
Abstract
G protein-coupled receptors (GPCRs) are excellent drug targets exploited by majority of the Food and Drug Administration-approved medications, but when modulated, are often accompanied by significant adverse effects. Targeting of other elements in GPCR pathways for improved safety and efficacy is thus an unmet need. The strength of GPCR signaling is tightly regulated by regulators of G protein signaling (RGS) proteins, making them attractive drug targets. We focused on a prominent RGS complex in the brain consisting of RGS7 and its binding partners Gβ5 and R7BP. These complexes play critical roles in regulating multiple GPCRs and essential physiological processes, yet no small molecule modulators are currently available to modify its function. In this study, we report a novel high-throughput approach to screen for small molecule modulators of the intramolecular transitions in the RGS7/Gβ5/R7BP complex known to be involved in its allosteric regulation. We developed a time-resolved fluorescence energy transfer-based in vitro assay that utilizes full-length recombinant proteins and shows consistency, excellent assay statistics, and high level of sensitivity. We demonstrated the potential of this approach by screening two compound libraries (LOPAC 1280 and MicroSource Spectrum). This study confirms the feasibility of the chosen strategy for identifying small molecule modulators of RGS7/Gβ5/R7BP complex for impacting signaling downstream of the GPCRs.
Introduction
G protein-coupled receptors (GPCRs) control a range of fundamental cellular processes across all tissue types. Thus, it is not surprising that the majority of pharmacological interventions target GPCRs. 1 In a prototypic series of events, binding of extracellular ligands to GPCRs induces them to activate heterotrimeric G proteins composed of αβγ subunits by promoting the exchange of guanosine diphosphate (GDP) to guanosine triphosphate (GTP) on Gα. This triggers subunit rearrangement, freeing Gα-GTP and Gβγ to engage with effector proteins. The signaling is terminated by the hydrolysis and conversion of Gα-GTP to Gα-GDP, whereupon Gαβγ subunits reassociate to form an inactive heterotrimer. 2 The intrinsic rate of Gα GTPase activity is much slower than the observed modulation of many biological responses such as ion channel regulation. 3 Therefore, inactivation of G proteins is facilitated by the GTPase-accelerating protein (GAP) activity provided by regulators of G protein signaling (RGS) proteins. 4
There are more than 30 known RGS genes encoded in mammalian genomes, with diverse cell- and tissue-specific expression patterns. The range of RGS influences is further extended by observations that different members of the family display varying selectivity in their ability to regulate different classes of the Gα subunits. 5,6 From the drug development perspective, RGS proteins may be considered genetically encoded GPCR antagonists that endogenously limit the extent of GPCR signaling events in a specific manner. Thus, the rationale for targeting them is largely similar to the development of positive allosteric modulators of GPCRs, particularly in situations where benefits are expected from the increased signaling to the endogenously released agonists under physiological conditions. Further benefits from targeting signal processing steps downstream from GPCRs (i.e., RGS) are in its potential to bias signals and selectively enhance only some of the reactions initiated by the GPCRs, thereby possibly avoiding adverse effects associated with synthetic ligands triggering multiple signaling pathways.
Despite significant rationale for targeting RGS proteins, former efforts have been rather limited. To date, only RGS4 has been successfully targeted by HTS campaigns from which small molecule inhibitors have emerged. 7 –11 Importantly, the lead compound shows considerable selectivity and nanomolar potency and has been successfully used to interrogate cellular physiology and disease mechanisms. 12,13 RGS4 is a small protein containing a single catalytic RGS domain. Thus, the strategy used for its inhibition was geared toward disruption of its interactions with the substrate: active Gαq. 14,15 Given substantial diversity of the RGS family both in terms of their structural organization and regulation, drug discovery efforts will likely benefit from the development of alternative approaches for designing assays reporting effects of small molecule modulators of RGS function.
A case in point is RGS7, a major RGS protein with widespread expression in the nervous system with critical roles in regulating learning/memory, pain, and addiction. 16,17 At the molecular level, RGS7 is known to negatively regulate signaling mediated by μ-opioid (MOR) and GABAB receptors. 17 A substantial body of evidence indicates that in vivo RGS7 exists as a macromolecular complex with auxiliary subunits that tightly regulate its function. 18,19 RGS7 forms a constitutive complex with type 5 G protein beta subunit (Gβ5) that plays an essential role in controlling complex expression. 20,21 In addition, it recruits small palmitoylated protein R7 binding protein (R7BP) 22,23 that localizes the complex to the plasma membrane and provides allosteric control influencing RGS catalytic activity and Gα selectivity. 23 –27 The physiologic consequences of eliminating R7BP and Gβ5 subunits generally resemble those associated with RGS7 loss, yet exhibit distinct features. For instance, knockout of Gβ5 in mice results in hypersensitivity to MOR and GABAB receptor activation, and dysregulates downstream synaptic plasticity and activation of inhibitory ion channels. 16,17,28 –30 Knockout of RGS7 also enhances morphine-related analgesia and reward, while exacerbating withdrawal symptoms 31,16 ; R7BP knockout mice display enhanced opiate analgesia, yet the withdrawal is unaffected. 32,33
These observations suggest that different subunits in the RGS7/Gβ5/R7BP complex regulate various aspects of receptor signaling and targeting allosteric intrasubunit transitions in this complex may be exploited by small molecules to mimic endogenous regulatory mechanisms. Such a strategy would be distinct from previous efforts centered on the disruption of RGS binding with its Gα substrates. Given the lack of any reported small molecule modulators for RGS7 complex, the case for developing first-in-class probes using our innovative approach grounded in biology is strong. In this study, we report the development of a biochemical HTS assay to identify conformational changes induced by compounds bound to the entire trimeric complex comprising full-length proteins: RGS7 with Gβ5 and R7BP. We utilized time-resolved fluorescence energy transfer (TR-FRET) to detect the effects of compounds on the RGS7/Gβ5/R7BP complex. The assay shows high sensitivity and excellent statistics (Z', signal to background) during validation in its screening against two commercial libraries of pharmacologically active compounds.
Materials and Methods
Materials
Chemicals were purchased either from Sigma, including the LOPAC 1280 compound library, or from MicroSource Discovery (Spectrum Library). Anti-GST-Tb (#61GSTTLA), anti-GST-d2 (61GSTDLA), anti-6xHis-Tb (61HI2TLA), and anti-6xHis-d2 (61HISDLA) were purchased from Cisbio, reconstituted in water following the manufacturer's instructions, and stored in aliquots at −20°C. The 384-well low-volume white plates where purchased from Greiner (#784075) and cleaned with an ionizing air gun (Simco-Ion, Hatfield, PA) immediately before experiments.
Expression and Purification of RGS7 and Gβ5
RGS7 was co-expressed with Gβ5 in Sf9 insect cells through baculovirus-mediated delivery and the recombinant complexes were purified by HisTALON™ Superflow Cartridge (Clontech Laboratories, Inc.) chromatography, utilizing His-tag present at the N termini of RGS7 proteins as described previously. 22 Briefly, insect cells containing recombinant RGS7/Gβ5 complex were lysed in buffer A (20 mM HEPES [pH 8], 300 mM NaCl, 10 mM imidazole [pH 8], 5 mM β-mercaptoethanol, and 1% [v/v] glycerol) containing EDTA-free complete protease inhibitor tablets (Roche) by sonication. The recombinant RGS7/Gβ5 complex containing supernatant was clarified by centrifugation at 32,000 rpm for 30 min and loaded onto pre-equilibrated HisTALON Superflow Cartridge with buffer A. The protein complex was eluted using 250 mM imidazole gradient. Protein complex was analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and fractions containing RGS7/Gβ5 complex were pooled, dialyzed, and loaded onto MonoQ column and eluted with 1 M NaCl gradient. The eluted complex was further purified using Hiload 26/60 Superdex 200 column (GE Healthcare), which was pre-equilibrated with buffer B (20 mM HEPES pH 8, 200 mM NaCl, and 1 mM DTT). Peak fractions were analyzed by SDS-PAGE, pooled, and concentrated to 1 mg/mL.
Expression and Purification of R7BP
The coding sequence of Mus musculus R7BP (residues 47–231) was amplified by polymerase chain reaction (PCR). The PCR product was cloned into the pGEX-6P-1 Escherichia coli expression vector downstream from a glutathione S-transferase (GST) tag followed by the PreScission Protease site. The recombinant vector containing R7BP coding region was confirmed by sequencing and was transformed into the BL21(DE3) E. coli strain induced by 0.4 mM isopropyl thiogalactoside at OD600 = 0.6 for 16–18 h at 18°C. The cells were harvested, resuspended in buffer C (20 mM Tris [pH 8], 300 mM NaCl, 1 mM DTT, and 1% [v/v] glycerol) containing complete protease inhibitor tablets (Roche), disrupted by sonication, and clarified by centrifugation at 32,000 rpm for 30 min. The cell lysate was loaded onto a pre-equilibrated GSTPrep FF 16/10 (GE Healthcare) with buffer C and eluted with 15 mM glutathione. The eluted protein was further purified using Hiload 26/60 Superdex 75 column (GE Healthcare), which was pre-equilibrated with buffer B. The purity of the protein was analyzed by SDS-PAGE, pooled, and concentrated to 1 mg/mL.
Assay Conditions
All reagents were diluted in the same buffer, which was prepared immediately before experiments and consisted of 20 mM HEPES pH 8.0, 200 mM NaCl, 1 mM DTT, and 0.1% BSA. The buffer was filtered through a 0.22 μm membrane before use (Corning, #431098). Automated liquid handling was performed using an Aurora Flying Reagent Dispenser. Dimethyl sulfoxide (DMSO) and test compounds were plated using a BiomekNXP (BeckmanCoulter) equipped with a pintool head (V&P Scientific).
Plate Reader
Plates were read on a PerkinElmer EnVision plate reader with the LANCE/DELFIA mirror module optics: UV2 (TRF) 320 excitation filter, APC 665 emission filter 1, and Europium 615 emission filter 2. The reader protocol was optimized for plate dimensions and 6.5 mm read height using a 50 μs delay and 400 μs window time.
Data Analysis
The TR-FRET ratio was calculated by dividing 665 raw data by 615 raw data. Each assay plate included four controls: two negative control groups, one low control, and one high control (Fig. 3A). Negative control wells were deficient for either 6xHis-RGS7/Gβ5 or GST-R7BP, which served to represent the background TR-FRET. The low control wells contained all reagents for optimal signal, which represented zero TR-FRET inhibition. The high control wells contained untagged-R7BP to achieve maximal TR-FRET inhibition. Percent inhibition was calculated by normalization to high and low control wells. Hits were classified as exceeding a defined cutoff value from the sample field (average ±3 × standard deviation) and we applied a stringent rubric demanding that hits also exceed the cutoff value in both channels of raw data (615 and 665 nm). This calculation for hit classification was only applied to the sample field and therefore the control groups were appropriately utilized to represent quality control of the assay. IC50 values were calculated by curve-fitting data in GraphPad Prism 6 with the following equation: log(agonist) versus response—variable slope (four parameters). Signal to background calculations were performed from raw TR-FRET ratio defining the average DMSO sample field or low control as the signal and the negative controls as background. Z' calculations from raw TR-FRET ratio were performed from high and low control data as described. 34
Results
Development and Optimization of a Novel Biochemical Assay Reporting Changes in Interactions Between Recombinant RGS7/Gβ5 and R7BP by TR-FRET
We first began with proof-of-concept experiments to gauge assay design and feasibility in detecting the interactions within RGS7 complex in vitro. This utilized recombinant full-length RGS7/Gβ5 and R7BP proteins in combination with TR-FRET toolbox antibodies conjugated with a terbium (Tb) donor and d2 acceptor (Fig. 1A). We strategically positioned antibody recognition tags proximal to binding regions of functionally recognized conformational transitions. Our rationale was that by docking antibodies in close proximity to RGS7/Gβ5 and R7BP interaction determinants, we could detect transition states within the macromolecular complex. RGS7 and R7BP utilize their N-terminal domains to comprise the binding surface with each other. 35 Therefore, a 6xHis tag was placed on the N-terminus of RGS7 and this was co-expressed with Gβ5 in insect Sf9 cells and the resulting complex was purified. The two subunits were easily identified under denaturing conditions on a Coomassie stained SDS gel, which also demonstrated homogeneity of the preparation (Fig. 1B). Similarly, recombinant R7BP was produced as an N-terminal fusion with a GST in E. coli and purified to homogeneity (Fig. 1B).
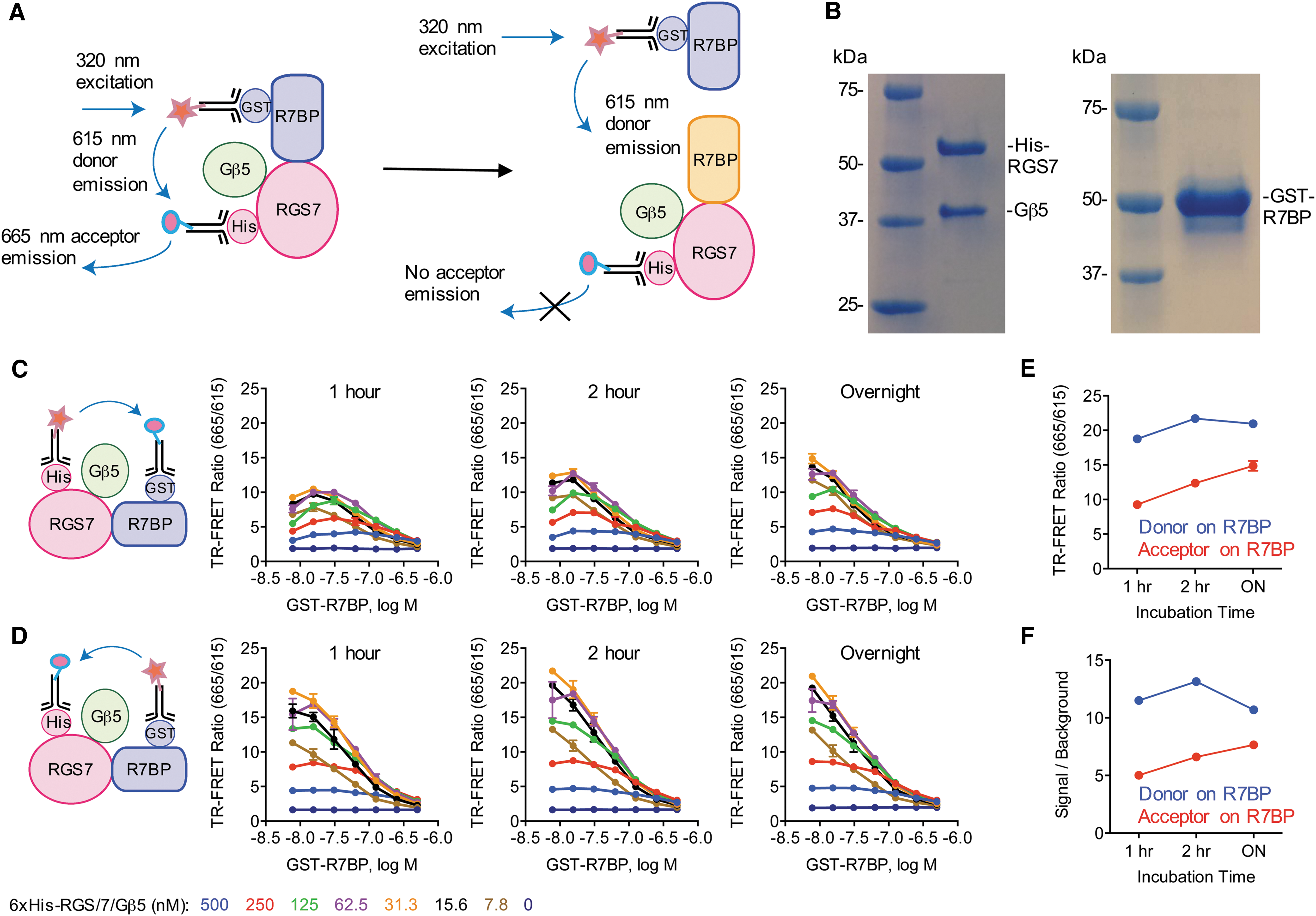
Development of a strategy to study interactions of RGS7/Gβ5 with R7BP using a TR-FRET assay in 384-well format.
Using the toolbox antibodies and proteins described above, we set up a simple mix and read assay eliminating the need for complicated washing and incubation steps. We focused our initial efforts on optimizing the ratios of RGS7/Gβ5 to R7BP to yield robust complex detection. Using the binding affinity of this interaction of ∼13 nM as a starting point, 19 we performed serial dilutions of our recombinant proteins ranging from ∼5 to ∼500 nM. We then added TR-FRET antibodies placing the donor fluorophore on 6xHis-RGS7/Gβ5 (anti-His-Tb; 0.4 nM) and acceptor on GST-R7BP (anti-GST-d2; 6.7 nM). A 1 h of room temperature incubation was sufficient to enable detection of the complex (Fig. 1C). The TR-FRET ratio was unchanged following 2 h of incubation, suggesting steady-state equilibrium had already been achieved. To test the stability of the proteins under these working conditions, the same plates were read after an overnight incubation (∼18 h), which showed only a slight improvement in TR-FRET ratio.
FRET efficiency is reliant on respective donor and acceptor fluorophore distance and dipole orientation. As there is currently no crystal structure available on RGS7 or R7BP, spatial arrangement of the FRET pairs remains elusive. We therefore sought to examine if switching the Tb donor to R7BP and d2 acceptor to RGS7 would impact FRET efficiency. This was achieved by repeating the experiment with the utilization of anti-GST-Tb (0.8 nM) and anti-His-d2 (6.7 nM) antibodies. Strikingly, this led to a nearly twofold increase in TR-FRET ratio after 1 h of room temperature incubation (Fig. 1D). As before, this ratio was largely unchanged after extending the incubation out to 2 h and these complexes were also stable following an overnight incubation.
Optimal results were achieved with 31.3 nM 6xHis-RGS7/Gβ5 and 7.8 nM GST-R7BP, regardless of which protein the donor and acceptor FRET pairs were coupled to. However, under equivalent conditions, we observed a greater TR-FRET ratio with the orientation of Tb donor on R7BP and d2 acceptor on RGS7 compared with the opposing scenario of Tb donor on RGS7 and d2 acceptor on R7BP (Fig. 1E). Furthermore, this combination also increased the signal to background ratio and was therefore used in all subsequent experiments (Fig. 1F).
We next verified protein binding specificity as well as the range of the TR-FRET readout in the assay. For this purpose, we removed the GST tag from R7BP, generating an untagged version of R7BP, which was utilized in the competition experiments as it binds to RGS7 without being recognized by Tb antibodies, therefore producing no TR-FRET signal (Fig. 2A). Indeed, the addition of untagged-R7BP decreased the TR-FRET ratio in a dose-dependent manner both at 2-h (Fig. 2B) and after overnight incubation (Fig. 2C). To further validate signal inhibition specificity, we performed concentration response studies by increasing the amount of GST-R7BP in the presence of untagged R7BP in the reaction. These experiments showed that increasing the concentrations of GST-R7BP increased the IC50 of untagged-R7BP inhibition in a linear manner, confirming that changes in TR-FRET signal originate specifically from modulation of RGS7-R7BP association. Collectively, this demonstrates successful development of an in vitro biochemical assay to probe interactions between full-length RGS7/Gβ5 and R7BP.
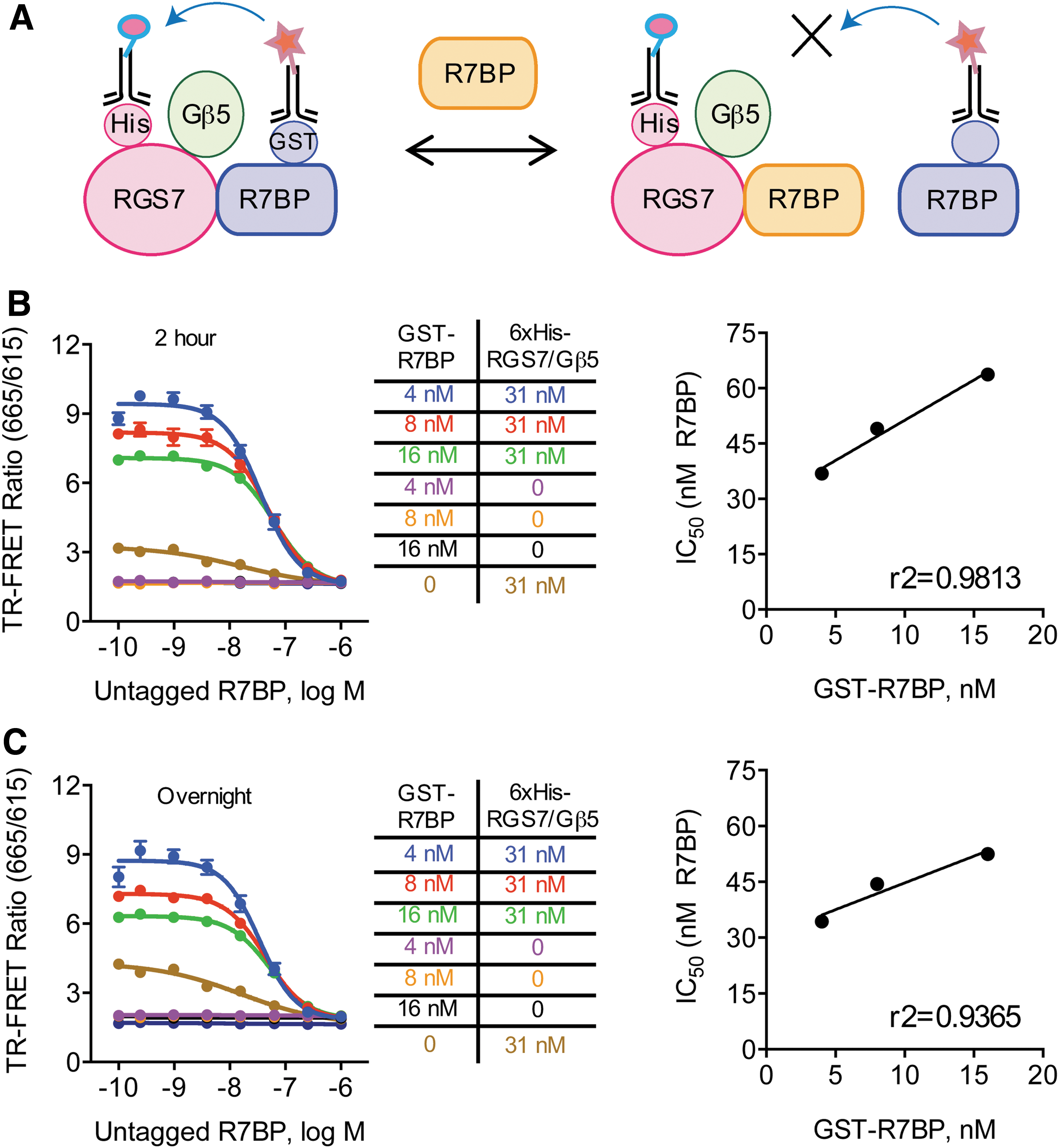
Validation of signal specificity of RGS7/Gβ5 interaction with R7BP.
Benchmarking Assay Performance Using Automated Systems
Our next step was to automate the assay on a high-throughput platform. Reagent dispensing was performed in three stages with the goal of introducing GST-R7BP and 6xHis-RGS7/Gβ5 simultaneously to the reaction well after delivery of potential screening compounds (Table 1). Our logic was to prevent a “preincubation” of RGS7 with R7BP and thus the first stage utilized automated reagent dispensing for adding antibodies and buffer. In the second stage, a pintool was utilized to deliver DMSO (note that the pintool dispensing mechanism requires volume in the well). Finally, the third stage employed automated reagent dispensing of recombinant proteins: GST-R7BP, 6xHis-RGS7/Gβ5, and untagged-R7BP, where appropriate. Based on observations during assay optimization, we chose to incubate plates overnight at room temperature to further streamline assay logistics.
Protocol
RGS, regulators of G protein signaling; GST, glutathione S-transferase; FRD, Flying Reagent Dispenser; DMSO, dimethyl sulfoxide; TR-FRET, time-resolved fluorescence energy transfer.
We validated this approach on two different days. Each day, experiments were performed with a DMSO plate, control response curve (CRC) plate, and another DMSO plate in the same sequence order. Our stringent data analysis pipeline entailed examining the raw signals from both channels (615 and 665 nm) as well as the normalized TR-FRET ratio (% inhibition). We defined a cutoff value from the sample field (average ±3 × standard deviation) and classified hits as wells in the sample field that exceeded the cutoff in both raw channels as well as the ratiometric % inhibition calculation. Control groups indicate assay performance and were therefore not subjected to cutoff classification. A representative DMSO plate from the first day highlighted the assay performance and absence of false positives (Fig. 3A). The CRC plate also demonstrated robust dose-dependent inhibition of GST-R7BP interaction with 6xHis-RGS7/Gβ5 by untagged-R7BP (Fig. 3B). Repetition of the assay on the second day showed a near-identical performance (Fig. 3C, D). We examined the consistency by quantifying several assay parameters. We first plotted the CRC dose–response inhibition facilitated by untagged-R7BP (Fig. 3E). Each replicate was very similar and the average IC50 was 726 ± 49 nM for R7BP (Fig. 3F). Although the signal to background ratio dropped on day 2, intraday variability was consistent and the overall signal was strong (Fig. 3G). Finally, we consistently observed excellent Z' values for each plate (Fig. 3H). Taken together, our assay statistics support robust performance of the assay in the automated platform compatible with the goal of identifying activators and inhibitors of the R7BP-RGS7 interaction.

Assay reproducibility on an automated platform.
Identification of Small Molecule Modulators of RGS7/Gβ5 Interaction with R7BP from LOPAC 1280 Library
To test the validity of our approach for the identification of small molecule compounds, we screened the LOPAC library containing 1,280 compounds. The LOPAC compounds were tested at 12.4 μM and signal was read after overnight incubation at room temperature. To fully test the robustness of the assay, we performed the screen in triplicate. Consistent with validation experiments, the DMSO plates used during the screen showed minimal false positives (Fig. 4A). The CRC control plates also exhibited dose-dependent inhibition of TR-FRET with an IC50 of 551 ± 112 nM R7BP (Fig. 4B). Analysis of the scatter plot of the test compounds revealed a bidirectional regulation of interactions between RGS7 and R7BP (Fig. 4C). Some compounds exhibited a positive score, similar to the effect of untagged-R7BP, and were thus considered putative “inhibitors.” However, the majority of compounds had generated a negative score and by analogy were considered putative “activators.”
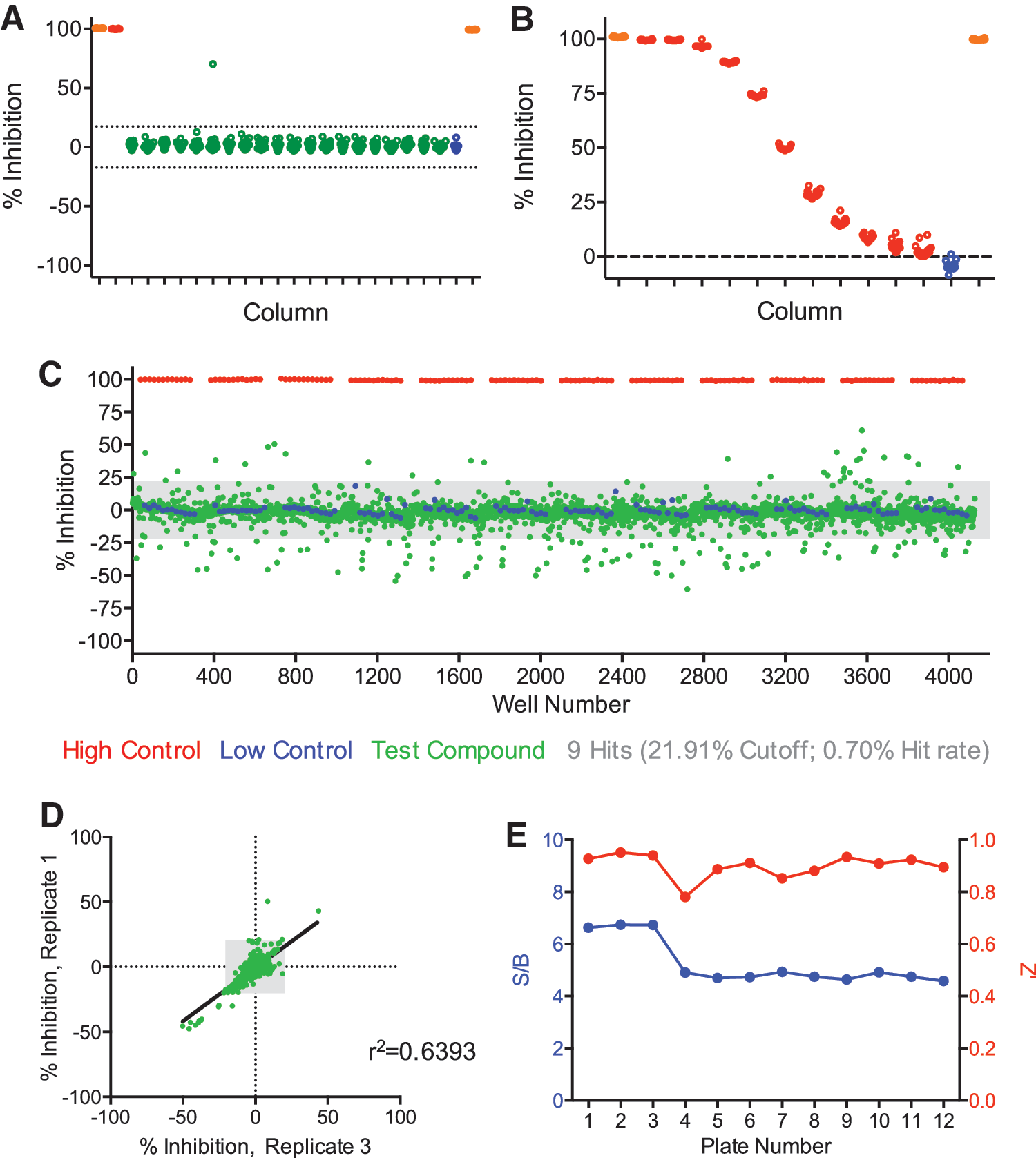
Validation of the assay by screening LOPAC1280 library.
Hits were identified, both inhibitors and activators, from the same calculated cutoff value (21.91) complied of all data across the entire sample field of compounds (criteria = average ±3 × standard deviation). Collectively, we identified nine hits that were reproduced across all three LOPAC replicates (0.70% hit rate). The bidirectional modulation of RGS7-R7BP interaction is illustrated through correlation analysis of the LOPAC compounds (Fig. 4D). Among these, only one hit generated a positive inhibition score and was classified as an inhibitor of RGS7-R7BP (0.08% hit rate), whereas the other eight hits generated a negative inhibition score and were classified as activators (0.63% hit rate). The quality of the screen was again satisfactory as we observed a stable signal to background ratio as well as an excellent Z' value across each plate (Fig. 4E).
Identification of Small Molecule Modulators of RGS7/Gβ5 Interaction with R7BP from MicroSource Spectrum Collection
The outcome of the LOPAC screen warranted additional testing of the assay for screening a larger collection of compounds. Therefore, we next screened the MicroSource Spectrum collection that contains 2,320 pharmacologically active compounds. The compounds were tested at the same nominal concentration as the LOPAC library. Given the strong assay performance and correlation of hits between plates from the LOPAC trial, we deemed that screening the library only in duplicate would be sufficient. In our screen with the Spectrum collection, we again observed only one false positive (0.31% rate) in our DMSO control plate (Fig. 5A). The CRC plate showed a dose-dependent decrease in TR-FRET (IC50 = 508 ± 154 nM R7BP), similar to prior experiments (Fig. 5B). The scatter plot data of the test compounds again revealed a bidirectional regulation of interactions between RGS7 and R7BP (Fig. 5C). As calculated above, a cutoff value of 43.78 (criteria = average ±3 × standard deviation) identified 25 total hits (1.08% hit rate). Correlation analysis between the replicates was utilized to identify hits both in the positive and negative direction (Fig. 5D). We found 3 inhibitors (0.13% hit rate) and 22 activators (0.95% hit rate) of RGS7-R7BP complex. Notably, the assay continued to exhibit a very stable signal to background ratio across all plates, along with an excellent Z' value (Fig. 5E). Overall, we have successfully demonstrated the utility of our RGS assay for high-throughput screening of compound libraries on an automated platform.
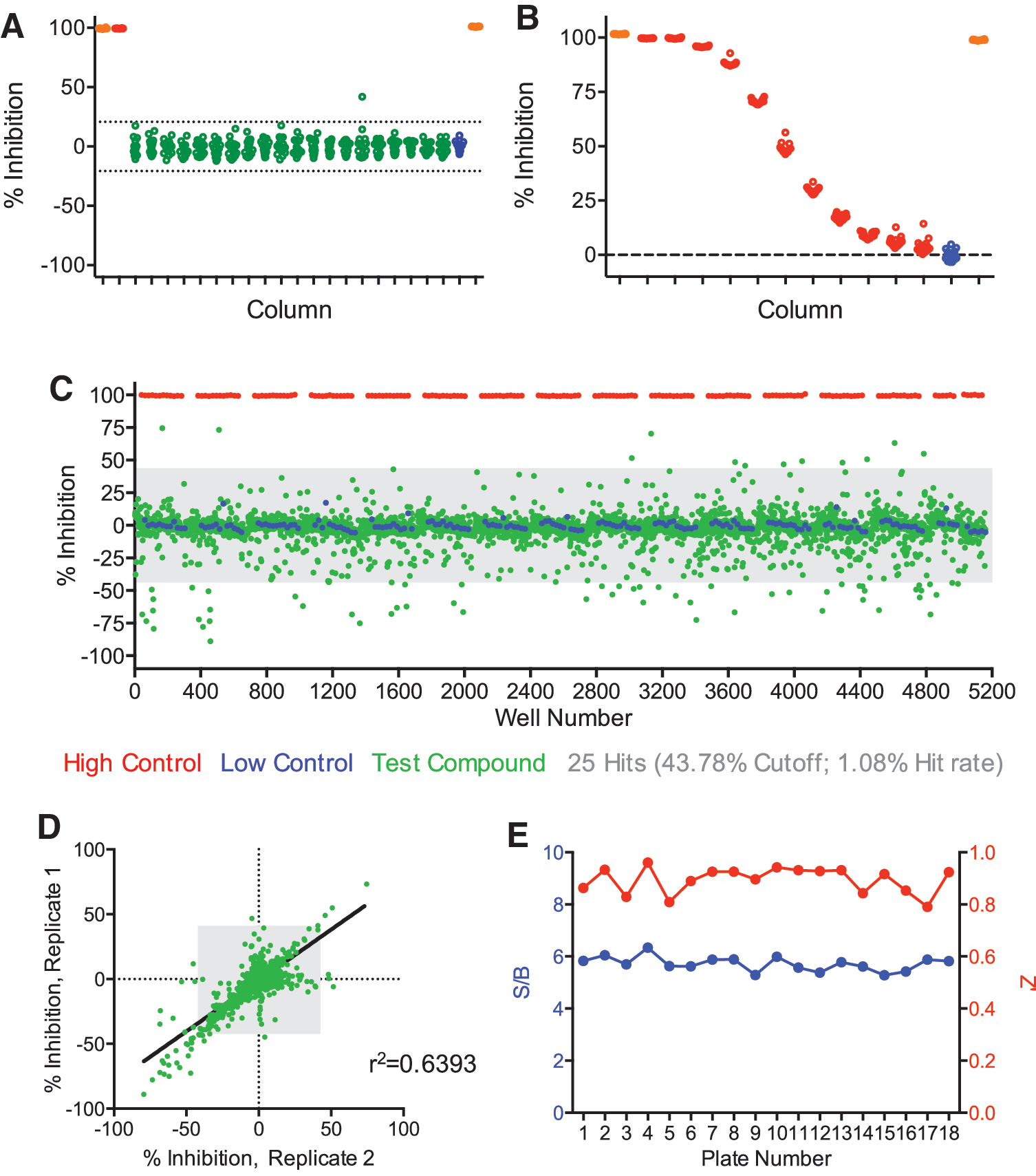
Validation of the assay statistics from duplicate assay of MicroSource Spectrum library.
Discussion
Although considerable progress has been made in the development of small molecule modulators of RGS proteins, 14,36,37 we are still in the infancy of developing an in vitro compound toolbox for numerous members of the family and translating these efforts toward in vivo applications. One hurdle has been the implementation of robust screening approaches. In this study, we describe a novel biochemical assay for high-throughput identification of in vitro modulators of macromolecular complex transitions between RGS7/Gβ5/R7BP. This approach utilizes full-length recombinant proteins for increased translational efficiency toward orthogonal cell-based validations. The assay can be performed rapidly using automated reagent dispensing and liquid handling. By applying a stringent data analysis rubric, this assay demonstrated extremely robust assay statistics, had a high level of sensitivity, and generated very few false positives. Moreover, we demonstrated screening feasibility by testing two libraries of active pharmacological compounds that encompass both a diverse range of chemical structures as well as in vivo drug targets.
An important feature of targeting transition states within the RGS macromolecular complex was a high level of sensitivity that enabled hit identification in both the positive and negative direction of TR-FRET ratio change. Therefore, we were able to detect a spectrum of modulation by small molecules over the RGS7/Gβ5/R7BP complex. To generate a robust change in signal, we think that the assay hits need to induce substantial conformation changes within the R7BP/RGS7/Gβ5 complex, which translate into changes in FRET pair orientations either enhancing or lowering energy transfer efficiency.
The observed changes in FRET efficiency induced by these small molecules underscore their putative biological impact on the RGS complex, which likely serve as either activators or inhibitors of RGS activity. The conceptual theme of RGS activators is grounded in the observation that RGS7 binding with R7BP enhances catalytic activity toward its physiological substrates Gαo and Gαi. 25,26 Therefore, it is plausible that endogenous RGS7 activity could be potentiated through modulation by an appropriate small molecule. Indeed, peptide fragments that bind RGS7 have been identified as allosteric modulators of catalytic activity in solution. 38 On the other hand, inhibitors of RGS activity would provide a means to reduce GAP activity toward Gαi/o, which would prolong downstream signaling events. Given the physiological roles of R7BP/RGS7/Gβ5 complex, such pharmacotherapies could be useful in contextual learning and memory 17 or in the avoidance of opioid tolerance and dependence. 16,32
Although our assay may have sensitivity to reveal R7BP/RGS7/Gβ5 modulators, it is important to note that the directionality in TR-FRET ratio change will likely have no correlation in terms of classifying small molecules as “activators” or “inhibitors.” Therefore secondary assays examining the mechanisms of identified hits will assist in binning their classification as well as defining their utility. One such assay could be analysis of RGS GAP activity in vitro 39 –41 ; however, these may be limited by laborious use of radioactive material or the requirement for using a mutated Gα. Therefore, an orthogonal cell-based approach capable of reporting RGS7 activity may be better suited for hit confirmation. One such system with a power to further quickly eliminate false positives utilized Bioluminescence Resonance Energy Transfer to monitor transitions in G proteins assisted by RGS proteins upon termination of GPCR signaling. 25,42 Thus, a cell-based follow-up would offer the advantage of probing off-target effects, compatibility with endogenous RGS complex, and viability for sustained cellular penetration.
Collectively, the advent of increasing the number of druggable targets in clinically relevant signaling pathways will be beneficial to the medical community. The presentation of our assay here is step toward this future. We present the first approach toward direct modulation of R7 RGS protein complexes with a novel rationale of targeting interaction transitions between complex subunits. Therefore in addition to drug screening, this assay can also be adapted toward uncovering interaction mechanisms with other proteins that interact R7BP/RGS7/Gβ5 such as receptors (GPR158) 43 or ion channels (GIRK). 28,33
Footnotes
Acknowledgments
B.S.M. designed experiments, performed the experiments, organized the data, and wrote the article. D.N.P. performed purification of 6xHis-RGS7/Gβ5 and GST-R7BP, and untagged-R7BP. F.M. designed experiments and organized data. J.F. designed experiments and organized data. L.S. designed experiments and organized data. T.P.S. designed experiments and organized data. K.A.M. designed the study, analyzed data, and wrote the article. We would like to thank Smitha Kota and Joseph Kissil (The Scripps Research Institute) for providing the MicroSource Spectrum compound library. This work was supported by National Institutes of Health Grants DA041207 (to B.S.M.), DA042746 (to K.A.M. and L.S.), and DA026405 (to K.A.M.).
Disclosure Statement
J.F. is employed by Cisbio US, who was manufacturer of the TR-FRET antibodies used in this work. The authors declare that no competing financials interests exist.