
Abstract
Discovering of new and effective antibiotics is a major issue facing scientists today. Luckily, the development of computer science offers new methods to overcome this issue. In this study, a set of computer software was used to predict the antibacterial activity of nonantibiotic Food and Drug Administration (FDA)-approved drugs, and to explain their action by possible binding to well-known bacterial protein targets, along with testing their antibacterial activity against Gram-positive and Gram-negative bacteria. A three-dimensional virtual screening method that relies on chemical and shape similarity was applied using rapid overlay of chemical structures (ROCS) software to select candidate compounds from the FDA-approved drugs database that share similarity with 17 known antibiotics. Then, to check their antibacterial activity, disk diffusion test was applied on Staphylococcus aureus and Escherichia coli. Finally, a protein docking method was applied using HYBRID software to predict the binding of the active candidate to the target receptor of its similar antibiotic. Of the 1,991 drugs that were screened, 34 had been selected and among them 10 drugs showed antibacterial activity, whereby drotaverine and metoclopramide activities were without precedent reports. Furthermore, the docking process predicted that diclofenac, drotaverine, (S)-flurbiprofen, (S)-ibuprofen, and indomethacin could bind to the protein target of their similar antibiotics. Nevertheless, their antibacterial activities are weak compared with those of their similar antibiotics, which can be potentiated further by performing chemical modifications on their structure.
Introduction
In May 2016, a nightmare had come true, after China, researchers in the United States reported first case of resistant bacteria that overcame one of the strongest antibiotics found in our arsenal, 1 which poses a real threat for global health. Unfortunately, finding new and effective antibiotics is very challenging. Mainly, because of the fact that most of the antibiotics are natural or semisynthetic compounds that comprise complex structures and unique chemical properties, 2 which make the process of finding compounds with these features very hard. 3 Recently, the development of chemoinformatics and bioinformatics made a breakthrough in the discovery process, paving the way to virtual screening (VS) and its methods. 4 In general, two main approaches exist for VS, the first one is the ligand-based virtual screening (LBVS), which relies on molecular similarity to find new drugs, 5 taking benefit from the general assumption that similar compounds may have similar biological activity. 6 The second approach is structure-based virtual screening (SBVS), which depends on the knowledge of target's structure and characteristics. 7 The most commonly used method in SBVS is molecular docking. 8 This method works by modeling the interaction between a chemical compound and a protein at the atomic level to predict the behavior of the compound in the binding site of target proteins. 9,10
In this study, LBVS was applied using rapid overlay of chemical structures (ROCS) software (ROCS 3.2.0.4; OpenEye Scientific Software, Santa Fe, NM;
This research used the Food and Drug Administration (FDA)-approved drugs as database as a response to the call by NIH associate director for science policy to find new uses for the current approved drugs. 12
Most of the FDA-approved drugs have simple structure and low molecular weight. Hence, the structure simplicity of the antibiotic and its broad antibacterial activity spectrum were considered to select one representative antibiotic from each of 12 antibiotic chemical families; except for quinolones, one antibiotic from each generation was included (the fourth generation was not investigated). Also, for beta-lactams, one member from each subfamily was considered.
Furthermore, clavulanic acid that is a broad-spectrum beta-lactamase inhibitor was included because evidence suggests that it has minimal antibacterial activity in vitro. 13
To verify ROCS results, disk diffusion susceptibility test was applied according to Clinical Laboratory Standards Institute document M02-A12. 14 Nevertheless, the antibacterial activity tests such as disk diffusion, minimum inhibitory concentration, or minimum bactericidal concentration do not give any explanation about the mechanism of action of antibacterial activity for the tested compound. However, regarding this study, the similarity between the active candidates and the antibiotics gives a good probability that candidates could bind to the same antibiotic targets. Hence to prove that, the candidates have to be tested on isolated targets or use more advanced methods, which would require special instruments and high skills in addition to high costs. 15,16 Luckily, the development of molecular docking has presented a fast, cheap, and easy way to predict the binding of a compound to a target receptor. 17
In this study, OpenEye HYBRID,
18
which is a SBVS software, was used (OEDOCKING 3.2.0.2; OpenEye Scientific Software;
Materials and Methods
Chemoinformatics
First, the simplified molecular-input line-entry system (SMILES) for the 1,991 FDA-approved drug (as the compounds database) and 17 selected antibiotics (see List of Selected Antibiotics section) were extracted from DrugBank v4.5.0 approved drugs Structure Data Format file 19 for the former and downloaded from PubChem 20 for the latter.
Second, the SMILESs of compounds were processed by OpenEye OMEGA (OMEGA 2.5.1.4; OpenEye Scientific Software, Santa Fe, NM.
By default, OMEGA does not apply a number of rotatable bonds cutoff but it limits the amount of time spent generating conformers for each molecule by 120 s. Also, conformers with strain energies >10 kcal/mol threshold are rejected, and minimum root mean square distance (RMSD) between two conformers below which they are considered as duplicates is set at 0.5 Å.
In addition, OMEGA was able to create an ensemble of stereoisomers for compounds that had unspecified stereocenters (up to five centers for each molecule) before passing the molecules for conformer generation. The previous step was unnecessary to the antibiotics because the isomeric SMILESs were available.
Third, ROCS was used to screen the resulted conformers of the compounds database to find compound conformers that are structurally similar to the selected antibiotics. The TanimotoCombo score, calculated by ROCS, was used to rank the candidates. This score is a combination of molecular volume similarity (shape) and chemical types of atoms similarity (color) between two compounds, with a full mark of 2 (1 for shape and 1 for color). Finally, the candidate drugs were chosen according to TanimotoCombo score and their availability in the local market.
List of Selected Antibiotics
Ampicillin (CID: 6249), azithromycin (CID: 447043), cephradine (CID 38103), chloramphenicol (CID: 5959), ciprofloxacin (CID: 2764), clavulanic acid (CID: 5280980),
Antibacterial Activity Test
To determine the antibacterial activity of the candidate drugs (List of Candidate Drugs section), the Kirby–Bauer disk diffusion susceptibility method was used, which is a simple and straight forward method to measure the activity of chemicals based on the radius of inhibition zone surrounding the chemical loaded disk on the Mueller–Hinton agar plate. 21,22
The Kirby–Bauer disk diffusion susceptibility method protocol was applied according to Clinical Laboratory Standards Institute document M02-A12, 14,20 barring the exception of the nonantibiotic diffusion disk preparation step.
First, the 4 mm thick Mueller–Hinton agar (HiMedia Laboratories, India) was prepared in 90 × 15-mm plates (Citotest, China) according to the manufacture recipe. Second, to prepare disks containing 500 and 1,000 μg from each drug, a specific amount of each drug was dissolved in 1 mL of suitable solvent (acetone, methanol, ethanol from ChamLab, Syria; or distilled water) (see the List of Candidate Drugs with Their Solubility section). Third, specific volumes from each drug solution were loaded on 6 mm blank diffusion disks (Whatman, United Kingdom) using micropipettes (Dragon Laboratory Instruments, China) and were dried at 37°C. Also, to rule out any antibacterial effect that results from the solvents, control disks from each solvent were prepared. Fourth, the agar plates were inoculated separately with 0.5 McFarland suspension of Escherichia coli (identified microscopically by Gram stain, eosin methylene blue agar from HiMedia Laboratories 23 ), and Staphylococcus aureus (identified microscopically by Gram stain, catalase, and coagulase tests 23 ) was taken from the microbiology laboratory in Arab International University, and the prepared drug disks plus the reference antibiotic disks (see List of Reference Antibiotic Disks and Their Load of Antibiotic section) were placed on the inoculated plates. Finally, the plates were incubated for 24 h at 37°C and the zones of inhibition were measured for each candidate drug (Table 1).
Compound Similarities and Antibacterial Activities Results
The bacterial susceptibility profile, against antibiotics, was retrieved from manufacturing companies.
Candidate's average inhibition zone in millimeter (mm) at 500/1000 μg. The standard deviations of all measurements were between 1.41 and 0.70 mm.
Resistant.
Used in standardized disk form (see List of Reference Antibiotic Disks and Their Load of Antibiotic section for quantities).
Susceptible.
Intermediate.
The compound is not available for test.
Set for docking purpose.
Unknown because the bacteria susceptibility against these antibiotics has not been set by the manufacturing company.
−, negative.
List of Candidate Drugs with Their Solubility in 1 mL of Solvent
Acetone 98%:25 mg Hydrochlorothiazide (Changzhou Pharmaceutical Factory, China).
Ethanol 98%:50 mg aspirin (Novacyl, France), 50 mg diclofenac sodium (Aarti Drugs Ltd., India), 50 mg drotaverine hydrochloride (Ra Chem Pharma Ltd., India), 50 mg ephedrine (Taj Pharmaceuticals Limited, India), 50 mg flurbiprofen (FDC Limited, India), 50 mg ibuprofen (Shasun, India), 50 mg indapamide (Ami Life Sciences Pvt. Ltd., India), 25 mg indomethacin (AS Joshi & Company, India), 50 mg ketoprofen (Yibin Hope Pharmaceutical, China), 12 mg lamotrigine (Divi's Laboratories Limited, India), 50 mg pseudoephedrine (Taj Pharmaceuticals Limited), 50 mg salbutamol sulfate (FDC Limited), 50 mg salicylic acid (Merck KGaA, Germany), and 50 mg warfarin sodium (Hubei Ocean BiotechCo., Ltd., China).
Ammonia 25%:50 mg theophylline (Taj Pharmaceuticals Limited).
Methanol 98%:25 mg clopidogrel bisulphate (IOL Chemicals and Pharmaceuticals Limited, India), 50 mg gliclazide (Zhejiang Jiuzhou Pharmaceutical Co., Ltd., China).
NaOH 5%:50 mg allopurinol (Zhejiang Ruiqing Pharmaceutical, India), 50 mg furosemide (AMRI, India).
Distilled water:25 mg betahistine (Kinfonpharma, China), 50 mg anhydrous caffeine (BASF, Germany), 50 mg captopril (Shandong Weifang Pharmaceutical, China), 50 mg guaifenesin (Granules India Limited, India), 50 mg lidocaine (Neha Pharma Pvt. Ltd., India), 50 mg metformin (Aarti Drugs Ltd.), 50 mg metoclopramide (SL Drugs and Pharmaceuticals, India), 50 mg niacin (Aarti Drugs Ltd.), 50 mg ondansetron (Shodana, India), 50 mg phenylephrine (Taj Pharmaceuticals Limited), 50 mg piracetam (Jiangxi Yuehua Pharmaceutical, China), 25 mg pregabalin (Changzhou Pharmaceutical Factory), 50 mg pyridoxine hydrochloride (Excellent Health Products Co., Ltd., China), 50 mg thiamine hydrochloride (Excellent Health Products Co., Ltd.).
List of Reference Antibiotic Disks and Their Load of Antibiotic
Azithromycin 15 μg, meropenem 10 μg, nalidixic acid 30 μg, and tetracycline 30 μg from Bioanalyse (Turkey). Chloramphenicol 30 μg, ciprofloxacin 5 μg, gentamicin 10 μg, and levofloxacin 5 μg from Abtek (United Kingdom).
Molecular Docking Protocol
First, the complex of the antibiotic or its relative (same chemical family) and its target receptor was downloaded from Protein Data Bank (PDB)
The docking type performed by HYBRID is rigid and thereby cannot assign flexible chains in receptors.
Finally, OpenEye VIDA 4.3.0.4 was used to show the predicted binding pose in the receptor-binding pocket compared with the binding pose of the crystal ligand.
List of Crystal Structures of Protein–Ligand Complexes
2XCS: “The 2.1 Å crystal structure of S. aureus gyrase complex with GSK299423 and DNA.” 25 Resolution: 3.35 Å, protein name: DNA gyrase, type II topoisomerase (catalytic activity: ATP-dependent breakage, passage, and rejoining of double-stranded DNA). Gene names: gyrB (Ordered Locus Name: SA0005) and gyrA (Ordered Locus Name: SA0006).
Description: The structure 2XCS has in total four chains. Two identical polypeptide (L)s: B, D each one has length of 692 residues that form two subunits A and B of DNA gyrase. Two polydeoxyribonucleotides E and F, each one has length of 20 residues. Chains F, E, B, and D in complex with GSK299423.
2XCT: “The twinned 3.35 Å structure of S. aureus gyrase complex with ciprofloxacin and DNA.” 25 Resolution: 3.35 Å. Protein name: DNA gyrase, type II topoisomerase (catalytic activity: ATP-dependent breakage, passage, and rejoining of double-stranded DNA). Gene names: gyrB (Ordered Locus Name: SA0005) gyrA (Ordered Locus Name: SA0006).
Description: The structure 2XCT has in total 12 chains. Four identical polypeptides (L): B, D, S, and U, each one has a length of 692 residues that form four subunits: two A and two B of DNA gyrase. Four polydeoxyribonucleotides: E, F, V, and W, each one has a length of eight residues. Four polydeoxyribonucleotides: G, H, X, and Y, each one has a length of 12 residues and in complex with ciprofloxacin. Each of the chains B, D, F, G, S, U, and W binds manganese ion (Mn2+).
4ALI: “Crystal structure of S. aureus FabI in complex with NADP and triclosan (P1).” 26 Resolution: 2.1 Å. Protein name: NAD(P)H-dependent trans-2-enoyl acyl carrier protein (ACP) reductase. Oxidoreductase. (Catalytic activity: an acyl-[ACP]+NADP+ = a trans-2,3-dehydroacyl-[ACP]+NADPH). Gene name: fabI (Ordered Locus Names: b1288, JW1281).
Description: The structure 4ALI has in total eight identical polypeptide(L) chains: A, B, C, D, E, F, G, and H, each chain has a length of 282 residues and is in complex with triclosan and NADP+. Each of the chains A, B, C, E, F, and G is in complex with glutamic acid.
Results
To find drugs similar to the selected 17 antibiotics, the compounds of FDA-approved drug database that have TanimotoCombo score equal to or >1 and available from the commercial vendors were considered as candidates in our investigation. As shown in Table 1 (or Supplementary Table S1; Supplementary Data are available online at
E. coli has resisted four reference antibiotics (chloramphenicol, ciprofloxacin, levofloxacin, and, nalidixic acid) and S. aureus is susceptible to all tested reference antibiotics except for gentamicin.
Furthermore, E. coli also resisted all the candidates except for mild effect showed by lamotrigine (anticonvulsant). In contrast, nine compounds showed activity toward S. aureus. The active candidates ranked by their inhibition zone size are diclofenac, a nonsteroidal anti-inflammatory drug (NSAID), drotaverine (antispasmodic), flurbiprofen (NSAID), ibuprofen (NSAID), indomethacin (NSAID), metoclopramide (antiemetic), niacin (vitamin B3), allopurinol (uric acid lowering agent), and captopril (antihypertensive).
As shown in Table 1, ROCS did not find any nonantibiotic FDA-approved drug similar to azithromycin, fusidic acid, gentamicin, tetracycline, and vancomycin. Mainly, because all the TanimotoCombo scores calculated between those antibiotics and the nonantibiotic FDA-approved drugs were <1. Therefore, this confirms that those antibiotics have unique chemical structures (Supplementary Table S1) as mentioned previously in the introduction.
In general, ranking of compounds by ROCS TanimotoCombo score was different than their inhibition zone's ranking. Hence, a negative association between score and antibacterial activity could be noted, such as thiamine having good score but failed in antibacterial test or when indomethacin, drotaverine, flurbiprofen, ibuprofen, and diclofenac have lower TanimotoCombo scores than the other candidates but came up with good results in antibacterial test. Nevertheless, some compounds with relatively high score such as captopril and niacin have a detectable antibacterial effect and others laid on the score edge of the cutoff such as clopidogrel, gliclazide, and lidocaine had no antibacterial activity at all.
To give a possible explanation for the antibacterial activity showed by the active candidates, OpenEye HYBRID was used to dock the candidates that achieved an inhibition zone ≥10 mm, and they were indomethacin, drotaverine, ibuprofen, flurbiprofen, and diclofenac. In fact, those antibacterial active candidates were found to be similar to ciprofloxacin, levofloxacin, and nalidixic acid, which all belong to quinolones's chemical family that targets DNA gyrase. 27 Thus, the docking can be done on one receptor site for all. However, Bax et al. discovered that DNA gyrase could be inhibited by interacting with another site other than the classical site of quinolones, 25 where authors demonstrated that the inhibitor (GSK299423) was located between the DNA and a transient noncatalytic cavity on the twofold axis at the DNA gyrase A dimer interface, and was closed to the active sites and fluoroquinolone binding sites. 25 Hence, active candidates were docked on this new site. Moreover, diclofenac showed the best antibacterial action between the candidates, which is worth to be investigated more. As shown in Table 1, diclofenac shares similarity with triclosan. Therefore, docking it to triclosan receptor may give a different explanation to its activity.
Next, to elaborate the understanding of activity observed, the docking of most potent antibacterial activity candidates, drotaverine and diclofenac, was examined using OpenEye VIDA.
The three quinolone antibiotics and the active candidates were docked to all sites in 2XCS (one site) and 2XCT (four sites). Only triclosan and diclofenac were docked to all sites in 4ALI (eight sites). For each crystal, the results of site with best HYBRID scores were retained.
Minimum RMSDs between the ligand in crystal and the HYBRID's docked ligand (retrieved from the crystal) were 0.31, 0.75, and 0.6 Å for GSK299423, ciprofloxacin, and triclosan, respectively (Figs. 1 and 2). Hence, the rigid docking using HYBRID could reflect possible molecular ligand position into the active site. 28
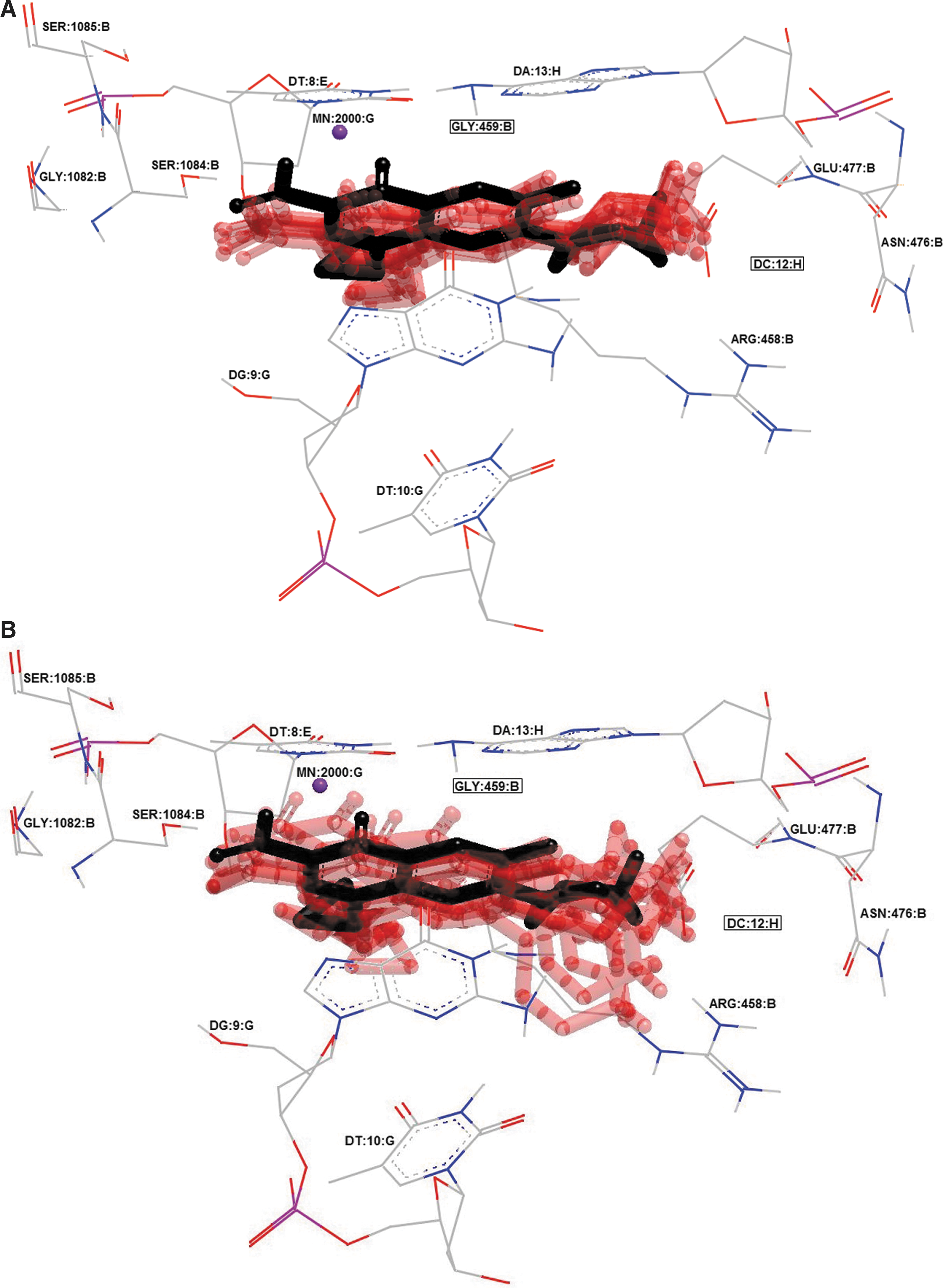
Cluster of 10 best docking poses (transparent red) of ciprofloxacin in 2XCT crystal.
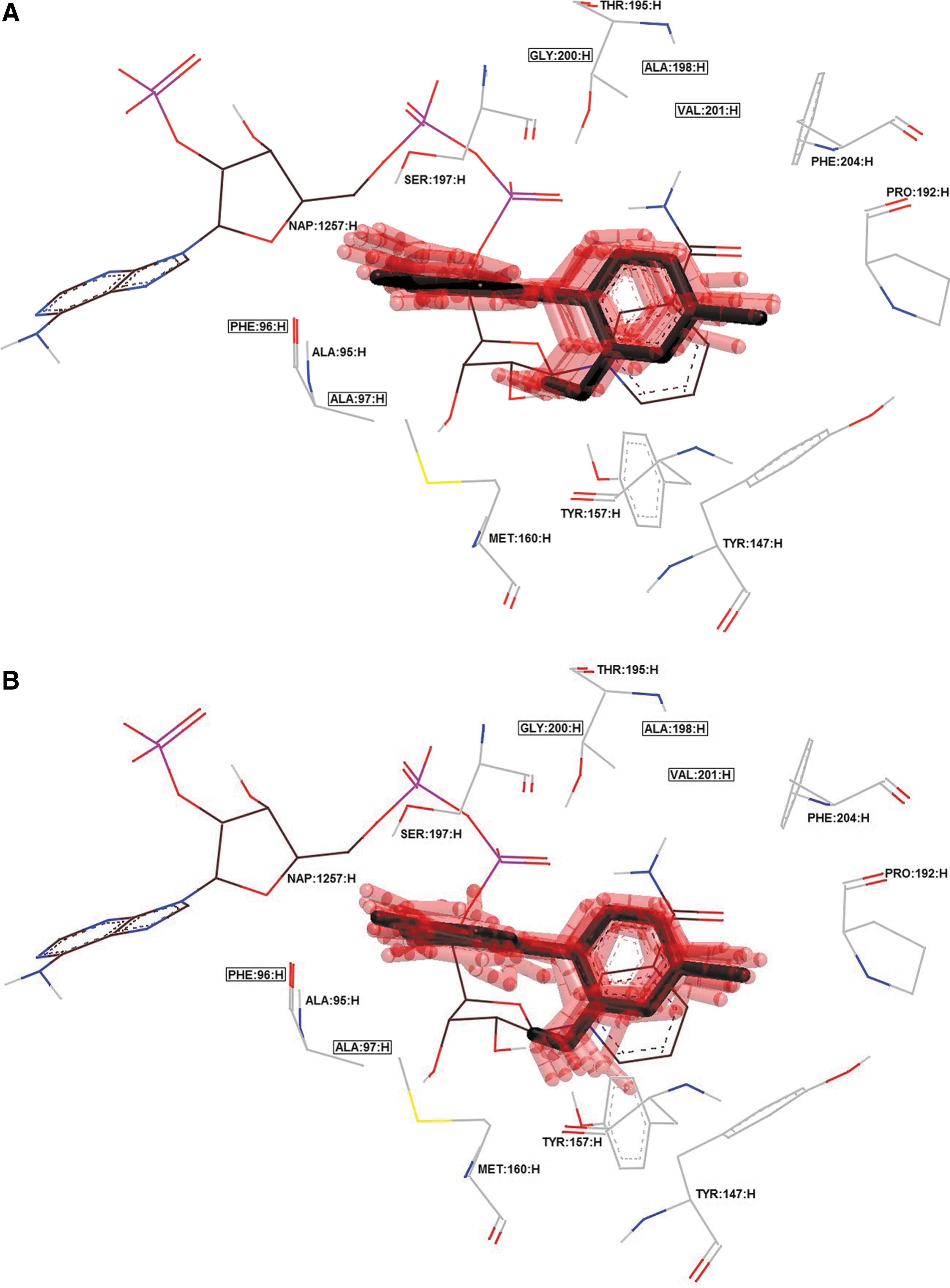
Cluster of 10 best docking poses (transparent red) of triclosan in 4ALI crystal.
In general, when the active candidates were docked to 2XCT, their HYBRID scores were better than their equivalents with 2XCS. Hence, only docking of 2XCT results was retained.
Table 2 represents the docking results of the three quinolone antibiotics and their similar active candidates to 2XCT crystal complex 25 S. aureus gyrase with ciprofloxacin and DNA complex. It is worth mentioning that the minimum RMSD between ciprofloxacin in crystal and the conformer generated for protonated ciprofloxacin using the default conformer generator library of OMEGA was 1.52 Å, with difference in the orientation of the –COO− group that prevented the generated conformer of protonated ciprofloxacin from forming a chelator bond with Mn2+ in the 2XCT crystal. Therefore, protonated ciprofloxacin in the 2XCT crystal was used as a base of bond comparison.
Candidates Binding Mode Predicted by Hybrid in Comparison with Protonated Ciprofloxacin in 2XCT Crystal
Candidates are arranged by descending order of hybrid score.
Metal–chelator interaction.
Ligand heavy atom is within 4 Å of the protein heavy atom.
Protonated.
Because the default conformer generator library of OMEGA was used, the conformer generated for protonated ciprofloxacin was different from the protonated ciprofloxacin in the 2XCT crystal.
Only the results of the S isomers are shown because the results of R isomers are weak and insignificant to show.
Comparing with the protonated ciprofloxacin in the 2XCT crystal, all compounds have made a hydrogen bond with SER1084B except for protonated drotaverine. Likewise, all the compounds form a chelating bond with MN2000G except for drotaverine (neutral or protonated), protonated indomethacin, (S)-flurbiprofen, and (S)-ibuprofen. Moreover, among all the candidates, only drotaverine (neutral or protonated) has made a hydrogen bond with ARG458B. Unlike ciprofloxacin in the crystal, (S)-flurbiprofen, (S)-ibuprofen, and indomethacin formed hydrogen bond with ARG1122D. Finally, regarding the interactions with nucleotides, neutral drotaverine and (S)-flurbiprofen made a hydrogen bond with DG9G, levofloxacin, ciprofloxacin, and nalidixic acid form a hydrogen bond with DT8E.
The binding model of drotaverine, ciprofloxacin, and gyrase-DNA is depicted in Figure 3, which revealed that drotaverine fits well in the ciprofloxacin binding pocket and is even more close to nucleotide DT10G and amino acids such as ASN476B, GLY1082B, GLU477B, and SER1085B. Similar to ciprofloxacin, drotaverine forms two hydrogen bonds with SER1084B (2.94 Å) and ARG458B (2.3 Å), and two π–π interactions between aromatic ring in ligand and pyrimidine ring in DG9G (3.67 Å), and DA8E (3.55 Å). Contrary to ciprofloxacin, drotaverine has one different hydrogen bond with DG9G (3.10 Å). Also, 4-carbonyl and 3-carboxyl groups in ciprofloxacin anchor Mn2+, but, even that Mn2+ is close to drotaverine (3.55 Å), these bonds are absent.
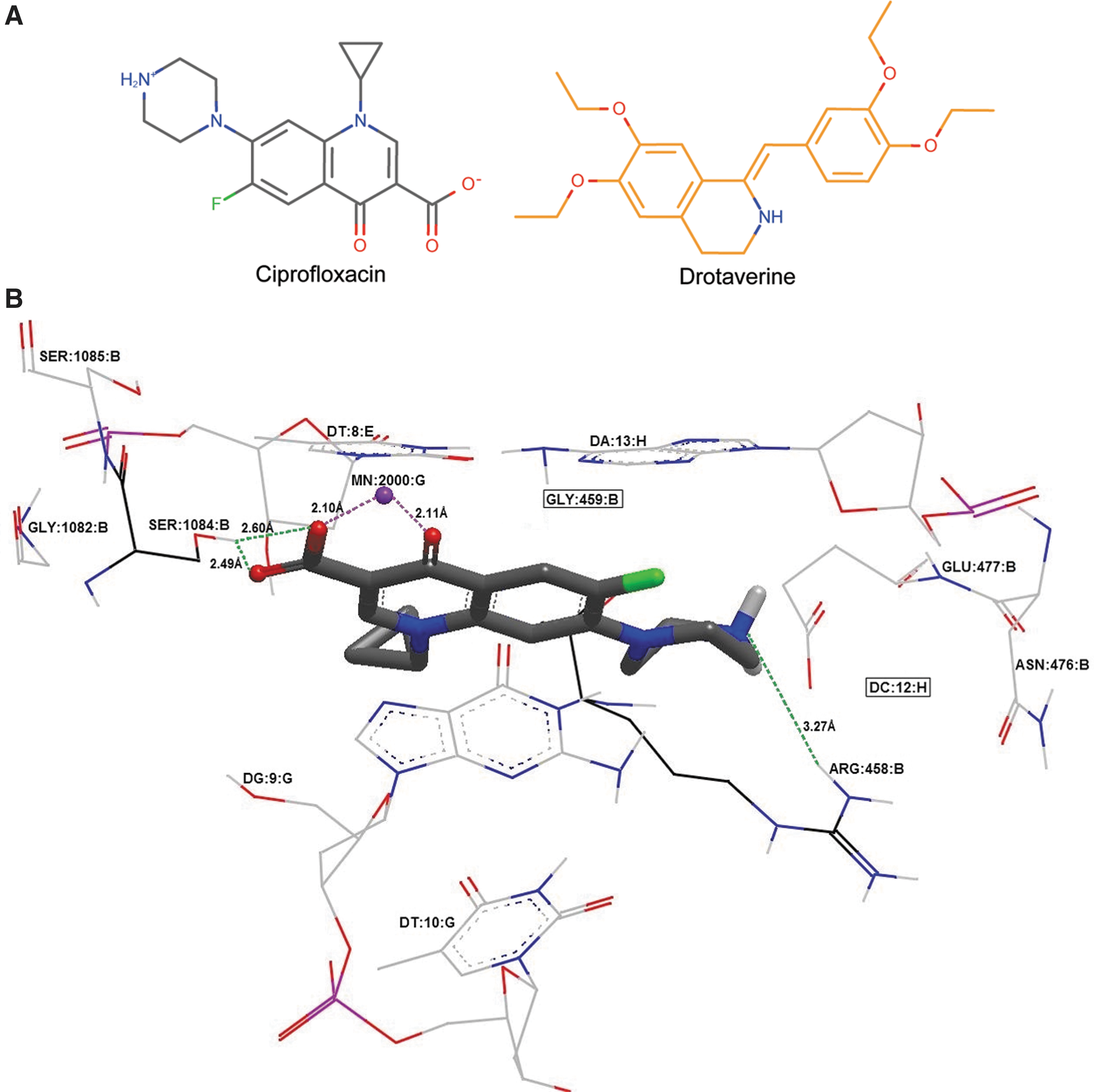
Drotaverine docked by HYBRID in gyrase from Staphylococcus aureus DNA–ciprofloxacin complex (2XCT.pdb crystal).

Table 3 represents the docking of triclosan and diclofenac (neutral or protonated) to the site of enoyl-ACP reductase from S. aureus based on enoyl-ACP reductase–NADP+-glutamate-triclosan complex structure; 4ALI crystal. 26 It is worth to note that the RMSD of the triclosan conformer generated by the default conformer generator library of OMEGA was 0.39 Å, which is near the RMSD of the docked triclosan (retrieved from crystal).
Candidates Binding Mode Predicted by Hybrid in Comparison with Triclosan in 4ALI crystal
Candidates are arranged by descending order of hybrid score.
Ligand heavy atom is within 4 Å of the protein heavy atom.
The docked triclosan has made the same binding mode of triclosan found in 4ALI crystal.
Protonated.
Obviously, triclosan and diclofenac (neutral or protonated) bind to the receptor but with some differences in interactions. First, diclofenac (neutral or protonated) binds to SER197H whereas triclosan binds to TYR157H. Second, the three compounds bind to NAP1257H but with different binding modes. Furthermore, the protonated diclofenac differs slightly from neutral diclofenac, whereby diclofenac has an extra hydrogen bond with ALA95H.
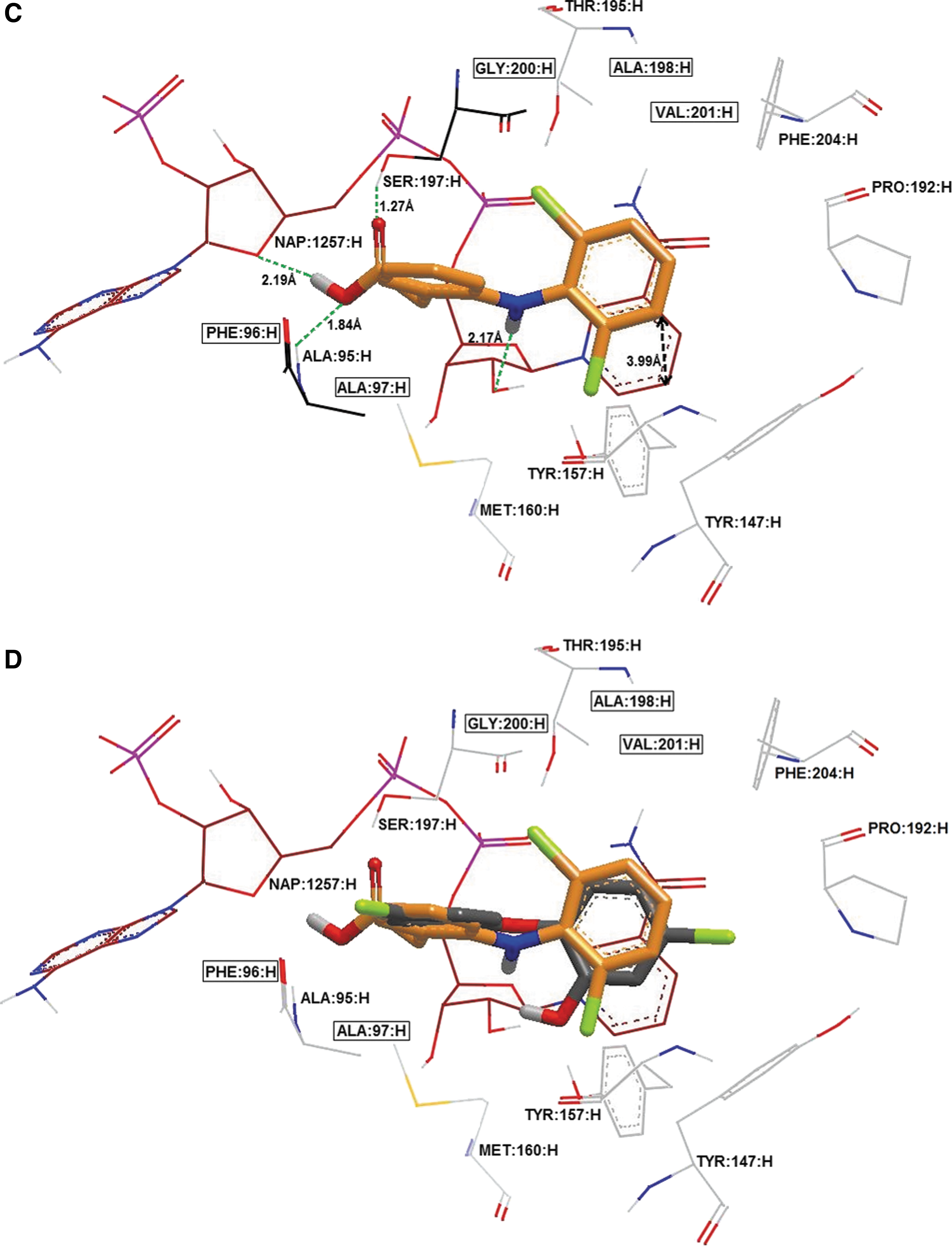
To take a closer look, the binding model of diclofenac, triclosan, and enoyl-ACP reductase–NADP+ is depicted in Figure 4, which revealed that diclofenac is well filled in the triclosan-binding pocket. Similar to triclosan, diclofenac forms hydrogen bond with the ribose ring that carries the nicotinamide moiety in NADP+ (2.17 Å), and a π–π interaction between (2,6-dichloroanilino) ring in ligand and nicotinamide in NADP+ (3.99 Å). But differing from triclosan, diclofenac has three additional hydrogen bonds with ALA95H (1.84 Å), SER197H (1.27 Å), and with the ribose ring that carries the adenine moiety (2.19 Å).
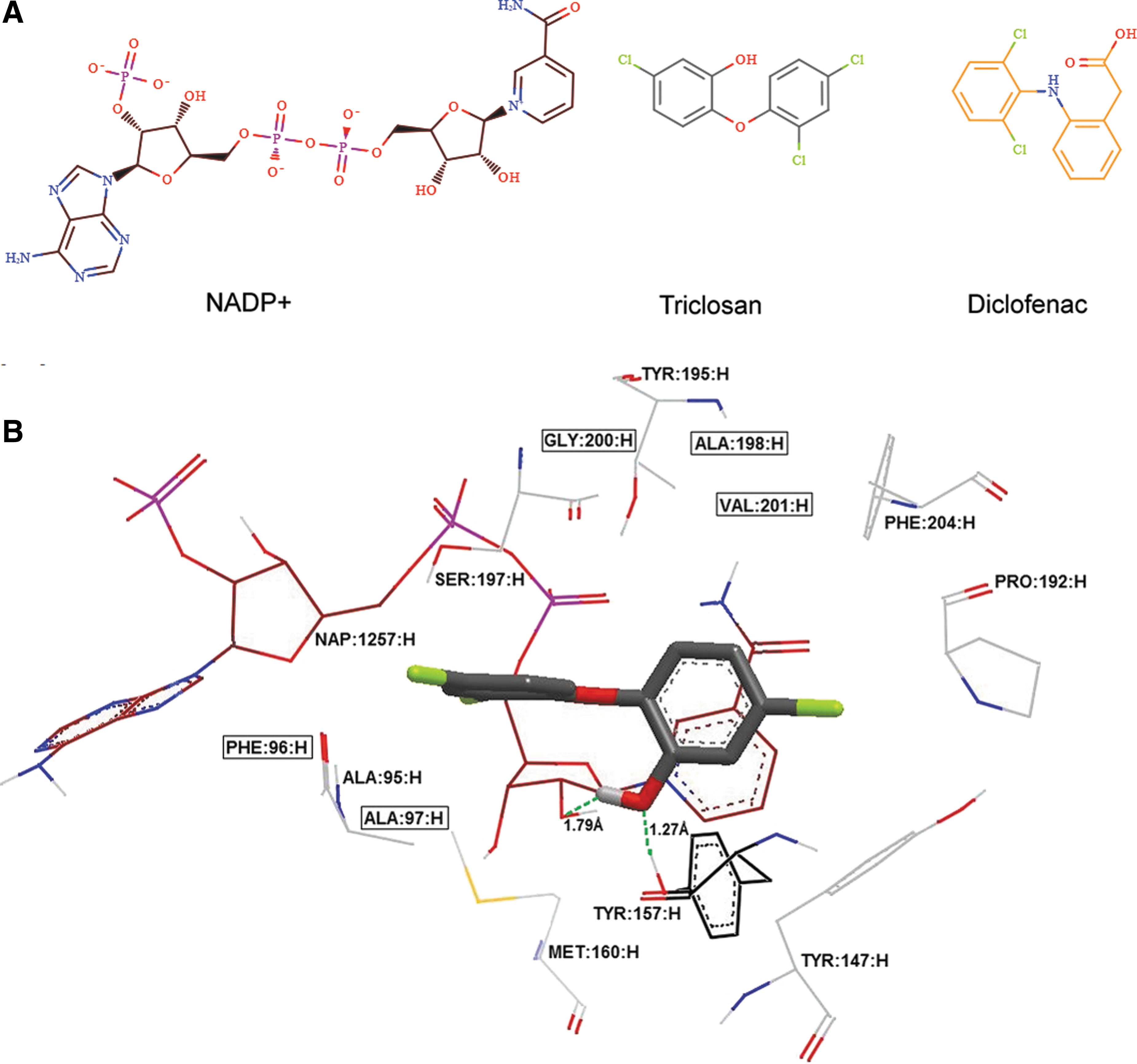
Diclofenac docked by HYBRID in enoyl ACP reductase from S. aureus based on enoyl-ACP reductase–NADP+-glutamate-triclosan complex structure (4ALI.pdb crystal).
Discussion
Overall, the ranking of compounds by ROCS TanimotoCombo score was different than their inhibition zone's ranking. Nevertheless, 10 of 34 tested candidates showed antibacterial activity, which was considered as good success rate.
The antibacterial activities reported in this article for diclofenac, flurbiprofen, ibuprofen, indomethacin, lamotrigine, niacin, allopurinol, and captopril were reported previously in the literature against a variety of bacterial strains. 29 –35
In contrast, the observed negative antibacterial effects of aspirin, metformin, and caffeine disagree with the findings of some literature, 36 –38 respectively. However, the antibacterial effects of drotaverine and metoclopramide, reported in our study, have not been identified before.
To interpret the mentioned results, there are important factors to be considered such as bacterial species and strains, candidate compound's physiochemical properties, ligand–receptor interaction and the type of the bioassay.
First, Gram-negative and Gram-positive bacteria differ in cell wall composition and structure. Therefore, they differ in influx and efflux of chemicals into/out of the cell. In addition, there are differences among bacterial strains of the same species, which may be interfering in the response to antibacterial activity. 39 Second, most of the tested candidates are small molecules with low molecular weights. Therefore, they are below the size exclusion limit of these two species' barriers. 40,41 However, the problems came up with lipo/hydrophilicity properties of the compounds especially with Gram-negative bacteria. According to Nikaido, 40 each of hydrophilic and lipophilic compounds has to pass a different barrier to get into the cell. For instance, hydrophilic compounds pass slowly and with difficulty through the porins' narrow channels; lipophilic compounds penetrate through the lipid bilayer slowly because of lipopolysaccharides. Also, they can pass through the porins, but much slower. In contrast, according to Lambert, 41 the cell wall of Gram-positive bacteria does not restrain the penetration of polar compounds.
Third, the penetration of bacterial barriers does not necessarily result in an effect; the compounds have to make suitable interactions with the targeted receptor to produce an effect. 42 Finally, it is worth taking into consideration the factors that affect the Kirby–Bauer disk diffusion, especially hydrophilicity and molecular weight of the compound, because they determine the diffusion characteristic of the compound in the agar medium. 21,22
Recently, the docking method has been used widely to model the ligand–receptor interactions to explain the therapeutic activity of many drugs. For instance, Xiao et al. 27 used docking to justify the action of their novel DNA gyrase inhibitor. In relation to this study, HYBRID has successfully predicted that levofloxacin, ciprofloxacin, and their protonated forms as the best binders to the 2XCT receptor. In addition, HYBRID suggests that indomethacin, ibuprofen, flurbiprofen, and diclofenac (or their protonated forms) have binding models similar to the quinolones. Moreover, drotaverine has the same pose of ciprofloxacin plus an additional hydrogen bond with the active site. However, the missing chelator bond with Mn2+ prevents drotaverine from achieving a binding mode very similar to ciprofloxacin.
The differences between the antibacterial activity results and the ranking of compounds by HYBRID could be explained by different factors related to physical and chemical properties of compounds, targeted protein, or the docking algorithm used in the docking. As an example, HYBRID predicts similar, but not identical, binding mode of ciprofloxacin because it uses the conformers generated by OMEGA as a rigid structure without any changes.
The success of diclofenac to bind to DNA gyrase gave a convincing explanation to the finding of Dastidar et al., 29 which concludes experimentally that diclofenac inhibits DNA synthesis but with an unknown mechanism back then. Moreover, as diclofenac shares similarity with triclosan, docking it to 4ALI 26 complex could justify further its good antibacterial activity. Indeed, the docking results showed that diclofenac could bind to the triclosan target receptor in 4ALI crystal, with the same binding pose of triclosan plus four hydrogen bonds with the active site (Fig. 3).
The antibacterial action shown by the drugs in this study cannot be compared with the action of the reference antibiotic both in matter of potency and in matter of doses. For instance, at the best scenarios, the dose required for nalidixic acid to produce 13 mm inhibition zone against S. aureus is ∼17 times lower than the dose required for diclofenac to produce the same zone (Table 1), making diclofenac much less potent than nalidixic acid. Moreover, from a therapeutic perspective, the antibacterial activities shown by the mentioned drugs are weak and not appropriate for off-label use as antibiotics, because they may require higher concentrations to produce sufficient antibacterial effects in humans than the concentration needed for their original indication, which poses a threat to exaggerate their side effects. 43 Despite that, a new study in 2016 suggests use of ibuprofen for uncomplicated urinary tract infections instead of antibiotics. 44
There are two potential uses to get benefit from the antibacterial activity for nonantibiotic drugs. First, they could be used as prophylactic agents from infections. For instance, diclofenac has been shown to protect mice from infection with virulent Salmonella typhimurium. 29 Second, they could be used in combination with other antibiotics to get a synergistic effect. Again, diclofenac is also an example of this use, a study showed that it is capable of producing synergism with streptomycin against Mycobacterium smegmatis. 45 However, the chance for resistance induction by antibacterial action of the nonantibiotic drugs to certain antibiotics should be taken into account. 43,46
Finally, to make full use of the antibacterial activity of nonantibiotic compounds, it is necessary to make modifications on their chemical structures to potentiate the antibacterial activity to the maximum and to get rid of the original biological activity to avoid potential side effects. In fact, thiazide diuretics is a famous example of a new drug emerging from chemical modification, thiazide has come after enhancing the diuretic action of sulfanilamide, which has been noticed to promote the excretion of sodium, potassium, and water. 47
Conclusion
The structure similarity method used in this research has narrowed down an initial pool of nearly 2,000 FDA-approved drugs to 34 candidates for performing the antibacterial tests, which resulted in detection of 10 compounds with antibacterial activities, whereby only drotaverine and metoclopramide displaying new, previously unknown activities, equaling success rate of 29.4%. The docking method gave an explanation for the antibacterial activity of the most active candidates, because it turned up that those candidates were able to bind to the targeted receptor of their similar antibiotics with a similar binding mode. Moreover, the docking results of diclofenac showed that its antibacterial activity likely resulted from the dual action against DNA gyrase and enoyl ACL reductase (Fabl). The antibacterial activities of the nonantibiotic drugs already mentioned are weak compared with those of real antibiotics, which make them not suitable to be used as antibiotics against infections. However, carrying out chemical modification on their structure to potentiate their antibacterial activity could provide the most beneficial use for these drugs. Regarding drotaverine and diclofenac, the addition of a highly electronegative or chelator atom in the correct position for the former and alternation of the position of one chloride residue for the latter promise to obtain effective antibacterial agents.
Footnotes
Acknowledgments
We like to thank OpenEye Scientific Software (Santa Fe, NM) for the free academic license to use its software. The authors would like to thank all the staff of the pharmacy faculty in the Arab International University, especially Wafika Zarzour, Rajwa Jbeily, Marwan Zabibi, Maher Al-Kuotayni, Abdul Wahab Allaf, Adel Nofal, Jamal Darwecha, Ahmad Ganama, Duraa Shareet, Batol Sati, Marah Shaheen, and Aline Saferian. Furthermore, we like to thank Habib Abboud, Luobna Darwish, Waill Alhourani, Abeer Al-Kafri, and Khanom Mudawar for their help in providing the materials needed for the research.
Disclosure Statement
This work was funded by Arab International University (private university), Daraa, Syrian Arab Republic. The authors declare no conflict of interest regarding the publication of this article.