIm][OctSO4], whereas it was lowest in ACN.
We investigated whether the disruption of substrate binding resulted from structural alteration of the enzyme by determining its thermal stability. No significant difference in Tm was observed in 10% DMSO (ΔTm = 1.1°C) and a slightly lower thermostability (ΔTm = 2.2–3.0°C) was observed in DMF, MeOH, and ACN, reflecting slight structural destabilization (Table 4). We note that thermal denaturation was performed in only 5% DMF rather than 10% DMF; the latter conditions were inconclusive as multiple minima were observed. Thus, the secondary structure of R67 DHFR is at most slightly destabilized by those cosolvents.
The thermal denaturation curves in as low as 5% [BMIm][OctSO4] were uninterpretable because the fluorescence signal decreased as the temperature increased, where the fluorescence signal should increase as the protein unfolds. This could be caused by the propensity of ≥1% [BMIm][OctSO4] to form micelles. 44 We note that the Tm values reported here by DSF with SYPRO-Orange in 50 mM potassium phosphate, pH 8.0, differ from values reported using differential scanning calorimetry in different conditions (50 mM acetic acid +50 mM MES +100 mM Tris +10 mM β-mercaptoethanol, pH 5.0, and 5 mM dithiothreitol). 45 That acidic pH was used to monitor the pH-dependent dissociation of the R67 DHFR intimate dimers into monomers rather than reporting on denaturation of tetrameric species, as investigated here.
A further potential contributor to the altered kinetic parameters is different accessibility of solvent molecules into the active site that could interfere with catalysis. Interestingly, the observed variations of KM NADPH and KM DHF for R67 DHFR are consistent with earlier studies regarding the perturbation of solvent composition on the catalysis of this enzyme where NADPH and DHF bind the same interface in the symmetric active site of R67 DHFR. 4,46 In that work, Chopra et al. 4 showed that R67 DHFR binds NADPH using a dry interface, in contrast to DHF that interacts with R67 DHFR less directly using a wet interface through water contacts. The binding of NADPH to R67 DHFR generates the net release of 38 water molecules, whereas binding of DHF to R67 DHFR is accompanied by the net uptake of water. 4 R67 DHFR active site forms an hourglass-shaped tunnel that bisects the tetramer and fits one molecule of NADPH and one of DHF. The tunnel is 24 Å long, 11 Å wide at the central narrows, and 18 Å wide near each end of the tunnel. 47 Thus, a large population of solvent molecules occupies the voluminous R67 DHFR active-site cavity.
If water is involved in a binding interaction, perturbation of water content should alter its affinity for the enzyme. 46 The large and mostly hydrophobic active site of R67 DHFR could be accessible to cosolvent molecules, perturbing the water content. The cosolvent molecules could enter completely or partially in R67 DHFR active site as their lengths vary from 1.5 to 12.1 Å (water <MeOH ≈ ACN <BF4 − <DMF <DMSO <BMIm+ <OctSO4 −). Replacement of water molecules by cosolvent could reduce the water contacts required for DHF binding, but would have little effect on the dry interface of NADPH binding. 4 Indeed, the greater observed variations in KM DHF (5- to 36-fold) than for KM NADPH (one- to sixfold) in the presence of cosolvents are coherent with alteration of the water content by the addition of those cosolvents, consistent with the study by Chopra et al. 4
In the case of hDHFR, only small decreases were observed for all k cat/KM in organic cosolvents (two- to sixfold) (Table 3). As observed for R67 DHFR, the catalytic efficiency of hDHFR was the highest in MeOH, followed in this case by DMF and DMSO, and is lowest in ACN and in [BMIm][BF4]. The small increases observed in KM for hDHFR might be explained by different accessibility of solvent molecules into the active site to interfere with catalysis. In contrast to R67 DHFR, the hDHFR active site cleft is protected by three loops that allow adoption of occluded and closed conformations upon substrate binding, formation of intermediates, and release of products. 48,49 Those loops limit access of solvent molecules into the active site during the catalytic cycle, where one of the loops remains in a closed conformation. 48,49 At the widest point of the loop movements, the active site cleft opens by only ∼3 Å. 49 This distance would allow water, MeOH, or ACN to enter the active site, but would not suffice to allow the larger DMF, DMSO, [BMIm][BF4], or [BMIm][OctSO4] molecules to enter. Thus, active site accessibility of cosolvents is probably more limited for hDHFR than R67 DHFR, consistent with the slighter effect on productive affinity for hDHFR than R67 DHFR.
Investigation of thermal stability revealed slight decreases in Tm in MeOH and ACN (ΔTm = 1.1–1.5°C). Greater differences were observed in DMSO, DMF, and [BMIm][BF4] (ΔTm = 2.5–4.4°C), reflecting structural destabilization (Table 4). We note that thermal denaturation was performed in only 5% DMF or [BMIm][BF4] because 10% of those cosolvents resulted in an anomalous fluorescence signal, as seen for DMF with R67 DHFR. Our results demonstrate that hDHFR is more susceptible to thermal destabilization by cosolvents than R67 DHFR. This is consistent with the lower thermostability of hDHFR (Tm = 40.1°C) relative to R67 DHFR (Tm = 53.7°C) under native conditions, suggestive in itself of greater thermosensitivity.
Overall, our results show that decreases in k cat/KM of both DHFRs in organic and IL cosolvents were mainly induced by increased KM, yet the mechanism underlying those changes differs for both DHFRs: cosolvents interfere with substrate binding to a greater extent in R67 DHFR, while they result in greater thermal instability of hDHFR.
In conclusion, the preferred cosolvent for R67 DHFR is MeOH, ACN, or [BMIm][OctSO4], while hDHFR is most compatible with MeOH, DMSO, DMF, [BMIm][BF4], or ACN. MeOH may offer the best compromise between retention of activity of both test enzymes (>50%) with good capacity to solubilize test compounds and to verify known IC50 values. DMSO and DMF remain reasonable alternatives to MeOH: although they reduced the activity of both DHFRs to a greater extent than MeOH, both DMSO and DMF solubilized almost all the selected compounds in millimolar range and were the only cosolvents tested that solubilized inhibitors
ILs were the least effective in solubilizing test compounds. Overall, the BMIm-based ILs investigated do not offer any clear advantage in screening hydrophobic compound libraries relative to organic cosolvents; while their low volatility can allow for longer working periods in open air than organic cosolvents, their higher cost is a further disadvantage. DMSO and DMF could thus be choice cosolvents for early-stage fragment screening where effective compound solubilization is a primary concern. MeOH, however, is suited to screening more hydrophilic or more advanced compound generations where lower compound concentrations are required and where maintenance of native-like enzyme properties becomes the primary concern.
Footnotes
Acknowledgments
The authors thank Daniela Quaglia for helpful discussions. This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), as well as PROTEO, the Québec Network for Research on Protein Structure, Function and Engineering, which is funded by les Fonds Québécois pour la Recherche sur la Nature et les Technologies (FRQ-NT). J.L.T., N.K., and D.B. received scholarships from PROTEO. J.L.T. received a scholarship from Faculté des études supérieures et post-doctorales de l'Université de Montréal (FESP) and S.M.J.A. received a scholarship from Groupe de recherche universitaire sur le médicament (GRUM)-FESP.
Disclosure Statement
No competing financial interests exist.